

Abstract
Next-generation therapies for chronic hepatitis B virus (HBV) infection will involve combinations of established and/or experimental drugs. The current study investigated the in vitro and in vivo efficacy of tenofovir disoproxil fumarate (TDF) and/or emtricitabine [(−)-FTC] alone and in combination therapy for HBV infection utilizing the HepAD38 system (human hepatoblastoma cells transfected with HBV). Cellular pharmacology studies demonstrated increased levels of (−)-FTC triphosphate with coincubation of increasing concentrations of TDF, while (−)-FTC had no effect on intracellular tenofovir (TFV) diphosphate levels. Quantification of extracellular HBV by real-time PCR from hepatocytes demonstrated the anti-HBV activity with TDF, (−)-FTC, and their combination. Combination of (−)-FTC with TDF or TFV (ratio, 1:1) had a weighted average combination index of 0.7 for both combination sets, indicating synergistic antiviral effects. No cytotoxic effects were observed with any regimens. Using an in vivo murine model which develops robust HBV viremia in nude mice subcutaneously injected with HepAD38 cells, TDF (33 to 300 mg/kg of body weight/day) suppressed virus replication for up to 10 days posttreatment. At 300 mg/kg/day, (−)-FTC strongly suppressed virus titers to up to 14 days posttreatment. Combination therapy (33 mg/kg/day each drug) sustained suppression of virus titer/ml serum (<1 log10 unit from pretreatment levels) at 14 days posttreatment, while single-drug treatments yielded virus titers 1.5 to 2 log units above the initial virus titers. There was no difference in mean alanine aminotransferase values or mean wet tumor weights for any of the groups, suggesting a lack of drug toxicity. TDF–(−)-FTC combination therapy provides more effective HBV suppression than therapy with each drug alone.
INTRODUCTION
Chronic hepatitis B has been a major target for the development of therapeutics for more than 20 years. Given that the pathogenesis of chronic hepatitis B virus (HBV) infection is immune mediated, early approaches attempted to boost immune responses against virus-infected hepatocytes by using alpha interferon and, later, pegylated interferon, but the success of these approaches has been limited (30, 38).
Another approach targeted the HBV-encoded DNA polymerase reverse transcriptase (RT). For example, lamivudine (3TC) (8, 16) resulted in the partial or total clearance of virus from blood and in the improvement of liver histology in most infected persons (22, 44). However, prolonged treatment was also associated with the appearance of drug-resistant mutants in up to 20% of subjects per year (4, 15, 45). Other drugs, such as adefovir (ADV) (19) and entecavir (ETV) (23, 33, 40, 46), were effective against 3TC-resistant mutants (47). Clevudine [CLV; 1-(2-fluoro-5-methyl-beta,l-arabinofuranosyl) uracil] has shown potent in vitro activity against HBV in HepG2.2.15 cells (11) and in clinical trials (2, 6, 11), but studies in the United States and Europe were discontinued due to clevudine-associated myopathy (12). The newer nucleoside analogs have lower rates of drug resistance, although rebound viremia often occurred after therapy was withdrawn. Tenofovir disoproxil fumarate (TDF; Viread), a prodrug of tenofovir (TFV), is also effective against HBV in cell lines supporting viral replication and in clinical trials (7, 14). However, most single nucleoside analog therapies do not suppress virus replication after the end of treatment and may result in the appearance of drug-resistant mutants, which limits their utility in chronic virus infection. Combination therapy of nucleoside analogs with emtricitabine [(−)-FTC; also called FTC or Emtriva] showed synergistic antiviral activity in the same culture system (26) and in clinical trials (13, 18, 21, 36, 42).
Clinical experience suggests that stronger and more prolonged suppression of virus replication is associated with a significantly decreased risk for disease progression and development of hepatocellular carcinoma (HCC) (9), as well as a decreased frequency of drug-resistant mutants (48). This provides a strong rationale for the development of simultaneous combination therapies to replace monotherapies and sequential combination therapies. With an ever expanding list of l-nucleosides [e.g., 3TC, (−)-FTC, CLV, l-deoxyribosylthymine, and l-deoxyribosylcytosine], acyclic phosphonates (e.g., ADV, TDF, and GS-3740, an isopropylalaninyl monoamidate phenyl monoester prodrug of tenofovir), and 2′-deoxyguanosine analogs (e.g., ETV), opportunities now exist to evaluate combination therapies using compounds with complementary chemistries and mechanisms of action. Recently, a multicenter study reported the efficacy and tolerance of the combination tenofovir disoproxil fumarate plus emtricitabine in persons with chronic HBV infection (42).
Part of the problem in developing new drugs against HBV is the lack of suitable in vivo models with sustained virus replication. It is impractical to test antiviral drug combinations in HBV-infected chimpanzees. Related hepadnaviruses exist in ground squirrels, woodchucks, and ducks (31, 32), and these systems have been used to evaluate new drugs against hepadnaviruses (25, 34), but the availability and handling of these models are limited. Alternatively, several groups have made HBV transgenic mice, but these have not been used extensively for preclinical characterization of lead compounds or for testing putative combination therapies (1, 5, 24, 35, 39).
Some years ago, researchers constructed HepAD38 cells in which HBV replication was under the control of the tetracycline (Tet) suppressor in HepG2 (human hepatoblastoma) cells. In the presence of Tet, virus replication was suppressed, while in the absence of Tet, very high titers of HBV were observed (29). Subsequent studies have shown the utility of this cell line for drug discovery and the development of lead compounds active against HBV (41). Previously, we demonstrated that HepAD38 cells transplanted subcutaneously into nude mice resulted in the development of viremia, and treatment of these mice with drugs active against HBV demonstrated significant antiviral activity in vivo, suggesting that this simple small-animal model could be used to assess new therapeutic approaches and combination therapies against wild-type (wt) HBV (17). The present study utilized the HepAD38 system to evaluate TDF and (−)-FTC alone and in combination therapy in vitro and in vivo.
(A portion of this work was presented at the HepDART 2011 Meeting, Koloa, Kauai, Hawaii, 4 to 8 December 2011 [4a].)
MATERIALS AND METHODS
Source of antiviral agents.
TDF was extracted from commercially available Viread obtained from the U.S. Department of Veterans Affairs pharmacy. TFV was obtained from the NIH AIDS Research and Reference Reagent Program (catalog number 10199; https://www.aidsreagent.org/index.cfm). (−)-FTC was synthesized in our laboratory and was greater than 99% pure. Nucleoside triphosphate analogs were synthesized as previously described (16, 37) and were >95% pure. 14C- or 3H-radiolabeled nucleosides were obtained from Moravek Biochemicals Inc. (Brea, CA).
TDF and (−)-FTC phosphorylation studies.
Due to the metabolic competition of nucleoside analogs for the same enzyme systems, drug-drug interactions were studied by determining the phosphorylation pathways of these nucleoside analogs. To achieve this, each nucleoside was radiolabeled with either 14C or 3H and coincubated with potential competing drugs in HepG2 cells at various concentrations (from 1 to 100 μM) for 4 h at 37°C in 5% CO2. Following each 4-h incubation, intracellular metabolites were extracted with 70% CH3OH, followed by subsequent high-pressure liquid chromatography analysis with a chromatograph coupled with a flow scintillation analyzer (FSA-625TR; PerkinElmer, Waltham, MA).
Cytotoxicity assay.
HepG2 cells (5 × 103 cells/well) were seeded in 96-well plates in the presence of increasing concentrations of test compounds alone or in combination and incubated at 37°C in a humidified 5% CO2 atmosphere for 5 days. Cell viability was then performed by an MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] reduction test using a CellTiter 96 nonradioactive cell proliferation assay (Promega, Madison, WI), according to the manufacturer's instructions. Cytotoxicity was expressed as the concentration of test compounds alone or in combination that inhibited cell growth by ≥50% (50% cytotoxic concentration [CC50]).
HBV quantitative PCR assay.
The HBV 7-day quantitative PCR assay was performed in HepAD38 cells as previously described (43). HBV replication in the HepAD38 cell line occurs under conditions that can be regulated by tetracycline (29). HepAD38 cells were seeded onto 96-well plates at a density of 5 × 104 cells/well and incubated for 2 days at 37°C in a humidified 5% CO2 atmosphere. On day 2, medium was removed and cells were washed with 1× phosphate-buffered saline (PBS). Compounds and controls were prepared in medium without tetracycline and added in duplicate at various concentrations. On day 7, total DNA was extracted using a DNeasy 96 blood and tissue kit (Qiagen, Valencia, CA), and HBV DNA was amplified by real-time PCR using primers specific for the highly conserved sequences complementary to the DNA sequences present in HBsAg, as described previously (37).
Assessment of drug efficacy in mice.
All studies were conducted under approval by Temple University's IACUC. Briefly, 1 × 107 HepAD38 cells were injected subcutaneously into a single site on the backs of nude mice (day 0). At day 21, mice with palpable tumors were bled and the initial levels of virus DNA were determined by real-time PCR. Mice were then randomly divided into different groups containing 5 mice each and kept on tetracycline from days 21 to 31. The tetracycline was then removed from the drinking water and the groups were treated with different doses of TDF, (−)-FTC, combination therapy [TDF plus (−)-FTC], or PBS from days 31 to 36. Drugs were administered intraperitoneally to ensure that each mouse received the same amount of drug at the same time as every other mouse in the same group. Mice were then taken off drug and observed until day 51, at which time they were all sacrificed. Blood samples were obtained on days, 21, 31, 36, 41, 46, and 51. Virus titers were determined at each time point by real-time PCR, and the results were plotted as the log of the virus titer. On day 51, blood samples were evaluated for alanine aminotransferase (ALT) levels using a commercially available kit (ALT/AST 50; Sigma Chemical Co., St. Louis, MO) and tumors were removed and weighed.
Statistical analysis.
Comparisons between data sets were evaluated using the Student t test, where significance was recognized when P was <0.05. The computer-simulated combination index (CI) for drug-drug interactions at 50, 75, 90, and 95% inhibition of HBV replication in HepAD38 cells was calculated as described previously (10).
RESULTS
Cellular pharmacology.
Intracellular levels of (−)-FTC triphosphate [(−)-FTC-TP] were determined in vitro from extracts of HepG2 cells incubated with radiolabeled nucleoside analog. Coincubation of (−)-FTC with the A analog TDF significantly increased (−)-FTC-TP levels in a concentration-dependent manner (Fig. 1A) (P < 0.01). In contrast, coincubation of TDF with (−)-FTC did not significantly alter TFV diphosphate (TFV-DP) levels (Fig. 1B). Although TDF does not compete with 2′-deoxyadenosine or guanosine (Fig. 1B), these observations suggest an additive or synergistic effect of TDF on the increased formation of 2′-deoxycytidine analog triphosphates such as (−)-FTC-TP (Fig. 1A). The effect of TDF upon intracellular levels of (−)-FTC-TP was also observed in decay studies using increasing concentrations of TDF (Fig. 1C). In these studies, the half-life of (−)-FTC-TP alone was 4.13 h, following increases to 5.03 h with 1 μM TDF, to 5.97 h with 10 μM TDF, and to 6.82 h with 30 μM TDF. In addition, these results demonstrate that (−)-FTC (Fig. 1B) did not cause any effect on the intracellular formation of TFV-DP. Given the high antiviral potency and high potential selection of (−)-FTC-resistant virus during treatment, these drug-drug interaction studies suggest that TDF plus (−)-FTC warrants further development as a possible combination therapy.
Fig 1.
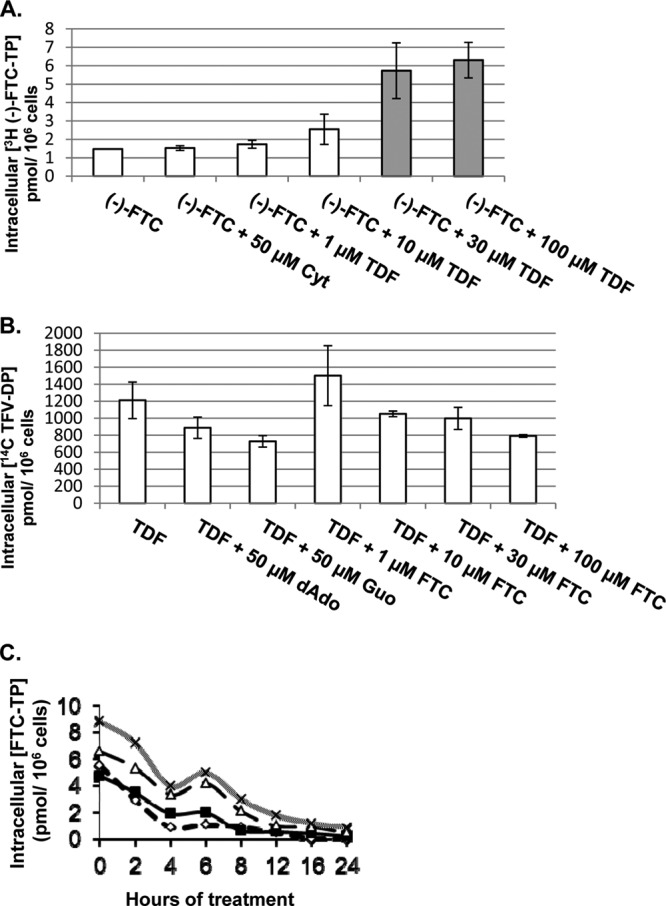
Drug-drug interaction assay. (A) Effect of TDF on intracellular levels of (−)-FTC-TP. HepG2 cells were treated with 10 μM (−)-FTC alone or in combination with controls (2′-deoxycytidine or cytidine) and various concentrations of TDF for 4 h. Gray bars, a significant increase in intracellular concentrations. (B) Effect of (−)-FTC on intracellular levels of TFV-DP. HepG2 cells were treated with 10 μM TDF alone or in combination with controls (2′-deoxyadenosine [dAdo] or guanosine [Guo]) and various concentrations of (−)-FTC for 4 h. (C) HepG2 cells were treated with 10 μM (−)-FTC alone (◊) or in combination with 1 μM TDF (■), 10 μM TDF (△), or 30 μM TDF (×) over 24 h in a decay study. Drugs were tested singly in triplicate, and each data point represents the mean value with a standard deviation of ≤20%. All concentrations shown are in μM. For the Student t test, two samples assuming equal variance were used to determine a P value of <0.05.
In vitro toxicity and anti-HBV activities of nucleosides in HepAD38 cells.
The 50% effective concentration (EC50) and EC90 of TDF, TFV, and (−)-FTC alone or in combination were determined in HepAD38 cells. Potent inhibition of HBV production was observed with (−)-FTC and TDF (Table 1). In a standard 5-day assay with HepG2 cells, toxicity was not detected for any of the anti-HBV compounds (Table 1). This was evident even among several drug combinations tested, suggesting that these drugs produced strong antiviral efficacy in the absence of toxicity.
Table 1.
In vitro anti-HBV activity and cytotoxicity of the three compounds used singly and in two-drug combinations in HepAD38 systema
| Compound | Ratio | EC50 (μM) | EC90 (μM) |
|---|---|---|---|
| TDF | NA | 0.01 (0.002–0.02) | 0.13 (0.03–0.25) |
| TFV | NA | 0.34 (0.11–0.93) | 3.72 (1.11–13.7) |
| (-)-FTC | NA | 0.05 (0.02–0.07) | 0.72 (0.18–1.42) |
| TDF + (−)-FTC | 1:1 | 0.01 (0.01–0.02) | 0.13 (0.01–0.18) |
| TDF + (−)-FTC | 1:2 | 0.01 (0.005–0.02) | 0.12 (0.07–0.22) |
| TDF + (−)-FTC | 1:5 | 0.02 (0.02–0.03) | 0.14 (0.13–0.15) |
| TDF + (−)-FTC | 1:10 | 0.02 (0.01–0.02) | 0.13 (0.11–0.15) |
| TFV + (−)-FTC | 1:1 | 0.02 (0.02–0.03) | 0.15 (0.12–0.18) |
| TFV + (−)-FTC | 2:1 | 0.33 (0.21–0.51) | 5.64 (2.04–15.6) |
| TFV + (−)-FTC | 5:1 | 1.00 (0.75–1.34) | 5.07 (3.47–7.40) |
All values represent the average of two experiments, and samples were performed in duplicate. EC50 and EC90, effective concentrations required to reduce HBV levels by 50% and 90%, respectively, on day 5. The values in parentheses indicate 95% confidence intervals generated by using the CalcuSyn program. The cytotoxic concentrations that inhibited cell growth by 50% (CC50s) in parent HepG2 cells were >10 μM for all compounds or compound combinations tested. Note that for the combination-drug studies, values were determined from standard curves for the first compound (either TDF or TFV). NA, not applicable.
Evaluation of drug interactions between (−)-FTC and TDF or TFV in HepG2 and HepAD38 cells.
HepAD38 cells were exposed to antiviral drugs for 5 days, and antiviral drug combination analyses were performed by the methodology described above. Data analyses were performed using the CalcuSyn program. Cytotoxicity was determined by MTT assay. The combination of (−)-FTC with TDF or TFV at ratio of 1:1 had a weighted average combination index of 0.7 for both combination sets (Table 2), indicating synergistic antiviral effects (since CIs of <1, 1, and >1 indicate synergism, an additive effect, and antagonism, respectively). In contrast, antagonism was observed when the ratio consisted of an increased amount of (−)-FTC relative to the amount of TDF or TFV (Table 2). No cytotoxic effects were observed with any of the combinations when tested at the highest concentrations.
Table 2.
Computer-simulated combination index (CI) of two-drug combinations at 50, 75, 90, and 95% inhibition of HBV replication in HepAD38 cellsa
| Drug combination | Ratio | CI at inhibition of: |
CIwt | |||
|---|---|---|---|---|---|---|
| 50% | 75% | 90% | 95% | |||
| TDF + (−)-FTC | 1:1 | 0.9 | 0.8 | 0.7 | 0.7 | 0.7 |
| TDF + (−)-FTC | 1:2 | 0.9 | 0.8 | 0.8 | 0.8 | 0.8 |
| TDF + (−)-FTC | 1:5 | 10.0 | 7.3 | 5.4 | 4.4 | 5.8 |
| TDF + (−)-FTC | 1:10 | 12.2 | 9.7 | 7.7 | 6.6 | 8.1 |
| TFV + (−)-FTC | 1:1 | 1.1 | 0.9 | 0.7 | 0.6 | 0.7 |
| TFV + (−)-FTC | 2:1 | 3.3 | 3.3 | 3.3 | 3.4 | 3.3 |
| TFV + (−)-FTC | 5:1 | 15 | 11 | 7.8 | 6.2 | 8.5 |
CI values were determined for a mutually exclusive interaction using the CalcuSyn program; a CI of <1, 1, or >1 indicates synergism, an additive effect, and antagonism, respectively. CIwt, weighted-average CI, which was assigned as (CI50 + 2CI75 + 3CI90 + 4CI95)/10, where CI50, CI75, CI90, and CI95 represent CIs for 50, 75, 90, and 95% inhibition, respectively (10).
Activity of TDF, (−)-FTC, and combination therapy in nude mice injected with HepAD38 cells.
Based upon the in vitro data presented above, TDF and (−)-FTC were further tested in a mouse model as monotherapy and combination therapies. Accordingly, nude mice injected with HepAD38 cells were prepared. After establishing viremia (21 days), mice were administered Tet for 10 days (days 21 to 31) and then switched to placebo (PBS), TDF, (−)-FTC, or combination therapy for another 6 days (days 31 to 36). Virus levels in the serum of mice were then observed for another 2 weeks (days 37 to 51) after treatment. A range of drug concentrations was used to obtain dose-response curves. The results for TDF monotherapy (Fig. 2A) demonstrated that at the higher doses of drug (33.3, 100, and 300 mg/kg of body weight/day), virus titers/ml serum remained depressed for up to 10 days after treatment compared to those in PBS-treated mice (P < 0.001 on days 41 and 46) but then rebounded strongly. This indicates that TDF maintains strong antiviral activity for some time after cessation of treatment. In Fig. 2B, the highest dose of (−)-FTC (300 mg/kg/day) suppressed virus titers/ml 3 or more log units for up to 2 weeks posttreatment compared to those in PBS-treated mice (P < 0.001 for days 41, 46, and 51). Rebound by day 51 was modest, being less than 1 log10 unit above the initial titer (at 21 days postinjection of HepAD38). Lower doses of (−)-FTC (33.3 and 100 mg/kg/day) suppressed the virus titer/ml serum during the period of treatment (days 31 to 36, inclusive), but once treatment was completed, virus titers rebounded about 1.5 log10 units over the following 2 weeks. At the lowest doses of (−)-FTC, inhibition of the virus titer/ml serum was modest (at 11.1 mg/kg/day) or absent (at 3.7 mg/kg/day) during the period of treatment, and by 2 weeks posttreatment, virus titers were about 2 log units higher than the initial titers. However, even at these lower doses of (−)-FTC (3.7 to 100 mg/kg/day), the increase in the virus titer/ml serum following cessation of therapy (1.5 to 2 log10 units) (Fig. 2B) was smaller than that observed among mice treated with PBS (4.5-log-unit rise in virus titer/ml serum) (P < 0.001 on days 36, 41, 46, and 51). Hence, both TDF and (−)-FTC demonstrated strong antiviral activity in vivo, and this activity was sustained for 10 to 14 days after cessation of therapy.
Fig 2.
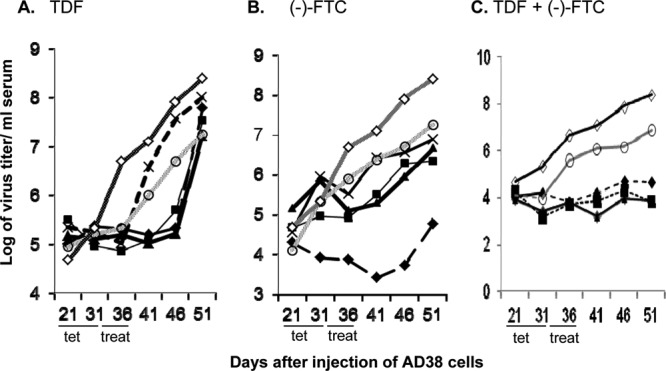
Comparison of TDF and (−)-FTC monotherapies or combination therapies at different drug concentrations. Dose-response curves for TDF (A), (−)-FTC (B), or the combination of both TDF and (−)-FTC (C) at 300 mg/kg/day (♦), 100 mg/kg/day (■), 33.3 mg/kg/day (▲), 11.1 mg/kg/day (×), or 3.7 mg/kg/day (○) for the indicated times (days 36 to 41 inclusive). Placebo treatment with PBS (◊) is also included for comparison. All mice were given Tet for 10 days (from days 21 to 31 after HepAD38 injection), treated daily for 6 days (day 31 to 36), and then followed off treatment for another 2 weeks (days 37 to 51). Each point represents the average HBV titer/ml serum from 5 mice in each group. The average values shown varied by no more than ±7.4%.
The antiviral activities of combination therapies are shown in Fig. 2C. At 300 mg/kg/day for each drug, combination therapy with TDF plus (−)-FTC suppressed virus levels to nearly the same extent through day 51 as therapy with (−)-FTC alone, while rebound was observed with TDF alone (Fig. 2A). Compared to PBS-treated mice, combination therapy suppressed the virus titer/ml serum between ∼3 log10 units on day 41 (P < 0.001) to 4.2 log10 units by day 51 (P < 0.001) (Fig. 2C). At 100 mg/kg/day, combination therapy suppressed the virus titer/ml serum throughout the period of observation (Fig. 2C).
In summary, with drug doses of 3.7 mg/kg/day, little difference between monotherapy, combination therapy, and placebo occurred (Fig. 2). When monotherapy and combination therapies are compared over the range of concentrations used, the antiviral effect of 300 mg/kg/day (−)-FTC in Fig. 2B is duplicated by combination therapy at 11.1 mg/kg/day in Fig. 2C, suggesting that the addition of TDF to (−)-FTC results in the use of 27-fold less (−)-FTC to achieve nearly the same antiviral effect. With regard to TDF, even 300 mg/kg/day did not suppress virus levels to the extent or for the duration that combination therapy down to 11.1 mg/kg/day did. These data suggest that combination therapy is more effective at inhibiting HBV during treatment and has more sustained antiviral activity following cessation of treatment than therapy with either drug alone at the same dose.
In vivo toxicity of combination therapy.
Although clinical trials with (−)-FTC and TDF alone or in combination (as for tenofovir-emtricitabine [Truvada]) demonstrate low toxicity, little is known about these combined modalities in animal models. Hepatotoxicity was determined by measuring serum ALT activities at the time of each blood draw (days 21, 31, 36, 41, 46, and 51) to assess toxicity. The results from day 51 for TDF or (−)-FTC monotherapy compared to those for (−)-FTC plus TDF combination therapy are shown in Fig. 3. The results show no statistical difference in mean ALT values (Fig. 3A). These results were indistinguishable from those obtained when ALT values were obtained at earlier times (data not shown). In addition, there was no statistical difference in the mean ALT values from day 21 to day 51 (data not shown), indicating that (−)-FTC plus TDF combination therapy was not toxic to the liver, at least by this criterion.
Fig 3.
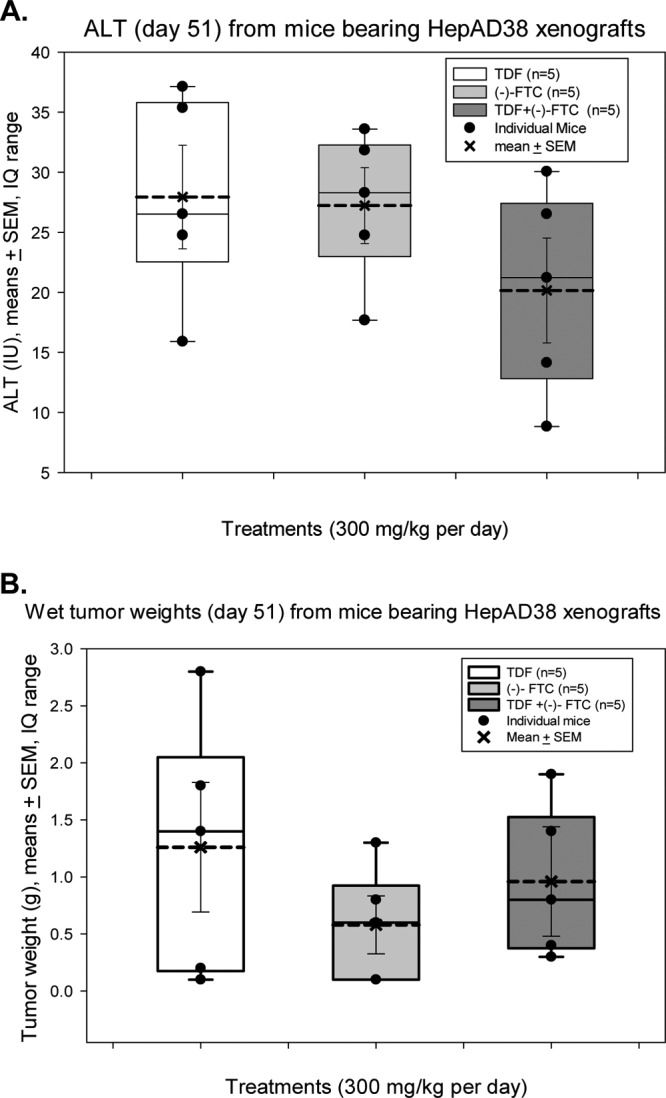
Drug toxicity assessment assays. (A) ALT measurements (means ± SEM) in serum from mice treated with 300-mg/kg/day doses of TDF (white bar), (−)-FTC (light gray bar), or a combination of TDF plus (−)-FTC (dark gray bar) at week 51 postinjection of HepAD38 cells. ALT values for mice injected with PBS or with TDF alone were indistinguishable from those reported here. (B) Wet weights of tumors recovered from nude mice bearing HepAD38 xenografts. Mice were treated with 300-mg/kg/day doses of TDF (white bar), (−)-FTC (light gray bar), or a combination of TDF plus (−)-FTC (dark gray bar). Tumor weights were evaluated at week 51 after subcutaneous injection of AD38 cells. Wet weights of tumors derived from mice injected with PBS or with TDF alone were indistinguishable from those reported here. The average is based upon data from 5 mice in each group. IQ, interquartile.
In addition, there was no significant difference in mean tumor wet weights when tumors were excised and weighed at day 51 (Fig. 3B), again suggesting little or no toxicity at different doses of monotherapy or combination therapy used.
At the end of the experiment, at day 51, livers and tumors from the mice were removed and stained with hematoxylin-eosin. Normal histology was observed in all mouse livers examined, and little to no necrosis was observed in the growing tumors (data not shown).
DISCUSSION
The development of multiple nucleoside and nucleotide analogs by academic institutions and biotechnological and pharmaceutical companies over the past 2 decades has provided new and powerful weapons against chronic hepatitis B. Each of these has been developed and many have successfully been marketed as monotherapies for HBV infections (20). However, continued use of these agents as monotherapy has also been associated with the appearance of drug-resistant mutations, and although the frequency of drug resistance is low with many of the newer agents (3, 6, 20, 21, 27, 28, 45), most have not been around long enough that their long-term use can be assessed. As is the case with treatment of HIV infection, it is likely that effective control/treatment of chronic hepatitis B will require the development of simultaneous combination therapies that can suppress virus replication to ultralow levels over an extended period of time with a minimal incidence of resistance. However, it is unlikely that these combined modalities using nucleoside analogs will eradicate HBV in humans. The novel nude mouse-HepAD38 model was designed as a rapid, in vivo system to assess multidrug toxicity and efficacy in the development of combination therapies (17).
Prior to in vivo assessment of multidrug combinations, various nucleoside inhibitors of HBV were evaluated in vitro using HepAD38 cells. Testing of nucleoside analogs individually showed that (−)-FTC and TDF were potent inhibitors of HBV (Table 1). Further, in vitro characterization showed that these drugs were not toxic to HepG2 cells (Table 1). These observations are consistent with these compounds reducing the virus titer due to their antiviral activities and not due to significant toxicity. Moreover, the computer-simulated CI showed that the TDF plus (−)-FTC and TFV plus (−)-FTC combinations demonstrated synergistic antiviral activity against HBV in HepAD38 cells (Tables 2) without toxicity, suggesting that these combinations would be efficacious in vivo.
The fact that (−)-FTC is a so-called l-nucleoside analog, while TDF is a flexible acyclic phosphonate, suggests that their modes of action of inhibition of both HBV reverse transcriptase and HBV DNA polymerase are distinct and that they may act in a complementary and perhaps additive or synergistic way. Results of cellular pharmacology studies with coincubation of TDF and (−)-FTC supported this hypothesis, as TDF increased the intracellular levels of (−)-FTC-TP in a dose-dependent manner (Fig. 1A).
In order to develop a meaningful combination therapy, an ideal model system must be robust enough to detect single and combination therapies that reduce virus titers by at least 3 to 4 log units. Previous results showed that the nude mouse-HepAD38 model is such a system (17). Accordingly, in order to determine the antiviral efficacy of TDF and (−)-FTC, dose-response curves were generated for each drug individually. As expected, each drug strongly suppressed the virus titer at high doses up to 10 days (for TDF) or 14 days [for (−)-FTC] postadministration (Fig. 2). Even the lowest dose of each drug used (3.7 mg/kg/day) partially suppressed the virus titer compared to that in placebo (PBS)-treated mice. When dose-response curves were generated with TDF plus (−)-FTC combination therapy, the combination was effective in suppressing virus titers at all doses except the lowest one (3.7 mg/kg/day) compared to those in mice treated with each drug alone or PBS (Fig. 2C). The finding that combination therapy with reduced levels of the individual drugs alone demonstrates antiviral efficacy validates the synergistic effects of (−)-FTC plus TDF in combination in vivo. Moreover, the observation that these drugs are apparently not toxic in vitro (Table 1) or in vivo (Fig. 3A and B) suggests that the reduction in virus titer is likely to be associated with the antiviral properties of the compounds used and not likely to be due to hepatotoxicity or the failure of tumors to grow in the nude mice.
Results from our studies further support the HepAD38 system as a valuable model to test parameters that may identify more efficient combination therapies for HBV and further development in human clinical trials. In particular, this work has important implications for the use of the product Truvada, which is a fixed-dose combination of TDF and (−)-FTC widely used for the treatment of HIV and is also used off-label in certain countries like France for the treatment of HBV infection. If the results in human clinical trials demonstrate the same synergy that was observed in the nude mouse-HepAD38 model herein, that synergy may provide future opportunities for the use of smaller amounts of drugs in combination to provide sustained antiviral suppression in a form that will be affordable in developing countries where HBV is endemic and current therapies are often too expensive. The possibility also exists that the use of three drugs will prove to be even better. For example, work is under way to test whether the combination described above, together with ETV, a 2′-deoxyguanosine analog, will provide even greater synergy in long-term suppression of the HBV titer in vivo. It can also be envisioned that unrelated drugs developed for other viral targets could be tested in this nude mouse-HepAD38 system. Furthermore, drug combinations effective against clinically relevant HBV mutants over the long term could also be evaluated in this in vivo system.
ACKNOWLEDGMENTS
This work was primarily supported by NIH grant R01-AI-076535 (to R.F.S. and M.A.F.). This work was partially supported by grants 5R37-AI-41980, 5R37-AI-25899, and 5P30-AI-50409 and the U.S. Department of Veterans Affairs (to R.F.S.), and by NIH grants CA48656, CA66971, and CA104025 (to M.A.F.).
Footnotes
Published ahead of print 17 September 2012
REFERENCES
- 1. Anderson AL, Banks KE, Pontoglio M, Yaniv M, McLachlan A. 2005. Alpha/beta interferon differentially modulates the clearance of cytoplasmic encapsidated replication intermediates and nuclear covalently closed circular hepatitis B virus (HBV) DNA from the livers of hepatocyte nuclear factor 1alpha-null HBV transgenic mice. J. Virol. 79:11045–11052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balakrishna Pai S, Liu SH, Zhu YL, Chu CK, Cheng YC. 1996. Inhibition of hepatitis B virus by a novel l-nucleoside, 2′-fluoro-5-methyl-beta-l-arabinofuranosyl uracil. Antimicrob. Agents Chemother. 40:380–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartholomeusz A, Locarnini SA. 2006. Antiviral drug resistance: clinical consequences and molecular aspects. Semin. Liver Dis. 26:162–170 [DOI] [PubMed] [Google Scholar]
- 4. Bartholomew MM, et al. 1997. Hepatitis-B-virus resistance to lamivudine given for recurrent infection after orthotopic liver transplantation. Lancet 349:20–22 [DOI] [PubMed] [Google Scholar]
- 4a. Bassit L, Zlotnick A, Feitelson M, Schinazi RF. 2011. A novel inhibitor of HBV capsid assembly interacts synergistically when combined with emtricitabine plus tenofovir disoproxil fumarate in liver cells. Global Antivir. J. 7(Suppl 1):108 [Google Scholar]
- 5. Brown JJ, et al. 2000. A long-term hepatitis B viremia model generated by transplanting nontumorigenic immortalized human hepatocytes in Rag-2-deficient mice. Hepatology 31:173–181 [DOI] [PubMed] [Google Scholar]
- 6. Buti M, Esteban R. 2003. Entecavir, FTC, L-FMAU, LdT and others. J. Hepatol. 39(Suppl 1):S139–S142 [DOI] [PubMed] [Google Scholar]
- 7. Butt AA. 2006. Tenofovir for chronic hepatitis B virus infection in HIV-coinfected patients. AIDS Read. 16:219–222 [PubMed] [Google Scholar]
- 8. Chang CN, et al. 1992. Deoxycytidine deaminase-resistant stereoisomer is the active form of (+/−)-2′,3′-dideoxy-3′-thiacytidine in the inhibition of hepatitis B virus replication. J. Biol. Chem. 267:13938–13942 [PubMed] [Google Scholar]
- 9. Chen CJ, et al. 2006. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 295:65–73 [DOI] [PubMed] [Google Scholar]
- 10. Chou TC. 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 58:621–681 [DOI] [PubMed] [Google Scholar]
- 11. Chu CK, et al. 1995. Use of 2′-fluoro-5-methyl-beta-l-arabinofuranosyluracil as a novel antiviral agent for hepatitis B virus and Epstein-Barr virus. Antimicrob. Agents Chemother. 39:979–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cobbold JF, et al. 2010. Hepatic lipid profiling in chronic hepatitis C: an in vitro and in vivo proton magnetic resonance spectroscopy study. J. Hepatol. 52:16–24 [DOI] [PubMed] [Google Scholar]
- 13. Cui L, et al. 1996. Effect of beta-enantiomeric and racemic nucleoside analogues on mitochondrial functions in HepG2 cells. Implications for predicting drug hepatotoxicity. Biochem. Pharmacol. 52:1577–1584 [DOI] [PubMed] [Google Scholar]
- 14. Delaney WE, IV, et al. 2006. Intracellular metabolism and in vitro activity of tenofovir against hepatitis B virus. Antimicrob. Agents Chemother. 50:2471–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dienstag JL, et al. 1999. Extended lamivudine retreatment for chronic hepatitis B: maintenance of viral suppression after discontinuation of therapy. Hepatology 30:1082–1087 [DOI] [PubMed] [Google Scholar]
- 16. Doong SL, Tsai CH, Schinazi RF, Liotta DC, Cheng YC. 1991. Inhibition of the replication of hepatitis B virus in vitro by 2′,3′-dideoxy-3′-thiacytidine and related analogues. Proc. Natl. Acad. Sci. U. S. A. 88:8495–8499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feitelson MA, Clayton MM, Sun B, Schinazi RF. 2007. Development of a novel mouse model to evaluate drug candidates against hepatitis B virus. Antivir. Chem. Chemother. 18:213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Furman PA, et al. 1992. The anti-hepatitis B virus activities, cytotoxicities, and anabolic profiles of the (−) and (+) enantiomers of cis-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine. Antimicrob. Agents Chemother. 36:2686–2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gilson RJ, et al. 1999. A placebo-controlled phase I/II study of adefovir dipivoxil in patients with chronic hepatitis B virus infection. J. Viral Hepat. 6:387–395 [DOI] [PubMed] [Google Scholar]
- 20. Gish RG. 2005. Clinical trial results of new therapies for HBV: implications for treatment guidelines. Semin. Liver Dis. 25(Suppl 1):29–39 [DOI] [PubMed] [Google Scholar]
- 21. Gish RG, et al. 2005. Safety and antiviral activity of emtricitabine (FTC) for the treatment of chronic hepatitis B infection: a two-year study. J. Hepatol. 43:60–66 [DOI] [PubMed] [Google Scholar]
- 22. Hunt CM, Brown NA, Rubin M. 1999. Lamivudine therapy of chronic hepatitis B. Adv. Exp. Med. Biol. 458:11–21 [DOI] [PubMed] [Google Scholar]
- 23. Innaimo SF, et al. 1997. Identification of BMS-200475 as a potent and selective inhibitor of hepatitis B virus. Antimicrob. Agents Chemother. 41:1444–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iyer RP, et al. 2004. Anti-hepatitis B virus activity of ORI-9020, a novel phosphorothioate dinucleotide, in a transgenic mouse model. Antimicrob. Agents Chemother. 48:2318–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Korba BA, et al. 1996. Liver-targeted antiviral nucleosides: enhanced antiviral activity of phosphatidyl-dideoxyguanosine versus dideoxyguanosine in woodchuck hepatitis virus infection in vivo. Hepatology 23:958–963 [DOI] [PubMed] [Google Scholar]
- 26. Korba BE. 1996. In vitro evaluation of combination therapies against hepatitis B virus replication. Antiviral Res. 29:49–51 [DOI] [PubMed] [Google Scholar]
- 27. Ladner SK, Miller TJ, King RW. 1998. The M539V polymerase variant of human hepatitis B virus demonstrates resistance to 2′-deoxy-3′-thiacytidine and a reduced ability to synthesize viral DNA. Antimicrob. Agents Chemother. 42:2128–2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ladner SK, Miller TJ, Otto MJ, King RW. 1998. The hepatitis B virus M539V polymerase variation responsible for 3TC resistance also confers cross-resistance to other nucleoside analogues. Antivir. Chem. Chemother. 9:65–72 [PubMed] [Google Scholar]
- 29. Ladner SK, et al. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 41:1715–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lok AS. 1994. Treatment of chronic hepatitis B. J. Viral Hepat. 1:105–124 [DOI] [PubMed] [Google Scholar]
- 31. Marion PL, Oshiro LS, Regnery DC, Scullard GH, Robinson WS. 1980. A virus in Beechey ground squirrels that is related to hepatitis B virus of humans. Proc. Natl. Acad. Sci. U. S. A. 77:2941–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mason WS, Seal G, Summers J. 1980. Virus of Pekin ducks with structural and biological relatedness to human hepatitis B virus. J. Virol. 36:829–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matthews SJ. 2006. Entecavir for the treatment of chronic hepatitis B virus infection. Clin. Ther. 28:184–203 [DOI] [PubMed] [Google Scholar]
- 34. Nicoll AJ, et al. 1998. Inhibition of duck hepatitis B virus replication by 9-(2-phosphonylmethoxyethyl)adenine, an acyclic phosphonate nucleoside analogue. Antimicrob. Agents Chemother. 42:3130–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ohashi K, et al. 2000. Sustained survival of human hepatocytes in mice: a model for in vivo infection with human hepatitis B and hepatitis delta viruses. Nat. Med. 6:327–331 [DOI] [PubMed] [Google Scholar]
- 36. Paff MT, Averett DR, Prus KL, Miller WH, Nelson DJ. 1994. Intracellular metabolism of (−)- and (+)-cis-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine in HepG2 derivative 2.2.15 (subclone P5A) cells. Antimicrob. Agents Chemother. 38:1230–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pas SD, Fries E, De Man RA, Osterhaus AD, Niesters HG. 2000. Development of a quantitative real-time detection assay for hepatitis B virus DNA and comparison with two commercial assays. J. Clin. Microbiol. 38:2897–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perrillo RP. 1993. Antiviral therapy of chronic hepatitis B: past, present, and future. J. Hepatol. 17(Suppl 3):S56–S63 [DOI] [PubMed] [Google Scholar]
- 39. Schinazi RF, Ilan E, Black PL, Yao X, Dagan S. 1999. Cell-based and animal models for hepatitis B and C viruses. Antivir. Chem. Chemother. 10:99–114 [DOI] [PubMed] [Google Scholar]
- 40. Sherman M, et al. 2006. Entecavir for treatment of lamivudine-refractory, HBeAg-positive chronic hepatitis B. Gastroenterology 130:2039–2049 [DOI] [PubMed] [Google Scholar]
- 41. Shi J, et al. 2003. N4-acyl-modified d-2′,3′-dideoxy-5-fluorocytidine nucleoside analogues with improved antiviral activity. Antivir. Chem. Chemother. 14:81–90 [DOI] [PubMed] [Google Scholar]
- 42. Si-Ahmed SN, et al. 2011. Efficacy and tolerance of a combination of tenofovir disoproxil fumarate plus emtricitabine in patients with chronic hepatitis B: a European multicenter study. Antiviral Res. 92:90–95 [DOI] [PubMed] [Google Scholar]
- 43. Stuyver LJ, et al. 2002. Antiviral activities and cellular toxicities of modified 2′,3′-dideoxy-2′,3′-didehydrocytidine analogues. Antimicrob. Agents Chemother. 46:3854–3860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Suzuki Y, et al. 1999. Histological changes in liver biopsies after one year of lamivudine treatment in patients with chronic hepatitis B infection. J. Hepatol. 30:743–748 [DOI] [PubMed] [Google Scholar]
- 45. Tipples GA, et al. 1996. Mutation in HBV RNA-dependent DNA polymerase confers resistance to lamivudine in vivo. Hepatology 24:714–717 [DOI] [PubMed] [Google Scholar]
- 46. Wolters LM, Hansen BE, Niesters HG, DeHertogh D, de Man RA. 2002. Viral dynamics during and after entecavir therapy in patients with chronic hepatitis B. J. Hepatol. 37:137–144 [DOI] [PubMed] [Google Scholar]
- 47. Xiong X, Flores C, Yang H, Toole JJ, Gibbs CS. 1998. Mutations in hepatitis B DNA polymerase associated with resistance to lamivudine do not confer resistance to adefovir in vitro. Hepatology 28:1669–1673 [DOI] [PubMed] [Google Scholar]
- 48. Yim HJ, et al. 2006. Evolution of multi-drug resistant hepatitis B virus during sequential therapy. Hepatology 44:703–712 [DOI] [PubMed] [Google Scholar]