

Abstract
The echinocandins caspofungin, micafungin, and anidulafungin, inhibitors of cell wall β-1,3-glucan synthesis, were recently elevated to first-line agents for treating infections due to the azole-refractory yeast Candida glabrata. In Candida albicans, echinocandin resistance is strictly associated with mutations in Fks1, a large integral membrane protein and putative β-1,3-glucan synthase, while mutations in both Fks1 and its paralog Fks2 (but not Fks3) have been associated with resistance in C. glabrata. To further explore their function, regulation, and role in resistance, C. glabrata fks genes were disrupted and subjected to mutational analysis, and their differential regulation was explored. An fks1Δ fks2Δ double disruptant was not able to be generated; otherwise, all three single and remaining two double disruptants displayed normal growth and echinocandin susceptibility, indicating Fks1-Fks2 redundancy. Selection on echinocandin-containing medium for resistant mutants was dependent on strain background: only fks1Δ and fks1Δ fks3Δ strains consistently yielded mutants exhibiting high-level resistance, all with Fks2 hot spot 1 mutations. Thus, Fks1-Fks2 redundancy attenuates the rate of resistance; further analysis showed that it also attenuates the impact of resistance-conferring mutations. Growth of the fks1Δ and, especially, fks1Δ fks3Δ strains was specifically susceptible to the calcineurin inhibitor FK506. Relatedly, FK506 addition or calcineurin gene CMP2 disruption specifically reversed Fks2-mediated resistance of laboratory mutants and clinical isolates. RNA analysis suggests that transcriptional control is not the sole mechanism by which calcineurin modulates Fks2 activity.
INTRODUCTION
In recent decades, the normally commensal yeast Candida glabrata has emerged as a common cause of life-threatening fungal infection, in large part a consequence of its intrinsically low susceptibility or resistance to widely used azole antifungals. The echinocandins caspofungin (CSF; FDA approved in 2001), micafungin (MCF; 2005), and anidulafungin (ANF; 2006) are lipopeptide inhibitors of β-1,3-glucan synthase and, hence, fungal cell wall synthesis (6, 8, 35). They are highly active versus most Candida species, including C. glabrata (32, 37). Consequently, recent guidelines have elevated echinocandins above azoles as first-line agents for treatment of C. glabrata infection (33).
Echinocandin resistance remains a rare event but appears to be increasing in response to increasing clinical use (38, 39, 40, 45). Resistance was initially described in clinical isolates of Candida albicans (8, 34) and associated with mutations in the essential gene FKS1 (also known as GSC1), encoding a large integral membrane protein and the putative β-1,3-glucan synthase (10, 29). To date, 18 distinct mutations in Fks1 of C. albicans or related species, Candida tropicalis and Candida krusei, have been reported, and all but 2 fall within an 8-residue region known as hot spot 1 (Fig. 1). In the genetic model Saccharomyces cerevisiae, resistance-conferring mutations selected in vitro are similarly limited to Fks1 except in fks1Δ disruptants in which comparable mutations occur in the paralog Fks2 (8, 21, 31, 34). Thus, in S. cerevisiae, Fks1 and Fks2 are functionally redundant, albeit only partially since fks1Δ strains grow slowly (19, 27). Furthermore, their expression in S. cerevisiae is differentially regulated: fks1 is constitutive, while fks2 is minimally expressed unless induced by glucose deprivation, pheromone, or calcium in a calcineurin-dependent manner (27). Indeed, the calcineurin inhibitor FK506 sensitivity is the basis for the gene name FKS1 (9). In wild-type (WT), mating-proficient S. cerevisiae, Fks1 is responsible for cell wall synthesis during vegetative growth, while Fks2 and a third paralog, Fks3, share this responsibility during sporulation (19, 20, 27).
Fig 1.

Fks1 and Fks2 hot spot 1 mutations associated with echinocandin resistance in clinical isolates of C. albicans, C. krusei, C. tropicalis (partial sequence), and C. glabrata and laboratory mutants of S. cerevisiae (2, 3, 4, 5, 7, 15, 16, 22, 24, 26, 28, 30, 31, 34, 39, 40, 45). Dots indicate identity at that position to the C. albicans sequence. Mutations are shown beneath the underlined wild-type residue. Δ, deletion.
C. glabrata is an evolutionarily close relative of S. cerevisiae (41), and its genome includes syntenic orthologs of FKS1, FKS2, and FKS3 (24). However, mating and sporulation have never been observed in this haploid yeast, and hence, the respective roles and regulation of these FKS genes are a matter of some interest. An intriguing clue has been provided by analysis of echinocandin resistance mutations in C. glabrata clinical isolates: in contrast to other Candida species and S. cerevisiae, these mutations involve both Fks1 and Fks2 (Fig. 1). Indeed, Fks2 mutations outnumber Fks1 mutations 2 to 1 in this yeast (50 versus 24, respectively) (3, 4, 5, 13, 16, 24, 28, 39, 40, 45). Furthermore, fks1 and fks2 are expressed at comparable levels in most strains examined (16).
Here we employed gene disruption, in vitro selection of resistant mutants, FK506 treatment, and expression analysis to explore the roles of fks1, fks2, and fks3 in C. glabrata growth and echinocandin susceptibility. Consistent with clinical data and in contrast to other yeasts, we observed that fks1 and fks2 are functionally fully redundant, with the latter a preferred target for resistance mutation. We also demonstrate how their differential regulation can be exploited to reverse Fks2-mediated resistance.
MATERIALS AND METHODS
Strains, media, and drugs.
C. glabrata strain 200989 (ura3 trp1Δ his3Δ) (25) was obtained from the American Type Culture Collection (Manassas, VA); the clinical isolates 20409 (24) and DPL23 to DPL41 (16) were previously described. The medium was 1% yeast extract, 2% peptone, and 2% dextrose (YPD) or, where indicated, synthetic defined (SD)-ura or SD-trp (dropout base [DOB] supplemented with complete supplement mixture [CSM]-ura or CSM-trp; Sunrise Science Products, San Diego, CA). ANF (Eraxis) was obtained from Pfizer (New Yori, NY), CSF (Cancidas) was obtained from Merck (Rahway, NJ), MCF (Mycamine) was obtained from Astellas (Deerfield, IL), terbinafine (Lamisil) was obtained from Novartis (East Hanover, NJ), and FK506 was obtained from Tecoland (Edison, NJ). Drug stocks were prepared in 100% dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO) and stored at −20°C. DNA primers (Table 1) were obtained from IDT (Coralville, IA).
Table 1.
DNA primers used in this study
| Application and primera | Sequence (5′–3′)b |
|---|---|
| Gene disruption | |
| CgFKS1c1-URA3F | TCTTTCATTTCTAGAGTTTTATCTTTTTTTTTTGCTCTTGT |
| TTGATATACATTCGCTATGTCGAAAGCTACATATAAGGA | |
| CgFKS1c635-URA3R | AGTGGTAGACAAAATTCTGATTGGATCTCTTAGAGATAGA |
| ATCAAGAAGTAGTATGATTCGGTATTTCACACCGCATAGG | |
| CgFKS2c1-URA3F | CAAGTCTCTATCAGCCAATAAAGGAATAAGAACAAGACAG |
| AAAAAGAAAATTCCAACCATGTCGAAAGCTACATATAAGG | |
| CgFKS2c1898-URA3R | ATTCCTAATTAGAAAAATTCTTGAAAATCATACTCAATTA |
| GGGGATTATCTATTGCCTCTTTAGTTTTGCTGGCCGCATC | |
| CgFKS3c1-TRP1F | AGTTTAGATATTCGTGAGACCAGTGAGGCCGCAGATTTAA |
| TTATATTGGAATTAGGCTAGAGAGTGCACCATAAACGAC | |
| CgFKS3c1841-TRP1R | TTATTGAGTTTCGGTGATTTCAATTCTTCAACTTTAAACG |
| GATCCATCACTCATTACTATTTCTTAGCATTTTTGACGAA | |
| CgCMP2c1-URA3F | TTGAATATATATTCTTGTTGGCACCGACAATATTTTCTAG |
| TGCTTATAAATAAAAGATGTCGAAAGCTACATATAAGG | |
| CgCMP2c576-URA3R | TAAATACGATATCAATATTATTATTTTGCTCTTGAGATGA |
| AAAATAATTCGTGAGTAGGATTAGTTTTGCTGGCCGCATC | |
| PCR screening and sequencing | |
| CgFKS1u198F | TCCCTATCGCTTAGGAAAAGT |
| CgFKS1c558F | GTTGCAGTCGCTACATTGCTA |
| CgFKS1c743R | TAGCGTTCCAGACTTGGGAA |
| CgFKS2u303F | AAGGACATCATACAGCGGTA |
| CgFKS2c594F | GGCCACTGTTTTATTCTTCTCG |
| CgFKS2c787R | GTAAATGTTCTCTGTACATGGA |
| CgFKS3u97F | ACTCTAATAACCTTGGAACTAG |
| CgCMP2u190F | CAGAGGATATCCCTTACCCA |
| CgCMP2c443R | TCAGGATCTCCACCAATCTC |
| CgFPR1u324F | AGCCCAAAATACGCCATTAAC |
| CgFPR1d84R | GTGATAAGGTTGTAAAGCGCA |
| ScURA3c117R | CTGCCCATTCTGCTATTCTG |
| ScTRP1c115R | TGGCAAACCGAGGAACTCTT |
| qRT-PCR | |
| CgFKS1c67F | TACCAACCAGAAGACCAACAGAATGG |
| CgFKS1c146R | TCACCACCGCTGATGTTTGGGT |
| CgFKS2c27F | CAATTGGCAGAACACCGATCCCAA |
| CgFKS2c172R | AGTTGGGTTGTCCGTACTCATCGT |
| CgFKS3c505F | GGGAGAGCACGTAAACGTAACTCAA |
| CgFKS3c671R | TTTGCTGCTGTAAGGTTAGTGGCG |
| CgURA3c210F | CGAGAACACTGTTAAGCCATTG |
| CgURA3c370R | CACCATGAGCGTTGGTGATA |
c, coding sequence; u, upstream (the number represents the nucleotide position of the 5′ end relative to the start codon); d, downstream (the number represents the nucleotide position of the 5′ end relative to the stop codon).
Underlined nucleotides indicate URA3 or TRP1 sequences from pRS416 or pRS414, respectively.
Construction of fksΔ and cmp2Δ disruptants.
The PRODIGE method was used to disrupt the C. glabrata orthologs of FKS1 and FKS2 (GenBank accession numbers XM_446406 and XM_448401, respectively) in strain 200989 essentially as described previously (12). Briefly, plasmid pRS416 (GenBank accession number U03450) was used as the template with Taq polymerase (New England BioLabs, Ipswich, MA) to amplify disruption cassettes consisting of the URA3 coding region fused to sequences immediately upstream of the FKS1 or FKS2 start codon (primer CgFKS1c1-URA3F or CgFKS2c1-URA3F, respectively) and sequences immediately downstream of FKS1 codon 635 or the FKS2 stop codon (CgFKS1c635-URA3R or CgFKS2c1898-URA3R, respectively). A cmp2Δ (GenBank accession number XM_449251) disruption cassette was similarly amplified (primers CgCMP2c1-URA3F and CgCMP2c576-URA3R). Since FKS3 is minimally expressed (16), in lieu of the PRODIGE method, a conventional disruption cassette was amplified with a plasmid pRS414 template (GenBank accession number U03448) which fused FKS3 upstream (primer CgFKS3c1-TRP1F) and downstream (CgFKS3c1841-TRP1R) sequences to TRP1 coding plus flanking sequences. Following transformation of these PCR products into strain 200989 with selection on SD-ura or SD-trp, colonies were streaked for isolation and screened by PCR with appropriate primer pairs (Table 1) and sequencing (see below).
Susceptibility assays.
MIC (≥80% inhibition relative to the drug-free control) was determined by broth microdilution in YPD medium with incubation at 35°C for 24 h as described previously (44). Where indicated, FK506 was added to cultures prior to aliquoting to the 96-well plates. Selected assays were repeated in RPMI 1640 (Sigma-Aldrich) supplemented with 2% dextrose, 0.165 M MOPS (morpholinepropanesulfonic acid) (pH 7), and CSM (Sunrise Science Products).
Selection and characterization of resistant mutants.
Mutant colonies were selected on echinocandin-containing agar medium overlaid with 1 × 108 cells of the indicated strain, followed by incubation at 35°C for 3 to 4 days, and the three largest colonies were streaked for isolation on drug-free plates. Susceptibility assays were used to identify stably resistant mutants, and the two exhibiting the highest MICs were further analyzed. Hotspot 1 regions of FKS1 and FKS2 were subsequently amplified (24), and PCR products were directly sequenced (Genewiz, South Plainfield, NJ) following treatment with exonuclease I plus thermosensitive alkaline phosphatase (New England BioLabs).
Mutants of the C. glabrata ortholog (GenBank accession number XM_448641) of S. cerevisiae FPR1 were selected on medium containing the synergistic combination of 16 μg/ml terbinafine plus 0.3 μg/ml FK506, followed by screening for unaltered terbinafine susceptibility; amplification and sequencing (Table 1) of two mutants identified an F106L substitution and a single base deletion within codon K78.
Expression analysis.
RNA was prepared from log-phase cultures (treated as indicated) using hot SDS-phenol extraction as previously described (44). FKS expression was assessed using one-step SYBR green quantitative reverse transcription-PCR (qRT-PCR) with the Mx3005P multiplex quantitative PCR system (Stratagene, La Jolla, CA), as previously described (14). Primers are listed in Table 1. Assays were performed in triplicate, and expression ratios were calculated using the Pfaffl method (36).
RESULTS
Disruption of C. glabrata Fks genes minimally affects growth and echinocandin susceptibility.
For these studies, we employed C. glabrata strain 200989 (25), a ura3 trp1Δ his3Δ derivative of CBS138 (ATCC 2001), the strain selected for genome sequencing (11). This auxotrophic strain grows well in standard YPD medium but not in RPMI, the standard for susceptibility assays in the clinical laboratory. Hence, YPD was routinely employed; representative susceptibility assays repeated in supplemented RPMI medium generated qualitatively similar results (not shown). Disruptants were generated with a ura3 or trp1 marker or a combination of the two. Compared to their 200989 parent, the single disruptants fks1Δ, fks2Δ, and fks3Δ had no apparent effect on growth; this was also true for the double disruptants fks1Δ fks3Δ and fks2Δ fks3Δ (data not shown). These C. glabrata results are similar to what has been described for S. cerevisiae, with the exception that fks1Δ disruptants of the latter yeast show a partial growth defect. In S. cerevisiae, fks1Δ fks2Δ double disruptants are synthetically lethal, indicating that these genes are redundant for an essential function (27). Similarly, repeated attempts to generate a C. glabrata fks1Δ fks2Δ double disruptant with trp1 and ura3 markers failed (data not shown).
The parent strain 200989 exhibited MICs for ANF, CSF, and MCF of 0.016, 0.06, and 0.016 μg/ml, respectively. None of the single or double disruptants exhibited a significant (>2-fold) change in susceptibility to any of the three echinocandins. This contrasts with the 16-fold increase in CSF susceptibility of an S. cerevisiae fks1Δ strain (21, 23).
Rates and impacts of resistance-conferring mutations are altered in fks1Δ strains.
Although fks disruption did not alter echinocandin susceptibility, we hypothesized that the loss of Fks1-Fks2 redundancy would alter the rate and magnitude of echinocandin resistance. To test this, 1 × 108 cells of each strain were spread on YPD plates containing echinocandin at concentrations corresponding to 16× the MIC (0.25, 1, and 0.25 μg/ml concentrations of ANF, CSF, and MCF, respectively). After 3 to 4 days incubation, it was apparent that there was considerable variation in the number and size of mutant colonies recovered (data not shown). The three largest colonies from each plate were streaked for isolation on drug-free YPD and then screened for resistance to the selection drug. ANF, CSF, and MCF susceptibilities were then determined for the two most-resistant mutants from each of the 6 strains and 3 selections. The results are summarized (Fig. 2) as fold changes in ANF, CSF, and MCF MICs relative to that of the wild-type parent. There was a marked difference between strains, as only fks1Δ strains (fks1Δ and fks1Δ fks3Δ) consistently yielded mutants exhibiting substantially increased MICs. Specifically, of the 12 fks1Δ mutants (i.e., 2 mutants for each of the 3 echinocandins for each of the 2 fks1Δ strains), 11 exhibited MIC increases of ≥64-fold. In contrast, only 2 of 12 mutants of fks2Δ strains and 0 of 12 mutants of wild-type and fks3Δ strains exhibited MIC increases of ≥64-fold. The difference in resistance rate and magnitude between fks1Δ strains and the wild-type or fks3Δ strains can be explained by Fks1-Fks2 redundancy and will be more directly examined below. Aspects of these data that elude explanation are the higher rate and magnitude of resistance in fks1Δ strains than in fks2Δ strains (see Discussion) and the failure to recover ANF-resistant mutants of the fks1Δ fks3Δ strain.
Fig 2.
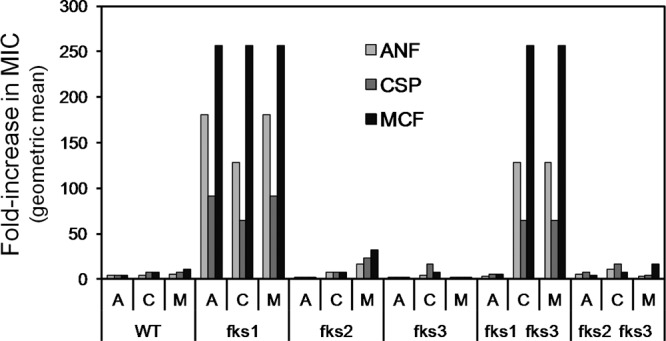
Differential effects of C. glabrata strain background on echinocandin resistance of laboratory-selected mutants. For each of the 6 strains and 3 selections, the 2 most-resistant mutants derived from the 3 largest colonies were assayed by broth microdilution for susceptibility to ANF (A), CSF (C), and MCF (M). The fold increase in MIC (relative to the wild-type parent of that strain), represented as the geometric mean for those 2 mutants, is shown.
Resistance-conferring mutations selected in vitro are similar to those reported in clinical isolates.
Genomic DNAs were prepared from 19 resistant mutants (selected to be representative but also to address the effects of redundancy), and their Fks1 or Fks2 mutations were identified by sequence analysis of PCR products. Sequencing results and MICs are summarized in Table 2. All 19 mutants exhibited a substitution or deletion within hot spot 1, consistent with previous analyses of resistant clinical isolates (Fig. 1). The Fks2 mutations F659Δ (11 mutants) and S663F (3 mutants) were repeatedly identified; these, along with Fks1-S629P (1 mutant), were previously reported in clinical isolates (7, 16, 45). The Fks1 mutations F625Δ (2 mutants), F625C, and P633T were novel, although clinical isolates with equivalent mutations in C. glabrata Fks2 or C. krusei Fks1 were previously reported (Fig. 1).
Table 2.
Echinocandin susceptibilities of C. glabrata wild-type and fksΔ strains and their echinocandin-selected mutants
| Strain | Mutant | Mutationa |
MIC (μg/ml) |
|||
|---|---|---|---|---|---|---|
| Fks1 | Fks2 | ANF | CSF | MCF | ||
| WT parent | WT | WT | 0.016 | 0.06 | 0.016 | |
| A1, C2, M2 | WT | F659Δ | 0.03 | 0.12 | 0.06 | |
| A2 | WT | S663F | 0.12 | 0.25 | 0.06 | |
| C1 | F625Δ | WT | 0.12 | 2.0 | 0.25 | |
| M1 | S629P | WT | 0.25 | 2.0 | 0.5 | |
| fks1Δ | WT | 0.016 | 0.03 | 0.008 | ||
| A1, C1, C2, M2 | F659Δ | 2 | 2 | 2 | ||
| A2, M1 | S663F | 4 | 4 | 2 | ||
| fks2Δ | WT | 0.016 | 0.03 | 0.008 | ||
| C1 | F625C | 0.5 | 1 | 0.25 | ||
| M1 | F625Δ | 2 | >16 | 4 | ||
| fks1Δ fks3Δ | WT | 0.016 | 0.03 | 0.008 | ||
| C1, C2, M1, M2 | F659Δ | 2 | 2 | 2 | ||
| fks2Δ fks3Δ | WT | 0.016 | 0.03 | 0.008 | ||
| A1 | P633T | 0.25 | 1 | 0.12 | ||
Sequencing was restricted to the DNA encoding Fks1 residues 570 to 736 or Fks2 residues 608 to 779. WT, wild type.
Fks1-Fks2 redundancy attenuates the impact of resistance-conferring mutations.
Three pairs of mutants presented in Table 2 have equivalent mutations but differ in their wild-type versus fksΔ strain backgrounds. Mutants WT-C1 and fks2Δ-M1 have Fks1-F625Δ mutations, mutants WT-A2 and fks1Δ-M1 have Fks2-S663T mutations, and multiple mutants, including WT-C2 and fks1Δ-C2, have Fks2-F659Δ mutations. For all three pairs, the mutations had dramatically increased effects in the fksΔ backgrounds. Specifically, the Fks1-F625Δ mutation decreased echinocandin susceptibility 8- to 32-fold in the WT background and 128- to 512-fold in the fks2Δ backgrounds. Similarly, in the WT and fks1Δ backgrounds, the Fks2-F659Δ mutation decreased susceptibility 2- to 4-fold and 64- to 256-fold, respectively, and the Fks2-S663F mutation decreased susceptibility 4- to 8-fold and 128- to 256-fold, respectively. In other words, Fks1-Fks2 redundancy in the WT background attenuated the impact of these mutations 32- to 64-fold (medians for Fks1 and Fks2 mutations, respectively).
fks1Δ disruptants are specifically susceptible to the calcineurin inhibitor FK506.
As noted above, FKS1 in S. cerevisiae was first defined by the sensitivity of null mutants in that gene to FK506, a calcineurin inhibitor and widely used immunosuppressive agent (tacrolimus). Similarly, while FK506 (0.3 μg/ml) had no effect on the growth of the C. glabrata fks2Δ, fks3Δ, and fks2Δ fks3Δ strains, it inhibited growth (measured spectroscopically in liquid cultures) of the fks1Δ and fks1Δ fks3Δ strains 91% and >99%, respectively, relative to FK506-free controls at 24 h. However, by 48 h, the fks1Δ strain fully recovered, while inhibition of the fks1Δ fks3Δ strain was sustained, implying that Fks3 supported growth to a limited extent. Cultures and isolated colonies obtained following recovery from FK506 inhibition were retested and exhibited the identical patterns of FK506 inhibition (data not shown), indicating that this recovery was not mediated by selection for FK506-resistant mutants.
Fks2-mediated resistance is reversed by FK506.
Fks2 mutations outnumber Fks1 mutations 2 to 1 among echinocandin-resistant clinical isolates as noted above, and they also predominate following laboratory selection on echinocandin-containing medium (Table 2). In light of the FK506 susceptibility of C. glabrata fks1Δ strains (see above), we reasoned that resistance mediated by an Fks2, but not Fks1, mutation would be reversed by FK506. This was confirmed in susceptibility assays with 4 representative laboratory mutants and, importantly, 6 clinical isolates (Table 3).
Table 3.
Reversal of Fks2-mediated echinocandin resistance by FK506
| Straina | Fks1 | Fks2 | Fold decrease in MIC due to FK506b |
||
|---|---|---|---|---|---|
| ANF | CSF | MCF | |||
| 200989 | WTc | WT | 2 | 2 | 1 |
| 200989-C1 | F625Δ | WT | 1 | 1 | 1 |
| 200989-M1 | S629P | WT | 0.5 | 1 | 1 |
| DPL38 | F625S | WT | 2 | 1 | 1 |
| DPL39 | S629P | WT | 2 | 4 | 1 |
| DPL41 | D632G | WT | 1 | 2 | 1 |
| 200989-A1 | WT | F659Δ | 4 | 8 | 8 |
| 200989-A2 | WT | S663F | 8 | 8 | 8 |
| DPL23 | WT | F659Δ | 8 | 128 | 64 |
| DPL34 | WT | P667T | 4 | 4 | 8 |
| 20409 | WT | F659V | 4 | 8 | 4 |
See Table 2 for MICs of strain 200989 and its laboratory-derived mutants. The DPL (16) and 20409 (24) clinical isolates were previously described, and their MICs were reassayed for this study.
Echinocandin MICs were determined in the absence or presence of FK506 (0.3 μg/ml), and the fold decrease due to FK506 was calculated.
WT, wild type.
FK506 effects are mediated by C. glabrata orthologs of Cmp2 and Fpr1.
To confirm that FK506 reversal of Fks2-mediated resistance is mediated by calcineurin inhibition, the Cmp2 catalytic subunit gene was disrupted in wild-type 200989 and its C1 (Fks1-F625Δ) and A1 (Fks2-659Δ) derivatives (Table 2). There was no net effect of the cmp2Δ deletion on the echinocandin MICs exhibited by 200989 and 200989-C1, i.e., they remained susceptible and resistant, respectively. In contrast, the cmp2Δ mutation reversed the echinocandin resistance of 200989-A1 (ANF, CSF, and MCF MICs were reduced 4-, 8-, and 16-fold, respectively).
To inhibit calcineurin, FK506 must bind to the peptidyl-prolyl isomerase Fpr1 (1). Two null mutants of the C. glabrata Fpr1 ortholog (isolated by selecting for resistance to the highly synergistic combination of terbinafine plus FK506 and then screening for unaltered terbinafine susceptibility) and their wild-type parent were tested for susceptibility to CSF plus FK506; only the parent demonstrated increased (4-fold) susceptibility in the presence of FK506.
FK506 and cmp2Δ effects may be mediated at levels other than FKS2 expression.
In S. cerevisiae, FKS1 is abundantly expressed during normal vegetative growth while FKS2 and FKS3 are minimally expressed unless induced by Ca2+ or pheromone; this induction is blocked by FK506 or calcineurin mutation (27). These transcriptional effects are presumed to mediate the growth inhibition by FK506 of fks1 null mutants. In C. glabrata, in contrast, FKS2 is expressed at levels more comparable to those for FKS1 (FKS2/FKS1 ratios = 0.13 to 3.4), while again, FKS3 is expressed only minimally (16). Here we extended this analysis to examine the effects on C. glabrata FKS expression ratios of brief (30 min) exposure to CaCl2 (30 mM) or CSF (1 μg/ml). Effects of these treatments were compared in the presence and absence of FK506 (1 μg/ml) and in wild-type 200989 versus its cmp2Δ disruptant. Under all conditions tested, FKS3 was expressed at levels >100-fold below those of FKS1 (not shown). As shown in Table 4, CaCl2 treatment increased the FKS2/FKS1 ratio a modest 2.2-fold, a result which was blocked by FK506 treatment or cmp2Δ disruption. CSF exposure yielded a similarly modest 2.0-fold increase, which was only partially blocked by FK506 or the cmp2Δ mutation. Consistent with these results, we detected no effect of FK506 on the growth in SD-ura medium of the fks2Δ strain described above in which the URA3 coding sequence is expressed from the fks2 promoter (data not shown). These results raise the possibility that calcineurin regulates Fks2 activity in C. glabrata at levels other than transcriptional regulation.
Table 4.
FKS2/FKS1 expression ratios determined by qRT-PCR, and change relative to untreated strain 200989a
| Treatment | Expression ratio and change (Δ) of each strain relative to the untreated strain 200989 |
|||||
|---|---|---|---|---|---|---|
| 200989 |
200989 plus FK506 |
200989 cmp2Δmutant |
||||
| fks2/fks1 | Δ | fks2/fks1 | Δ | fks2/fks1 | Δ | |
| No treatment | 0.17 ± 0.03 | 1 | 0.14 ± 0.03 | 0.8 | 0.10 ± 0.02 | 0.6 |
| CaCl2 | 0.37 ± 0.04 | 2.2 | 0.10 ± 0.01 | 0.6 | 0.13 ± 0.02 | 0.8 |
| CSF | 0.33 ± 0.15 | 2.0 | 0.25 ± 0.04 | 1.5 | 0.20 ± 0.01 | 1.2 |
Log-phase cultures of wild-type strain 200989 (with or without 1 μg/ml FK506) and the 200989 cmp2Δ strain were untreated or treated with CaCl2 (30 mM) or CSF (1 μg/ml) for 30 min, followed by RNA extraction and qRT-PCR analysis of the FKS2/FKS1 expression ratios (means and standard deviations).
DISCUSSION
The genomes of ascomycetous yeasts, including S. cerevisiae, C. albicans, and C. glabrata, encode three Fks paralogs. In S. cerevisiae, they have primary roles in vegetative growth (Fks1) and spore wall formation (Fks2 and Fks3) (9, 19, 20, 27). However, the viability, albeit with a growth defect, of S. cerevisiae fks1Δ strains indicates that Fks1-Fks2 are partially redundant for vegetative growth. In C. albicans, this redundancy appears to be lacking, as fks1Δ strains are nonviable (10, 29). In this respect, C. albicans is more similar to fungi such as Aspergillus fumigatus and Cryptococcus neoformans, which encode a single essential Fks1 (17, 43). In contrast, in C. glabrata, Fks1 and Fks2 appear to be fully redundant for growth.
We speculate that this true redundancy reflects an evolutionary shift in C. glabrata Fks2 function in response to an apparent lack of sporulation in this organism and the potential benefits of Fks1-Fks2 redundancy. Consistent with this shift, resistance-conferring mutations commonly occur in Fks2 of C. glabrata clinical isolates (indeed, at twice the apparent frequency of Fks1 mutations), while they occur exclusively in Fks1 of wild-type S. cerevisiae and C. albicans. This shift is also consistent with the more equivalent levels of FKS1-FKS2 expression in C. glabrata (16) compared to FKS1-dominant expression in S. cerevisiae (27).
Beyond its evolutionary significance, this shift in C. glabrata Fks2 function is likely to have clinical implications. Specifically, Fks1-Fks2 redundancy reduced the rate and impact of resistance-conferring mutations. This was clearly evident in comparisons to fks1Δ strains, which generated mutants, all involving Fks2 hot spot 1, at higher frequencies and with the most elevated echinocandin MICs. This was less evident, however, in comparisons to fks2Δ strains, suggesting that Fks1 mutations are more likely to confer reduced fitness. Consistent with this, Garcia-Effron et al. (16) showed that hot spot mutations in Fks1 had a more significant effect on glucan synthase Vmax than mutations in Fks2.
An additional consequence of this redundancy in C. glabrata is that the echinocandin resistance level conferred by Fks1 or Fks2 mutation will likely depend on the relative expression of their genes, which in clinical isolates varies more than 20-fold (16). In support of this, the resistance conferred by Fks2 but not Fks1 mutation was reversed to various degrees following treatment with the calcineurin inhibitor FK506 or disruption of its catalytic subunit gene cmp2. Calcineurin is specifically required for Fks2 activity in C. glabrata, as evidenced by its toxicity to fks1Δ (particularly fks1Δ fks3Δ) strains but not fks2Δ strains. FK506 (tacrolimus) is a widely used immunosuppressant in transplant patients; these data imply that C. glabrata infections in these patients are less likely to develop echinocandin resistance, especially since this resistance, both clinically and in the lab, is more commonly due to Fks2 than Fks1 mutation. The potential influence on fungal infection of synergistic antifungal-immunosuppressant interaction has been noted (42). Of course, with respect to C. glabrata therapy, a more ideal calcineurin inhibitor would be nonimmunosuppressive while retaining the ability to block Fks2-mediated resistance.
In S. cerevisiae, the calcineurin requirement of fks1 null mutants is mediated at the level of FKS2 expression (27). In C. glabrata, our analysis similarly showed that FKS2 expression can be induced by Ca2+ or CSF and that this induction can be blocked to various extents by FK506 treatment or in a cmp2Δ strain. However, these effects were very modest compared to those reported for S. cerevisiae. This difference no doubt reflects the differences in the level of constitutive FKS2 expression in these two yeasts. Therefore, to explain the potent effects of FK506 on C. glabrata, alternative mechanisms to transcriptional regulation warrant consideration. For example, in S. cerevisiae, the mitogen-activated protein (MAP) kinase Smk1 negatively regulates Fks2 activity during spore wall formation, presumably through phosphorylation (18). If the Smk1 ortholog or other kinase has a similar negative effect on C. glabrata Fks2, the phosphatase activity of calcineurin may be required to restore its activity.
ACKNOWLEDGMENTS
This work was supported by grants to T.D.E. from the NIH (AI075272) and Pfizer and to D.S.P. from the NIH (AI069397) and Pfizer.
Footnotes
Published ahead of print 1 October 2012
REFERENCES
- 1. Arevalo-Rodriguez M, Wu X, Hanes SD, Heitman J. 2004. Prolyl isomerases in yeast. Front. Biosci. 9:2420–2446 [DOI] [PubMed] [Google Scholar]
- 2. Balashov SV, Park S, Perlin DS. 2006. Assessing resistance to the echinocandin antifungal drug caspofungin in Candida albicans by profiling mutations in FKS1. Antimicrob. Agents Chemother. 50:2058–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Castanheira M, et al. 2010. Low prevalence of fks1 hot spot 1 mutations in a worldwide collection of Candida strains. Antimicrob. Agents Chemother. 54:2655–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cleary JD, Garcia-Effron G, Chapman SW, Perlin DS. 2008. Reduced Candida glabrata susceptibility secondary to an FKS1 mutation developed during candidemia treatment. Antimicrob. Agents Chemother. 52:2263–2265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dannaoui E, et al. 2012. Candida spp. with acquired echinocandin resistance, France, 2004-2010. Emerg. Infect. Dis. 18:86–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Denning DW. 2003. Echinocandin antifungal drugs. Lancet 362:1142–1151 [DOI] [PubMed] [Google Scholar]
- 7. Desnos-Ollivier M, et al. 2008. Mutations in the fks1 gene in Candida albicans, C. tropicalis, and C. krusei correlate with elevated caspofungin MICs uncovered in AM3 medium using the method of the European Committee on Antibiotic Susceptibility Testing. Antimicrob. Agents Chemother. 52:3092–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Douglas CM. 2001. Fungal β(1,3)-d-glucan synthesis. Med. Mycol. 39(Suppl 1):55–66 [DOI] [PubMed] [Google Scholar]
- 9. Douglas CM, et al. 1994. The Saccharomyces cerevisiae FKS1 (ETG1) gene encodes an integral membrane protein which is a subunit of 1,3-β-d-glucan synthase. Proc. Natl. Acad. Sci. U. S. A. 91:12907–12911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Douglas CM, et al. 1997. Identification of the FKS1 gene of Candida albicans as the essential target of 1,3-beta-d-glucan synthase inhibitors. Antimicrob. Agents Chemother. 41:2471–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dujon B, et al. 2004. Genome evolution in yeasts. Nature 430:35–44 [DOI] [PubMed] [Google Scholar]
- 12. Edlind TD, et al. 2005. Promoter-dependent disruption of genes: simple, rapid, and specific PCR-based method with application to three different yeast. Curr. Genet. 48:117–125 [DOI] [PubMed] [Google Scholar]
- 13. Garcia-Effron G, et al. 2010. Novel FKS mutations associated with echinocandin resistance in Candida species. Antimicrob. Agents Chemother. 54:2225–2227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garcia-Effron G, Katiyar SK, Park S, Edlind TD, Perlin DS. 2008. A naturally occurring proline-to-alanine amino acid change in Fks1p in Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis accounts for reduced echinocandin susceptibility. Antimicrob. Agents Chemother. 52:2305–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garcia-Effron G, Kontoyiannis DP, Lewis RE, Perlin DS. 2008. Caspofungin-resistant Candida tropicalis strains causing breakthrough fungemia in patients at high risk for hematologic malignancies. Antimicrob. Agents Chemother. 52:4181–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garcia-Effron G, Lee S, Park S, Cleary JD, Perlin DS. 2009. Effect of Candida glabrata FKS1 and FKS2 mutations on echinocandin sensitivity and kinetics of 1,3-β-d-glucan synthase: implication for the existing susceptibility breakpoint. Antimicrob. Agents Chemother. 53:3690–3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ha YS, Covert SF, Momany M. 2006. FsFKS1, the 1,3-β-glucan synthase from the caspofungin-resistant fungus Fusarium solani. Eukaryot. Cell 5:1036–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang LS, Doherty HK, Herskowitz I. 2005. The Smk1p MAP kinase negatively regulates Gsc2p, a 1,3-beta-glucan synthase, during spore wall morphogenesis in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 102:12431–12436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Inoue SB, et al. 1995. Characterization and gene cloning of 1,3-beta-d-glucan synthase from Saccharomyces cerevisiae. Eur. J. Biochem. 231:845–854 [DOI] [PubMed] [Google Scholar]
- 20. Ishihara S, et al. 2007. Homologous subunits of 1,3-beta-glucan synthase are important for spore wall assembly in Saccharomyces cerevisiae. Eukaryot. Cell 6:143–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson ME, Katiyar SK, Edlind TD. 2011. New Fks hot spot for acquired echinocandin resistance in Saccharomyces cerevisiae and its contribution to intrinsic resistance of Scedosporium species. Antimicrob. Agents Chemother. 55:3774–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kahn JN, et al. 2007. Acquired echinocandin resistance in a Candida krusei isolate due to modification of glucan synthase. Antimicrob. Agents Chemother. 51:1876–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Katiyar SK, Edlind TD. 2009. Role for Fks1 in the intrinsic echinocandin resistance of Fusarium solani as evidenced by hybrid expression in Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 53:1772–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Katiyar S, Pfaller M, Edlind T. 2006. Candida albicans and Candida glabrata clinical isolates exhibiting reduced echinocandin susceptibility. Antimicrob. Agents Chemother. 50:2892–2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kitada K, Yamaguchi E, Arisawa M. 1995. Cloning of the Candida glabrata TRP1 and HIS3 genes, and construction of their disruptant strains by sequential integrative transformation. Gene 165:203–206 [DOI] [PubMed] [Google Scholar]
- 26. Laverdiere M, et al. 2006. Progressive loss of echinocandin activity following prolonged use for treatment of Candida albicans oesophagitis. J. Antimicrob. Chemother. 57:705–708 [DOI] [PubMed] [Google Scholar]
- 27. Mazur P, et al. 1995. Differential expression and function of two homologous subunits of yeast 1,3-β-glucan synthase. Mol. Cell. Biol. 15:5671–5681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Messer SA, Jones RN, Moet GJ, Kirby JT, Castanheira M. 2010. Potency of anidulafungin compared to nine other antifungal agents tested against Candida spp., Cryptococcus spp., and Aspergillus spp.: results from the Global SENTRY Antimicrobial Surveillance Program (2008). J. Clin. Microbiol. 48:2984–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mio T, et al. 1997. Cloning of the Candida albicans homolog of Saccharomyces cerevisiae GSC1/FKS1 and its involvement in β-1,3-glucan synthesis. J. Bacteriol. 179:4096–4105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niimi K, et al. 2010. Clinically significant micafungin resistance in Candida albicans involves modification of a glucan synthase catalytic subunit GSC1 (FKS1) allele followed by loss of heterozygosity. J. Antimicrob. Chemother. 65:842–852 [DOI] [PubMed] [Google Scholar]
- 31. Ohyama T, Miyakoshi S, Isono F. 2004. FKS1 mutations responsible for selective resistance of Saccharomyces cerevisiae to the novel 1,3-β-glucan synthase inhibitor arborcandin C. Antimicrob. Agents Chemother. 48:319–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ostrosky-Zeichner L, et al. 2003. Antifungal susceptibility survey of 2,000 bloodstream Candida isolates in the United States. Antimicrob. Agents Chemother. 47:3149–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pappas PG, et al. 2009. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 48:503–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Park S, et al. 2005. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob. Agents Chemother. 49:3264–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perlin DS. 2007. Resistance to echinocandin-class antifungal drugs. Drug Resist. Updat. 10:121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45 doi:10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pfaller MA, et al. 2008. In vitro susceptibility of invasive isolates of Candida spp. to anidulafungin, caspofungin, and micafungin: six years of global surveillance. J. Clin. Microbiol. 46:150–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pfaller M, et al. 2011. Use of epidemiological cutoff values to examine 9-year trends in susceptibility of Candida species to anidulafungin, caspofungin, and micafungin. J. Clin. Microbiol. 49:624–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pfaller MA, et al. 2012. Frequency of decreased susceptibility and resistance to echinocandins among fluconazole-resistant bloodstream isolates of Candida glabrata. J. Clin. Microbiol. 50:1199–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pfeiffer CD, et al. 2010. Breakthrough invasive candidiasis in patients on micafungin. J. Clin. Microbiol. 48:2373–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roetzer A, Gabaldón T, Schüller C. 2011. From Saccharomyces cerevisiae to Candida glabrata in a few easy steps: important adaptations for an opportunistic pathogen. FEMS Microbiol. Lett. 314:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Singh N, Heitman J. 2004. Antifungal attributes of immunosuppressive agents: new paradigms in management and elucidating the pathophysiologic basis of opportunistic mycoses in organ transplant recipients. Transplantation 77:795–800 [DOI] [PubMed] [Google Scholar]
- 43. Thompson JR, et al. 1999. A glucan synthase FKS1 homolog in Cryptococcus neoformans is single copy and encodes an essential function. J. Bacteriol. 181:444–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vermitsky J-P, Edlind TD. 2004. Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrob. Agents Chemother. 48:3773–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zimbeck AJ, et al. 2010. FKS mutations and elevated echinocandin MIC values among Candida glabrata isolates from U.S. population-based surveillance. Antimicrob. Agents Chemother. 54:5042–5047 [DOI] [PMC free article] [PubMed] [Google Scholar]