

Abstract
Two primary prevention trials unexpectedly demonstrated adverse effects of supplemental β-carotene on lung cancer incidence in cigarette smokers. To elucidate the molecular mechanisms that might underlie these effects, we studied the immunohistochemical expression of cytochrome P450 (CYP) 1A1, 1A2, and 2E1, retinoic acid receptor-β (RAR-β), activated protein-1 (AP-1) elements, cyclin D1, and Ki67 in lung tumors and, when available, adjacent normal tissues obtained from incident cases in the Alpha-Tocopherol, Beta-Carotene Cancer Prevention (ATBC) Study. Archival lung tissue was available from 52 men randomized to receive 20 mg of β-carotene per day and 30 men randomized to the placebo arm, all of whom were diagnosed with incident non-small cell lung carcinoma during the course of the trial and subsequently underwent radical pulmonary resection. In normal appearing bronchial epithelium, positive staining for cyclin D1 was observed in 23% of cases in the β-carotene group and 0% of cases in the placebo group (based on only 3 of 13 versus 0 of 11 cases staining positively, however; p=0.04), with no differences in expression noted in lung tumor tissue (p=0.48). There were no statistically significant differences in Ki67 expression in normal or cancerous lung tissue between intervention groups, although a small increase in staining was noted among cases in the β-carotene versus placebo group (88% versus 71% of cases stained positive, respectively; p=0.13). Contrary to expectation, β-carotene supplementation had no apparent effect on RAR-β expression. These findings suggest that male smokers supplemented with β-carotene may have had an increased risk of lung cancer due to aberrant cell growth, although our results are based on a relatively small number of cases and require confirmation in other completed trials of β-carotene supplementation.
Keywords: β-carotene, immunohistochemistry, lung cancer, smoking, supplementation
Introduction
A vast amount of observational epidemiologic data has accumulated in support of a beneficial association between higher dietary intakes and circulating levels of β-carotene and lung cancer risk (1). Despite this evidence, controlled trials demonstrated either harmful (2–4) or no (5) effects of β-carotene supplementation on lung cancer incidence and mortality. In the Alpha-Tocopherol, Beta-Carotene Cancer Prevention (ATBC) Study, statistically significant increases in lung cancer incidence (16%) and overall mortality (8%) were observed among male smokers randomized to receive 20 mg supplemental β-carotene daily compared to those that did not receive β-carotene (2, 3). Similar results were obtained from the Beta-Carotene and Retinol Efficacy Trial (CARET), in which men and women randomized to a daily combination of β-carotene (30 mg) and retinyl palmitate (25,000 IU) experienced a 28% increase in lung cancer incidence and a 17% increase in total mortality over those receiving placebo (4). In both trials, the adverse effects were limited to heavier cigarette smokers (3, 6). While discrepancies between the observational and trial data have been discussed (7, 8), the biological mechanisms underlying these unexpected adverse effects have not been adequately elucidated.
A series of experiments conducted in ferrets have provided the strongest leads to date regarding the observed interactions among β-carotene, cigarette smoking, and lung tumorigenesis. Animals simultaneously fed pharmacological doses of β-carotene (equivalent to 30 mg/day in humans) and exposed to cigarette smoke for 6 months had increased expression of several phase I carcinogen-metabolizing enzymes, including cytochrome P450’s (CYP) 1A1 and 1A2, reduced retinoid signaling due to lower retinoic acid concentrations and retinoic acid receptor (RAR)-β expression, and increased indicators of cellular proliferation in comparison to animals that were not exposed to β-carotene and cigarette smoke (9–11).
In order to determine whether similar molecular alterations occur in lung tissue in humans, we examined protein expression in a series of non-small cell lung cancers (NSCLC) and adjacent normal tissues from smokers who received a β-carotene supplement or placebo for several years in the ATBC Study.
Materials and Methods
Study population
The ATBC Study was a randomized, double-blind, placebo-controlled, primary prevention trial that tested whether daily supplementation with β-carotene (20 mg) and/or vitamin E (50 mg dl-α-tocopheryl acetate) reduced the incidence of lung and other cancers. Details regarding study design, methods, participant characteristics, and compliance have been reported (12). Briefly, 29,133 Finnish male smokers ages 50–69 years old and meeting all eligibility criteria were successfully randomized to one of four intervention groups based on the 2×2 factorial design (α-tocopherol + β-carotene, α-tocopherol alone, β-carotene alone, or a placebo). The trial ended on April 30, 1993 after 5 – 8 years of active intervention (median = 6.1 years). The institutional review boards of both the National Public Health Institute of Finland and the U.S. National Cancer Institute approved the study, and written, informed consent was obtained from each participant.
Data collection
Prior to randomization, all subjects were asked to provide detailed demographic, smoking, and occupational information, give a medical history, complete a 276-item food use questionnaire, and provide an overnight fasting blood sample that was subsequently stored at −70°C until analyzed. A follow-up blood sample was collected at three years, and baseline and on-study concentrations of α-tocopherol, β-carotene, and retinol were determined using high-performance liquid chromatography (13).
Case ascertainment
Incident cancers of the lung (ICD-9 code 161) were identified through the Finnish Cancer Registry, which provides close to 100% case ascertainment nationwide (14). Administration of a chest radiograph at baseline ensured that men with apparent lung cancer were excluded from the trial, and periodic chest radiographs taken during the trial and at study exit facilitated ascertainment of lung cancer cases. All relevant medical records were obtained and reviewed independently by two study physicians. Pathology and cytology specimens were evaluated by two pathologists.
We selected a representative sample of trial-period NSCLC cases from the β-carotene alone (n=52; 70% of all surgical NSCLC cases in this subgroup) and placebo (n=30; 60% of all surgical NSCLC cases in this subgroup) intervention groups, all of whom underwent surgical resection without prior radiotherapy or chemotherapy, for immunohistochemical study. Formalin-fixed, paraffin-embedded tissue blocks were collected from hospitals where study participants underwent surgical resection. Blocks were then transferred to the Finnish Institute of Occupational Health (FIOH), where a 4-μm section was cut from each of 1–5 blocks per case and stained with hematoxylin-eosin for pathologic confirmation and assessment of the presence and proportion of tumor (relative to normal) tissue. Blocks containing 30% or more of tumor tissue were selected for this immunohistochemical study, and 10 4-μm consecutive sections were cut from a representative block for each case.
Antibodies and immunohistochemistry
Immunohistochemical procedures for all antibodies except RAR-β were performed on the Ventana NexES automated immunostainer platform at 37°C using the I-VIEW (Ventana Medical Systems, Tucson, AZ) enhanced DAB kit with the addition of an Avidin/Biotin blocking kit. Negative control sera was either mouse ascites with protein concentration equal to the primary antibody (for Ki-67, cyclin D1, and RAR-β) or primary antibody diluent (CYP1A1, CYP1A2, and CYP2E1).
CYP1A1, CYP1A2, and CYP2E1: Epitope retrieval (RDI, Concord, MA) required enzyme digestion in Pronase (Chemicon/Invitrogen, Carlsbad, CA) in PBS pH 7.6 at 0.5 mg/mL for 7 minutes at room temperature. Antibodies were incubated for 32 minutes (1:1000 for CYP1A1 and CYP1A2 and 1:75 for CYP2E1). CYP2E1 signal required an Amplifier kit (Ventana). Pancreas islet cells were used as positive controls for all three markers. Ki67: Epitope retrieval (Ventana) was performed in Biocare’s Decloacker chamber (Biocare Medical, Concord, CA) pressure cooker for 5 minutes at 125° C (22 psi). Prediluted Ki67 antibodies were incubated for 28 minutes. Normal tonsil tissue was used as a positive control. Cyclin D1: Sections were retrieved in 10mM sodium citrate/citric acid buffer, pH 5.8, for 5 minutes in Biocare’s Decloaking chamber at 125° C (22 psi) and subsequently incubated for 32 minutes in a prediluted antibody from Ventana; the signal was amplified with a kit containing rabbit anti-mouse heavy and light chains (Amplifier A) and mouse anti-rabbit heavy and light chains (Amplifier B). Breast carcinoma was used as the positive control. AP-1: The AP-1 antibody was directed against a C-terminal peptide of c-Jun and recognized both the c-Jun homodimer and the c-Jun/c-Fos heterodimer. Working citrate buffer retrieval for 10 minutes in the Decloaker and incubation at 1:100 for 20 minutes was required to detect the AP-1 antigen targeted by the Sigma-Aldrich (St. Louis, MO) primary in a colon carcinoma control. RAR-β: Antigens to RAR-β (LabVision/ThermoFisher, Fremont, CA) were retrieved in 10mM sodium citrate/citric acid buffer pH 5.8 for 15 minutes in Biocare’s Decloaking chamber at 125° C (22 psi). All incubations were performed at ambient temperature. Endogenous peroxidase was quenched with 3% aqueous hydrogen peroxide for 10 minutes and the primary antibody incubation lasted for 30 minutes at a concentration of 1:100. A Labvision UltraVision LP Polymer Detection Kit was used at half strength for detection and 5 minutes of Cardassian DAB (Biocare) visualized the bound antibody. Colon carcinoma was used as the positive control.
All slides were counterstained with Mayer’s hematoxylin for 2 minutes except for those stained for Ki-67, which received hematoxylin and eosin under a standard method.
After sections were stained with the appropriate antibody and control, two pathologists blinded to the trial intervention assignment were involved in producing the final scores for each specimen and antibody. In each tissue section, three separate ‘compartments’ where analyzed: tumor, normal airways, and normal type-I alveolar pneumocytes. Not all slides contained both normal and tumor tissue. Within each compartment, an intensity score between 0 and 3 was assigned to the staining based on evaluation of the entire slide. For percentage staining, random high-power fields (total magnification of 400×) were selected within the specified compartment and 200 cells per sample were counted, scoring each cell as positive or negative. The percentage scores were then binned as described below.
Labeling was restricted to nuclei for RAR-β, AP-1, Ki67, CYP1A2, and cyclin D1 and to the cytoplasm for CYP1A1 and CYP2E1. Cases that displayed immunostaining in more than 5% of counted cells in a particular target tissue (i.e., tumor, adjacent normal bronchioles, or adjacent normal pneumocytes) were considered as positive in that tissue. The extent of positive staining was categorized as 6–20%, 21–50%, and > 50% of cells; in alternative analyses, we scored expression as positive or negative only. A four-point scale was used to describe the intensity of immunostaining for all markers except Ki67 and CyclinD1 as follows: 0 (negative), 1 (weak positivity), 2 (moderate positivity), and 3+ (strong positivity). An index that integrated both the extent and intensity of staining was created by summing categorical scores for both measures (maximum possible score = 3 (staining in >50% of cells) + 3 (3+ intensity) = 6).
Statistical analysis
Statistical analyses were performed using SAS software version 8.02 (SAS Institute, Inc., Cary, NC). For each marker, we compared the extent and intensity of expression in both tumor and adjacent normal tissues across trial intervention groups. Wilcoxon rank sum and Fisher’s exact tests were utilized to examine differences in continuous (extent) and categorical (intensity) staining values, respectively, between the two intervention groups. To determine whether the expression of individual markers differed between tumor and surrounding adjacent normal lung tissues in the same histological section, we used the Wilcoxon signed rank test for matched pairs. To address potential confounding, Wilcoxon rank sum and Fisher’s exact tests were employed to test whether several factors, including age, smoking dose and duration, alcohol consumption, body mass index, pre-randomization serum nutrient levels, and history of non-malignant pulmonary disease, differed between the two intervention groups. In addition, correlations between the extent of staining for each marker and the aforementioned covariates were assessed with Spearman correlation coefficients. Stratification of results by lifestyle factors, histological type (squamous cell carcinoma, adenocarcinoma), intervention time, and baseline serum β-carotene concentrations was also performed. All reported p-values are 2-sided.
Results
Characteristics of men diagnosed with lung cancer during the course of the trial and from whom archival lung tissue specimens were collected and analyzed are shown in Table 1 by intervention assignment. There were no significant differences in age at randomization or diagnosis, smoking dose or duration, alcohol consumption, or baseline serum β-carotene concentrations between cases who received the β-carotene supplements and those given placebo. The duration of intervention was also similar in both groups, with most cases participating in the trial for at least 4 ½ years before diagnosis. As expected, men in the β-carotene group experienced an approximate 15-fold elevation in serum β-carotene concentrations after 3 years on-study, whereas there was virtually no change in blood concentrations in the placebo group. Squamous cell carcinoma was the most common histological type in this case series (49%), followed by adenocarcinoma (30%) and other tumor types (21%); this pattern did not differ by intervention assignment.
Table 1.
Lifestyle and clinical characteristics of lung cancer cases according to trial intervention group*
| Placebo (n=30) | β-Carotene (n=52) | p-value† | |
|---|---|---|---|
| Age at randomization, years | 62 (56–64) | 60 (56–65) | 0.93 |
| Age at diagnosis, years | 65 (61–67) | 64 (60–68) | 0.98 |
| Intervention time, days‡ | 1689 (601–2079) | 1716 (981–2243) | 0.30 |
| Cigarettes / day at baseline | 20 (15–25) | 20 (19-30) | 0.55 |
| Years smoked at baseline | 41 (38–47) | 41 (37–45) | 0.90 |
| Smoking cessation§ | 5 (17) | 9 (17) | 0.95 |
| Alcohol intake at baseline, g/day | 10.7 (5.5–18.6) | 10.7 (2.7–34.5) | 0.71 |
| Baseline serum β-carotene, µg/L | 167 (109–282) | 180 (102–247) | 0.95 |
| On-study serum β-carotene, µg/L║ | 150 (118–236) | 2804 (1785–3375) | <0.0001 |
| Histology | 0.81 | ||
| Squamous cell carcinoma | 14 (47) | 26 (50) | |
| Adenocarcinoma | 11 (37) | 14 (27) | |
| Other | 5 (16) | 12 (23) |
Median (interquartile range) for continuous variables and number (percent) for categorical variables
From the Wilcoxon rank sum test for continuous variables and Fisher’s exact test for categorical variables
Time from baseline to last study visit
Defined as having stopped smoking for at least 1 year during the trial
Measured 3 years after randomization
As expected, the extent of cyclin D1 and Ki67 staining was greater in tumor as compared with normal appearing pulmonary tissue in both intervention groups (Table 2, all matched-pair p-values < 0.05). CYP1A1 and CYP1A2 expression were also significantly higher in tumor tissue as compared with normal adjacent pneumocytes (all matched-pair p-values < 0.05), but not when compared with normal bronchiolar epithelium. Finally, RAR-β expression was higher in tumor as compared to both normal bronchiolar (matched-pair p-value=0.02) and normal pneumocytes (matched-pair p-value <0.0001) among men randomized to receive the β-carotene supplement, but was only greater in tumor versus normal pneumocytes (matched-pair p-value=0.02) in placebo group cases.
Table 2.
Number (%) of lung cancer cases according to extent of immunostaining for selected markers in tumor and adjacent uninvolved tissue, by trial intervention group
| Placebo |
β-Carotene |
p-value* | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Negative staining |
Positive staining | Negative staining |
Positive staining | |||||||||
| Marker | Total cases |
≤5% | 6–20% | 21–50% | >50% | Any positive |
≤5% | 6–20% | 21–50% | >50% | Any positive |
|
| AP-1 | ||||||||||||
| Tumor | 80 | 28 (100) | 0 | 0 | 0 | 0 | 49 (94) | 0 | 3 (6) | 0 | 3 (6) | 0.97 |
| Normal, bronchioles | 25 | 10 (100) | 0 | 0 | 0 | 0 | 15 (100) | 0 | 0 | 0 | 0 | 0.47 |
| Normal, pneumocytes | 37 | 15 (100) | 0 | 0 | 0 | 0 | 22 (100) | 0 | 0 | 0 | 0 | 0.99 |
| Cyclin D1 | ||||||||||||
| Tumor | 80 | 7 (25) | 9 (32) | 10 (36) | 2 (7) | 21 (75) | 20 (38) | 11 (21) | 18 (35) | 3 (6) | 32 (62) | 0.48 |
| Normal, bronchioles | 24 | 11 (100) | 0 | 0 | 0 | 0 | 10 (77) | 2 (15) | 1 (8) | 0 | 3 (23) | 0.04 |
| Normal, pneumocytes | 30 | 13 (100) | 0 | 0 | 0 | 0 | 13 (76) | 4 (24) | 0 | 0 | 4 (24) | 0.15 |
| CYP1A1 | ||||||||||||
| Tumor | 79 | 7 (25) | 2 (7) | 8 (29) | 11 (39) | 21 (75) | 12 (24) | 3 (6) | 19 (37) | 17 (33) | 39 (76) | 0.99 |
| Normal, bronchioles | 20 | 7 (70) | 0 | 0 | 3 (30) | 3 (30) | 8 (80) | 0 | 0 | 2 (20) | 2 (20) | 0.99 |
| Normal, pneumocytes | 41 | 11 (69) | 0 | 3 (19) | 2 (13) | 5 (31) | 20 (80) | 1 (4) | 2 (8) | 2 (8) | 5 (20) | 0.41 |
| CYP1A2 | ||||||||||||
| Tumor | 79 | 0 | 2 (7) | 7 (25) | 19 (68) | 28 (100) | 7 (14) | 4 (8) | 11 (22) | 29 (57) | 44 (86) | 0.19 |
| Normal, bronchioles | 20 | 2 (29) | 0 | 2 (29) | 3 (43) | 5 (71) | 3 (23) | 0 | 0 | 10 (77) | 10 (77) | 0.27 |
| Normal, pneumocytes | 38 | 2 (14) | 0 | 9 (64) | 3 (21) | 12 (86) | 5 (21) | 0 | 16 (67) | 3 (13) | 19 (79) | 0.52 |
| CYP2E1 | ||||||||||||
| Tumor | 80 | 1 (4) | 1 (4) | 4 (14) | 22 (79) | 27 (96) | 10 (19) | 3 (6) | 11 (21) | 28 (54) | 42 (81) | 0.07 |
| Normal, bronchioles | 32 | 3 (23) | 0 | 0 | 10 (77) | 10 (77) | 2 (11) | 0 | 0 | 17 (89) | 17 (89) | 0.67 |
| Normal, pneumocytes | 51 | 3 (16) | 0 | 0 | 16 (84) | 16 (84) | 2 (6) | 0 | 0 | 30 (94) | 30 (94) | 0.89 |
| Ki67 | ||||||||||||
| Tumor | 79 | 8 (29) | 9 (32) | 8 (29) | 3 (11) | 20 (71) | 6 (12) | 18 (35) | 23 (45) | 4 (8) | 45 (88) | 0.13 |
| Normal, bronchioles | 32 | 13 (93) | 1 (7) | 0 | 0 | 1 (7) | 17 (94) | 1 (6) | 0 | 0 | 1 (6) | 0.76 |
| Normal, pneumocytes | 47 | 17 (100) | 0 | 0 | 0 | 0 | 30 (100) | 0 | 0 | 0 | 0 | 0.38 |
| RAR-β | ||||||||||||
| Tumor | 79 | 14 (50) | 0 | 5 (18) | 9 (32) | 14 (50) | 22 (43) | 5 (10) | 9 (18) | 15 (29) | 29 (57) | 0.86 |
| Normal, bronchioles | 25 | 8 (73) | 0 | 0 | 3 (27) | 3 (27) | 12 (86) | 0 | 0 | 2 (14) | 2 (14) | 0.46 |
| Normal, pneumocytes | 42 | 18 (100) | 0 | 0 | 0 | 0 | 23 (96) | 1 (4) | 0 | 0 | 1 (4) | 0.42 |
From the Wilcoxon rank sum test for differences in the extent of marker staining (continuous values) between intervention groups.
Cyclin D1 was expressed in lung tumor tissue in 32 of 52 cases (62%) and 21 of 28 (75%) cases in the β-carotene and placebo groups, respectively (Table 2). Of note was the total absence of cyclin D1 expression in the adjacent normal bronchiolar epithelium in cases randomized to placebo, whereas reactivity was significantly higher in normal bronchioles (p-value=0.04) and suggestively higher in normal pneumocytes (p-value=0.15) in β-carotene group cases. Ki67 staining was virtually absent in the normal lung tissue, whereas most tumors expressed this proliferation marker, particularly in the β-carotene group [45 out of 51 cases (88%) as compared to those in the placebo group (20 out of 28 cases (71%)) (p-value=0.13)]. The immunohistochemical staining patterns for cyclin D1 and Ki67 from representative cases with both tumor and adjacent normal tissues present are shown in Figures 1 and 2.
Figure 1.
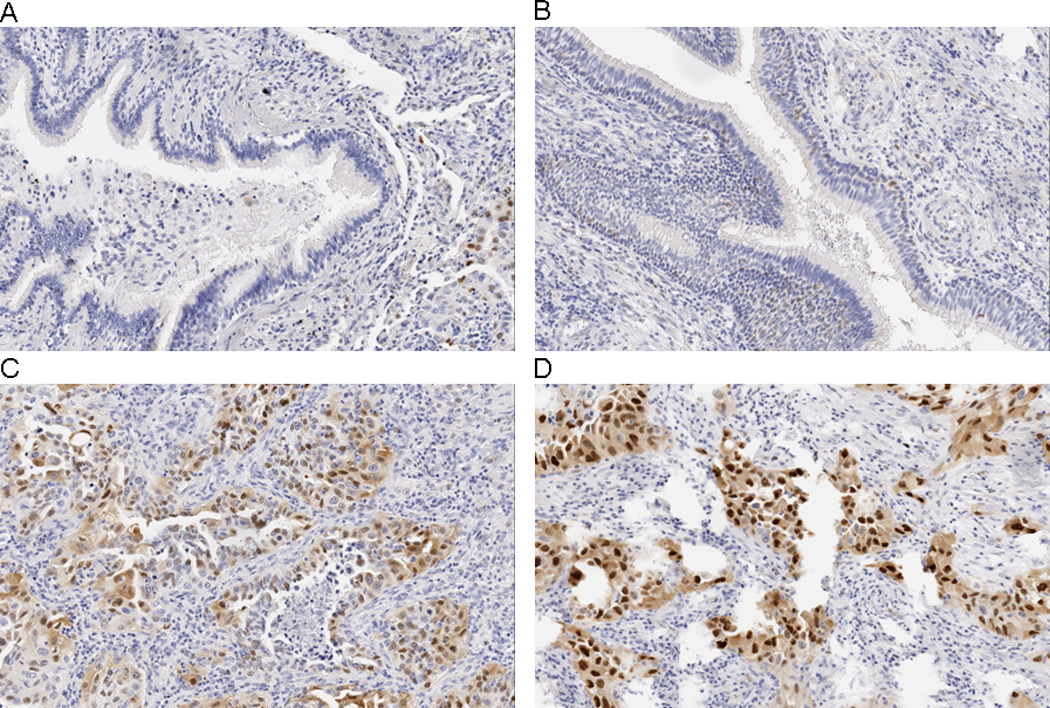
Immunohistochemical localization of cyclin D1 in A) normal bronchial epithelium, placebo group, B) normal bronchial epithelium, β-carotene group, C) lung tumor cells, placebo group, and D) lung tumor cells, β-carotene group. In Panel A, a bronchial lumen filled with mucus and lined by tall columnar ciliated epithelium is shown. Cyclin D1 immunoreactivity is essentially absent, with the exception of rare scattered nuclei. Panel B shows numerous positive-staining nuclei within the basal layer and in regions of pseudo-stratification of the columnar ciliated bronchiolar-type epithelium. Tumor cells in Panel C and Panel D demonstrate frequent strong nuclear positivity for cyclin D1 along with some diffuse cytoplasmic staining. In contrast, stromal cells and chronic inflammatory cells show rare cyclin D1 staining in these latter panels. All photomicrographs are 20× magnification.
Figure 2.
Immunohistochemical localization of Ki67 in A) normal bronchial epithelium, placebo group, B) normal bronchial epithelium, β-carotene group, C) lung tumor cells, placebo group, and D) lung tumor cells, β-carotene group. In Panels A and B, nuclear positivity for Ki67 is essentially absent in columnar ciliated bronchiolar-type epithelium. In Panel C, only scattered malignant cells show nuclear staining for Ki67, whereas numerous positive-staining nuclei are apparent in Panel D. Rare scattered inflammatory cells are also positive for Ki67 in Panel D. All photomicrographs are 20× magnification.
For CYP1A2 and CYP2E1, there were modest differences between the supplementation groups in the prevalence of positive staining in tumor tissue, with the placebo group showing somewhat greater positivity than the β-carotene group in each instance (p-values=0.19 and 0.07, respectively). RAR-β was not expressed in normal pneumocytes, was present in a small fraction of normal bronchiolar cells, and was expressed in at least half of the tumor samples, albeit with no differences between the β-carotene and placebo groups.
Results pertaining to the intensity of marker expression corresponded closely with those reported for the extent of positive staining (data not shown). Results from analyses using an index that integrated both parameters were also virtually identical to those reported in Table 2 (data not shown).
In order to address potential confounding of the aforementioned findings by demographic, lifestyle, and clinical factors, correlations between the extent of staining for each marker and age, smoking intensity and duration, baseline serum antioxidant concentrations, alcohol consumption, body mass index, and history of nonmalignant pulmonary conditions, including asthma, emphysema, and chronic bronchitis, were examined. The majority of markers were not significantly correlated with any of these factors in either tumor or normal tissues (Spearman p-values > 0.05), and none of the covariates examined differed by β-carotene supplementation status (data not shown).
In analyses stratified by the two primary histological types of lung cancer, differences in cyclin D1 expression in normal epithelial tissues between intervention groups were only evident in cases diagnosed with adenocarcinoma during the trial: 2 of 5 (40%) and 0 of 7 (0%) adenocarcinoma cases demonstrated positive cyclin D1 staining in normal bronchiolar epithelium in the β-carotene and placebo groups, respectively (p-value=0.06), and 3 of 8 (38%) and 0 of 8 (0%) normal pneumocytes expressed cyclin D1 in the β-carotene and placebo groups, respectively (p-value=0.10). In other stratified analyses, the difference in Ki67 tumor positivity between supplementation groups was apparent only among those cases with a longer trial intervention time (>4.7 years; 93% of cases in the β-carotene intervention group and 54% of cases in the placebo arm exhibited Ki67 tumor positivity, respectively; p-value = 0.0009). No notable effect modification was observed for any of the other markers.
Discussion
This investigation is nested within one of a small number of primary lung cancer prevention trials that can examine the biological mechanisms responsible for the adverse effects of long-term β-carotene supplementation in smokers. Consistent with results from prior animal studies, we found some evidence for more aberrant cell growth in the respiratory epithelium of smokers supplemented with β-carotene compared to those randomized to placebo. These changes were indicated by modest differences in the expression of cyclin D1 in normal lung tissues between the intervention groups. We also observed somewhat suggestive, yet statistically non-significant, increases in Ki67 expression in lung tumors among cases randomized to the β-carotene versus placebo group. By contrast, β-carotene supplementation had no effect on RAR-β, CYP1A1, or AP-1 expression in our study.
In the only other study to date that has examined similar hypotheses within the context of a randomized supplementation trial, Liu et al. found no significant differences in cyclin D1, proliferating cellular nuclear antigen (PCNA), RAR-β, or CYP1A1 staining in lung tumor tissues from a small subset of β-carotene versus placebo group cases within the Physicians’ Health Study (PHS) (15). It is important to note that, in contrast to ATBC and CARET, PHS showed no effect of β-carotene supplementation on lung cancer incidence (5), and it is therefore not surprising that the immunohistochemical results of Liu et al. were consistent with the main trial finding.
Cyclin D1 is an important regulator of cell cycle progression in normal cells, and its overexpression leads to shortened transition through the G1 phase and uncontrolled growth and proliferation (16). Ki67 is only expressed by proliferating cells and is therefore an established indicator of rapid cell growth (17). Several reports have shown that Ki67 can be detected in the normal appearing lung tissue of smokers (18–20), and a recent review concluded that Ki67 is an independent predictor of prognosis in NSCLC (21). Since proliferation is a direct result of more rapid progression through the cell cycle, we expected concordant staining between the two markers, which has been previously reported (22). Instead, we found that compared to the placebo group, men receiving β-carotene had higher cyclin D1, but not Ki67, expression in normal appearing lung epithelium and slightly higher Ki67, but not cyclin D1, expression in malignant tissue. Of note was our finding that only 6% of cases exhibited positive Ki67 immunostaining in the normal bronchiolar lung tissue, which is low when compared to several previous reports (18–20). Although this discrepancy may be attributed to our threshold for positive Ki67 immunostaining – greater than 5% of counted cells in a particular target tissue – lowering the cutpoint to >3% of cells staining positive yielded nearly identical results. The discordance between cyclin D1 and Ki67 staining suggests that negative regulators of cyclin D1, such as cyclin-dependent kinase (CDK) inhibitors (including the INK4 and Cip/Kip families of proteins (23)), may be active in lung tumor tissue, and that malignant cells are proliferating in a cyclin D1-independent manner. Additional evaluation of CDK inhibitors such as p21Cip1 and p27Kip1 would be highly informative in this regard. Alternatively, overexpression of cyclin D1 may be influencing processes unrelated to proliferation, or our findings could be due to chance.
In contrast to animal studies that showed diminished retinoid signaling – confirmed by decreases in the expression of retinoic acid receptor β (RARβ) and up-regulation of downstream targets, including activator protein-1 (AP-1) – and increased expression of CYP1A1 and CYP1A2 in ferrets supplemented with β-carotene and simultaneously exposed to tobacco smoke (10, 11), we did not observe an effect of β-carotene supplementation on RAR-β, AP-1, or CYP1A1 expression in the lungs of male smokers. Although we found that expression of CYP1A2 in lung tumor tissues was suggestively lower in β-carotene supplemented versus placebo cases, these differences were not statistically significant. These discrepancies could be due to subtle biological and anatomical differences between the two species and / or differences in study procedures, including specific antibodies used and route of administration of the carcinogenic agents. For example, while the entire ferret lung was exposed to tobacco smoke – yielding peripheral alveolar changes – cigarette smoke concentrates in the central airways in humans. Importantly, none of the ferrets in any exposure group developed lung tumors, although some developed squamous metaplasia (11) – a lesion common in the bronchi of smokers. Differences could also be due to the fact that all ATBC Study participants were smokers, whereas smoke-exposed and non-smoke-exposed ferrets with or without β-carotene supplementation were evaluated in aforementioned experiments.
Loss of expression of RAR-β – a nuclear receptor that mediates the effects of retinoic acid on normal epithelial cell growth and differentiation – is a common event in lung cancer precursor lesions and overt tumors (24–27), and is often ascribed to promoter methylation (28). Contrary to expectation, we observed that RAR-β was upregulated in lung tumors as compared to normal adjacent tissue. This may be explained by failure of our antibody to distinguish between the different RAR-β isoforms; this could be important since the β4 form appears to be oncogenic whereas the β2 variant acts as a tumor suppressor (26).
Our study benefited from the availability and examination of both lung tumor and adjacent normal tissue in many of the cases, although histologically normal appearing epithelial cells may not actually be truly normal due to field cancerization (29). The small sample size may have restricted our ability to detect modest, yet significant, differences in protein expression between the β-carotene and placebo groups; in a similar vein, all stratified analyses were considered completely exploratory. Also, a comparison group supplemented with β-carotene but not having lung cancer (e.g., from a bronchoscopy series) could have been informative but was not available for study. The semi-quantitative nature of the immunohistochemical techniques employed in our study is a known limitation (30, 31). Finally, paraffin-embedded tissue sections may show diminished immunoreactivity over time, which could lead to false-negative staining (32). In our study, decreased antigenicity due to extended storage could, for example, have masked subtle differences in Ki67 staining in the normal bronchial epithelium between trial intervention groups.
In summary, our findings indicate that the higher lung cancer rates observed in smokers randomized to receive supplemental β-carotene in two separate clinical trials may have been due to aberrant cell growth in the respiratory epithelium of these individuals, although our findings certainly require confirmation in CARET or other completed trials of β-carotene supplementation. Importantly, most of the molecular markers dysregulated by the combination of exposure to tobacco smoke and β-carotene supplementation in animal experiments, including RAR-β, were not similarly altered in our investigation. Identification of additional molecular markers of the adverse β-carotene effect on lung carcinogenesis is warranted.
Acknowledgement
Grant support: Intramural Research Program of the National Cancer Institute, NIH
The authors thank Dr. Anna Kannio and Ms. Tuula Suitiala for formalin-fixed, paraffin-embedded lung tissue preparation and quality control.
References
- 1.World Cancer Research Fund/American Institute for Cancer Research. Food, Nutrition, Physical Activity, and the Prevention of Cancer: a Global Perspective. Washington, DC: AICR; 2007. [Google Scholar]
- 2.The Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med. 1994;330:1029–1035. doi: 10.1056/NEJM199404143301501. [DOI] [PubMed] [Google Scholar]
- 3.Albanes D, Heinonen OP, Taylor PR, et al. Alpha-tocopherol and beta-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: effects of base-line characteristics and study compliance. J Natl Cancer Inst. 1996;88:1560–1570. doi: 10.1093/jnci/88.21.1560. [DOI] [PubMed] [Google Scholar]
- 4.Omenn GS, Goodman GE, Thornquist MD, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334:1150–1155. doi: 10.1056/NEJM199605023341802. [DOI] [PubMed] [Google Scholar]
- 5.Hennekens CH, Buring JE, Manson JE, et al. Lack of effect of long-term supplementation with beta carotene on the incidence of malignant neoplasms and cardiovascular disease. N Engl J Med. 1996;334:1145–1149. doi: 10.1056/NEJM199605023341801. [DOI] [PubMed] [Google Scholar]
- 6.Omenn GS, Goodman GE, Thornquist MD, et al. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and Retinol Efficacy Trial. J Natl Cancer Inst. 1996;88:1550–1559. doi: 10.1093/jnci/88.21.1550. [DOI] [PubMed] [Google Scholar]
- 7.Albanes D. Beta-carotene and lung cancer: a case study. Am J Clin Nutr. 1999;69:1345S–1350S. doi: 10.1093/ajcn/69.6.1345S. [DOI] [PubMed] [Google Scholar]
- 8.Meyskens FL, Jr, Szabo E. Diet and cancer: the disconnect between epidemiology and randomized clinical trials. Cancer Epidemiol Biomarkers Prev. 2005;14:1366–1369. doi: 10.1158/1055-9965.EPI-04-0666. [DOI] [PubMed] [Google Scholar]
- 9.Liu C, Russell RM, Wang XD. Exposing ferrets to cigarette smoke and a pharmacological dose of beta-carotene supplementation enhance in vitro retinoic acid catabolism in lungs via induction of cytochrome P450 enzymes. J Nutr. 2003;133:173–179. doi: 10.1093/jn/133.1.173. [DOI] [PubMed] [Google Scholar]
- 10.Wang XD, Liu C, Bronson RT, Smith DE, Krinsky NI, Russell M. Retinoid signaling and activator protein-1 expression in ferrets given beta-carotene supplements and exposed to tobacco smoke. J Natl Cancer Inst. 1999;91:60–66. doi: 10.1093/jnci/91.1.60. [DOI] [PubMed] [Google Scholar]
- 11.Liu C, Wang XD, Bronson RT, Smith DE, Krinsky NI, Russell RM. Effects of physiological versus pharmacological beta-carotene supplementation on cell proliferation and histopathological changes in the lungs of cigarette smoke-exposed ferrets. Carcinogenesis. 2000;21:2245–2253. doi: 10.1093/carcin/21.12.2245. [DOI] [PubMed] [Google Scholar]
- 12.The Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study Group. The alpha-tocopherol, beta-carotene lung cancer prevention study: design, methods, participant characteristics, and compliance. Ann Epidemiol. 1994;4:1–10. doi: 10.1016/1047-2797(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 13.Milne DB, Botnen J. Retinol, alpha-tocopherol, lycopene, and alpha- and beta-carotene simultaneously determined in plasma by isocratic liquid chromatography. Clin Chem. 1986;32:874–876. [PubMed] [Google Scholar]
- 14.Korhonen P, Malila N, Pukkala E, Teppo L, Albanes D, Virtamo J. The Finnish Cancer Registry as follow-up source of a large trial cohort--accuracy and delay. Acta Oncol. 2002;41:381–388. doi: 10.1080/028418602760169442. [DOI] [PubMed] [Google Scholar]
- 15.Liu C, Wang XD, Mucci L, Gaziano JM, Zhang SM. Modulation of lung molecular biomarkers by beta-carotene in the Physicians' Health Study. Cancer. 2009;115:1049–1058. doi: 10.1002/cncr.24061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knudsen KE, Diehl JA, Haiman CA, Knudsen ES. Cyclin D1: polymorphism, aberrant splicing and cancer risk. Oncogene. 2006;25:1620–1628. doi: 10.1038/sj.onc.1209371. [DOI] [PubMed] [Google Scholar]
- 17.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 18.Barsky SH, Roth MD, Kleerup EC, Simmons M, Tashkin DP. Histopathologic and molecular alterations in bronchial epithelium in habitual smokers of marijuana, cocaine, and/or tobacco. J Natl Cancer Inst. 1998;90:1198–1205. doi: 10.1093/jnci/90.16.1198. [DOI] [PubMed] [Google Scholar]
- 19.Lee JJ, Liu D, Lee JS, et al. Long-term impact of smoking on lung epithelial proliferation in current and former smokers. J Natl Cancer Inst. 2001;93:1081–1088. doi: 10.1093/jnci/93.14.1081. [DOI] [PubMed] [Google Scholar]
- 20.Mao JT, Fishbein MC, Adams B, et al. Celecoxib decreases Ki-67 proliferative index in active smokers. Clin Cancer Res. 2006;12:314–320. doi: 10.1158/1078-0432.CCR-05-1440. [DOI] [PubMed] [Google Scholar]
- 21.Pugsley JM, Schmidt RA, Vesselle H. The Ki-67 index and survival in non-small cell lung cancer: a review and relevance to positron emission tomography. Cancer J. 2002;8:222–233. doi: 10.1097/00130404-200205000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Mate JL, Ariza A, Aracil C, et al. Cyclin D1 overexpression in non-small cell lung carcinoma: correlation with Ki67 labelling index and poor cytoplasmic differentiation. J Pathol. 1996;180:395–399. doi: 10.1002/(SICI)1096-9896(199612)180:4<395::AID-PATH688>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 23.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 24.Martinet N, Alla F, Farre G, et al. Retinoic acid receptor and retinoid X receptor alterations in lung cancer precursor lesions. Cancer Res. 2000;60:2869–2875. [PubMed] [Google Scholar]
- 25.Picard E, Seguin C, Monhoven N, et al. Expression of retinoid receptor genes and proteins in non-small-cell lung cancer. J Natl Cancer Inst. 1999;91:1059–1066. doi: 10.1093/jnci/91.12.1059. [DOI] [PubMed] [Google Scholar]
- 26.Xu XC. Tumor-suppressive activity of retinoic acid receptor-beta in cancer. Cancer Lett. 2007;253:14–24. doi: 10.1016/j.canlet.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu XC, Lee JS, Lee JJ, et al. Nuclear retinoid acid receptor beta in bronchial epithelium of smokers before and during chemoprevention. J Natl Cancer Inst. 1999;91:1317–1321. doi: 10.1093/jnci/91.15.1317. [DOI] [PubMed] [Google Scholar]
- 28.Virmani AK, Rathi A, Zochbauer-Muller S, et al. Promoter methylation and silencing of the retinoic acid receptor-beta gene in lung carcinomas. J Natl Cancer Inst. 2000;92:1303–1307. doi: 10.1093/jnci/92.16.1303. [DOI] [PubMed] [Google Scholar]
- 29.Auerbach O, Stout AP, Hammond EC, Garfinkel L. Changes in bronchial epithelium in relation to cigarette smoking and in relation to lung cancer. N Engl J Med. 1961;265:253–267. doi: 10.1056/NEJM196108102650601. [DOI] [PubMed] [Google Scholar]
- 30.Walker RA. Quantification of immunohistochemistry--issues concerning methods, utility and semiquantitative assessment I. Histopathology. 2006;49:406–410. doi: 10.1111/j.1365-2559.2006.02514.x. [DOI] [PubMed] [Google Scholar]
- 31.Taylor CR, Levenson RM. Quantification of immunohistochemistry--issues concerning methods, utility and semiquantitative assessment II. Histopathology. 2006;49:411–424. doi: 10.1111/j.1365-2559.2006.02513.x. [DOI] [PubMed] [Google Scholar]
- 32.Bertheau P, Cazals-Hatem D, Meignin V, et al. Variability of immunohistochemical reactivity on stored paraffin slides. J Clin Pathol. 1998;51:370–374. doi: 10.1136/jcp.51.5.370. [DOI] [PMC free article] [PubMed] [Google Scholar]