

Abstract
The incidence of hospital-acquired infections with multidrug-resistant (MDR) Gram-negative pathogens is increasing at an alarming rate. Equally alarming is the overall lack of efficacious therapeutic options for clinicians, which is due primarily to the acquisition and development of various antibiotic resistance mechanisms that render these drugs ineffective. Among these mechanisms is the reduced permeability of the outer membrane, which prevents many marketed antibiotics from traversing this barrier. To circumvent this, recent drug discovery efforts have focused on conjugating a siderophore moiety to a pharmacologically active compound that has been designed to hijack the bacterial siderophore transport system and trick cells into importing the active drug by recognizing it as a nutritionally beneficial compound. MC-1, a novel siderophore-conjugated β-lactam that promotes its own uptake into bacteria, has exquisite activity against many Gram-negative pathogens. While the inclusion of the siderophore was originally designed to facilitate outer membrane penetration into Gram-negative cells, here we show that this structural moiety also renders other clinically relevant antibiotic resistance mechanisms unable to affect MC-1 efficacy. Resistance frequency determinations and subsequent characterization of first-step resistant mutants identified PiuA, a TonB-dependent outer membrane siderophore receptor, as the primary means of MC-1 entry into Pseudomonas aeruginosa. While the MICs of these mutants were increased 32-fold relative to the parental strain in vitro, we show that this resistance phenotype is not relevant in vivo, as alternative siderophore-mediated uptake mechanisms compensated for the loss of PiuA under iron-limiting conditions.
INTRODUCTION
The infectious disease community has reached a critical point; the prevalence of multidrug-resistant (MDR) pathogens has become a serious public health threat that is continuing to worsen as these bacterial species continue to evolve and acquire new antibiotic resistance determinants. While the pharmaceutical industry has recently provided clinicians with some useful antibiotics, such as linezolid, for the treatment of MDR Gram-positive infections, there is currently an overwhelming lack of therapeutic options in the war against MDR Gram-negative organisms (33). In addition, it is particularly alarming that the vast majority of antimicrobial compounds, whether marketed or currently in development, either have a narrow spectrum or lack efficacy against strains expressing certain resistance mechanisms. Serious nosocomial pathogens, such as Pseudomonas aeruginosa, Klebsiella pneumoniae, and Acinetobacter baumannii, wreak havoc in critical care institutions and intensive care units, preying on the elderly, the wounded, and the immunocompromised. Conventional antibiotic therapies, which were once efficacious against these pathogens, have been thwarted by resistance mechanisms that inactivate, exclude, or extrude the antibiotics from the cell (21, 34).
One of the major hurdles to designing an effective Gram-negative antibiotic is the requirement to cross the outer membrane. P. aeruginosa, K. pneumoniae, and A. baumannii can become resistant to certain β-lactam-type antibiotics by downregulating their major outer membrane porins (OprD, OmpK, and CarO, respectively), which limits compound entry into the Gram-negative periplasmic space (11, 20, 35). To address this widespread resistance phenotype, researchers have designed and developed siderophore-conjugated antimicrobial compounds (4, 14, 32). The incorporation of the siderophore allows for the hijacking of the normal siderophore-mediated iron uptake mechanisms used by pathogens such as P. aeruginosa, thereby avoiding the dependence on outer membrane porin expression for effective entry into cells. The redundancy of Gram-negative outer membrane receptors responsible for siderophore uptake suggests that a universal siderophore moiety could be recognized and transported by multiple species, irrespective of how distinct their native iron acquisition systems may be. This strategy is consistent with one taken recently by scientists at Basilea Pharmaceuticals, whose novel β-lactam BAL30072 also incorporates a siderophore moiety to promote uptake across the outer membrane (27). Once transport across the outer membrane is achieved, an effective antibiotic must resist the activity of efflux pumps and inactivating enzymes, such as β-lactamases, in order to reach its target (17, 28). Overcoming the latter challenge has proven to be extremely problematic for the development of a novel β-lactam, as the spectrum of activity of these enzymes is constantly increasing and subsequently destroying the utility of many marketed β-lactam antibiotics. Therefore, the strategic design of siderophore conjugates could be extended beyond outer membrane penetration to avoid other clinically relevant Gram-negative resistance mechanisms, such as efflux and β-lactamase-mediated hydrolysis.
We previously described MC-1 (Fig. 1), a novel siderophore-conjugated β-lactam antibiotic that is highly efficacious against MDR P. aeruginosa and extended-spectrum β-lactamase (ESBL)-producing members of the Enterobacteriaceae (13, 16). In the present report, we demonstrate that this compound is transported across the P. aeruginosa outer membrane through specific TonB-dependent outer membrane receptors and is therefore unaffected by porin downregulation. Additionally, we provide evidence that other significant Gram-negative antibiotic resistance mechanisms do not significantly alter the activity of MC-1 against MDR Gram-negative pathogens. Interestingly, while first-step resistance mutations affect the expression of a specific outer membrane siderophore receptor, we show that these mutations, while relevant in vitro, do not translate to corresponding levels of MC-1 resistance in vivo.
Fig 1.
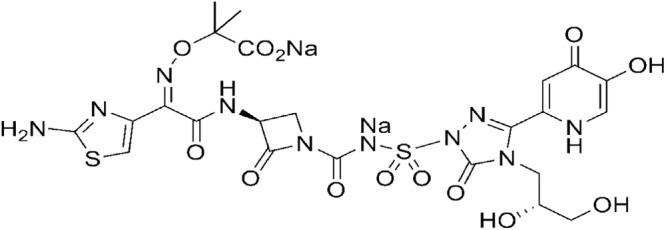
Structure of MC-1.
MATERIALS AND METHODS
Bacterial strains and media used.
Bacterial strains were routinely maintained on Luria-Bertani (LB) agar and broth. Antibiotics were purchased from Gibco and were supplemented, where appropriate, at the following concentrations: 15 μg/ml tetracycline (TET) and 15 μg/ml gentamicin (GEN) for Escherichia coli or 75 μg/ml GEN for P. aeruginosa. E. coli DH5α was used as the host strain to construct the isogenic β-lactamase library. P. aeruginosa PAO1 and the isogenic ΔfptA deletion mutant were kindly provided by M. Vasil (University of Colorado at Denver), and efflux pump panel strains were provided by H. Schweizer (Colorado State University). For mutagenesis experiments, E. coli HB101 harboring pRK2013 (12) was used as a helper strain in triparental matings, and Pseudomonas isolation agar (PIA; Difco) was used to select against the E. coli donors. MDR clinical isolates of P. aeruginosa were purchased from JMI Laboratories. MIC assays were routinely conducted according to CLSI guidelines in cation-adjusted Mueller-Hinton broth (MHB) unless otherwise indicated. An iron-deficient version of MHB was prepared by adaptation of a similar method described previously (26). First, 200 ml of 10× cation-adjusted MHB (Difco) was chelated with 20 g of Chelex 100 resin (Bio-Rad) for 6 h at room temperature with constant stirring, after which the entire mixture was transferred to dialysis tubing (12,000 to 14,000 molecular weight cutoff) and dialyzed against 1.8 liters of distilled water for 16 h at 4°C. After removal of the dialysis tubing, the dialysate was sterilized either by autoclaving or by filtration, and 3.6 ml of 1 M MgSO4 and 360 μl of 1 M CaCl2 were added to supplement the chelated Mg2+ and Ca2+.
Western blot analysis.
Total cell lysates were prepared from LB cultures grown overnight at 37°C. The cultures were normalized based on the optical density at 600 nm (OD600), and cell pellets were resuspended in 100 μl of phosphate-buffered saline (PBS) and 100 μl of 2× sample loading buffer (Bio-Rad). Samples were subjected to brief sonication followed by boiling for 5 min at 100°C. Fifteen-microliter aliquots of each sample were subjected to SDS-PAGE on a 4-to-20% Tris-glycine gel (Life Technologies). Separated proteins were transferred to nitrocellulose, blocked with 5% skim milk, and incubated with an OprD-specific polyclonal serum.
Construction of an isogenic β-lactamase library in E. coli.
An isogenic library of individual β-lactamases was constructed by first synthesizing a representative β-lactamase from each class and using that clone as a template for subsequent site-directed mutagenesis to construct each of the desired variants of the original gene. Point mutations that differentiated the variants within each class of β-lactamase were created based on the amino acid reference sequences available at www.lahey.org/studies. Each individual β-lactamase was cloned into pUCP26 (36) and transformed into E. coli DH5α, and the integrity of TET-resistant transformants was verified by DNA sequencing. β-Lactamase activity of each clone was assessed using the chromogenic cephalosporin CENTA (EMD Millipore) (3). MIC testing of this isogenic library was conducted according to CLSI guidelines (8, 9) using E. coli DH5α harboring empty pUCP26 as a negative control. The activity of MC-1 was compared with the siderophore-containing sulfactam compound BAL30072, along with a number of representative commercial antibiotics, to demonstrate the potential contribution of each β-lactamase to drug resistance. MIC50 and MIC90 values were calculated by identifying the drug concentration required to inhibit the growth of 50 and 90% of the total number of strains tested, respectively.
Cloning, expression, and purification of β-lactamases.
SHV-12, CTX-M-15, KPC-2, and TEM-1 β-lactamase-encoding genes were cloned into pET28a (Novagen) and transformed into E. coli BL21 (AI) cells. Transformants were grown at 37°C in terrific broth (Difco) containing 50 μg/ml kanamycin until the OD600 reached 0.6, and expression was induced with 0.2% l-(+)-arabinose (Sigma) overnight at 25°C. Cultures were harvested via centrifugation in a Beckman Avanti J-20 XPI centrifuge and JLA 8.1000 rotor at 7,000 rpm at 4°C for 20 min. The cell pellets were resuspended in buffer (50 mM potassium phosphate [pH 8.0], 500 mM sodium chloride, 10 mM imidazole, 10% glycerol, 1 mM magnesium chloride, 3 tablets of EDTA-free Complete protease inhibitor cocktail tablets [Roche], and 5 μl of Benzonase nuclease [Sigma]), and the proteins were liberated from the cells via a periplasmic lysis method involving 5 cycles of freezing the cell suspension with liquid nitrogen, followed by thawing in a water bath at 55°C. The soluble fraction was recovered by ultracentrifugation at 40,000 rpm for 40 min at 4°C. A 20-ml column of His-Select Ni resin (Sigma) was equilibrated in 50 mM potassium phosphate (pH 8.0) 500 mM sodium chloride, 10 mM imidazole, 10% glycerol (equilibration buffer), and the supernatant was loaded via gravity. The resin was washed with 10 column volumes (200 ml) of the equilibration buffer, followed by high-salt wash buffer (50 mM potassium phosphate [pH 8.0], 1.5 M sodium chloride, 10 mM imidazole, 10% glycerol, 20 mM β-mercaptoethanol). After another wash with equilibration buffer, elution buffer (50 mM potassium phosphate [pH 8.0], 500 mM sodium chloride, 250 mM imidazole, 10% glycerol) was added. Fractions were collected and analyzed both by SDS-PAGE and by using nitrocefin (Becton, Dickinson) to verify β-lactamase activity. The appropriate fractions were concentrated and loaded onto an S300 size exclusion column equilibrated with 50 mM potassium phosphate (pH 8.0), 400 mM sodium chloride, 5% glycerol. Purity was confirmed using SDS-PAGE, and a Cary50 UV/Vis spectrophotometer was used to determine the protein concentration from the predicted extinction coefficients for each enzyme.
ITC.
Proteins were dialyzed with 50 mM phosphate buffer, pH 7.2, and substrates were suspended in the same buffer. A VP-ITC (MicroCal) instrument was used to determine the molar enthalpy of hydrolysis (ΔH) for each β-lactamase (SHV-12, CTX-M-15, KPC-2, and TEM-1) and β-lactam (ampicillin and MC-1) combination. Each β-lactamase (ranging from 10 μM to 35 pM) was added to the isothermal titration calorimetry (ITC) cell (1.425 ml), and 10-μl injections of individual β-lactams (ranging from 4 mM to 300 μM) were performed. Following the first injection, the power decreases because of the heat generated by the hydrolysis reaction and returns to baseline when there is no more substrate to hydrolyze. The peaks from the thermograms were integrated using ORIGIN software (MicroCal). From this integration of the area of the peak and the known concentration of substrate, the molar enthalpy of hydrolysis was calculated. In a second experiment, the steady-state kinetic parameters were determined through 20 to 30 successive 10-μl injections of each β-lactam (ranging from 4 mM to 500 μM) into a catalytic concentration of β-lactamase (ranging from 4 μM to 75 pM) in the cell. In this experiment, the concentration of β-lactamase used does not cause significant changes in substrate levels over the 2- to 3-min duration of each injection, resulting in an accumulation of β-lactam concentration over time. The molar enthalpy of the hydrolysis reaction, together with the resulting thermogram of power versus time, allows for the calculation of the rate of hydrolysis for each injection. From the conversion to a plot of rate versus substrate concentration data performed by the software, the steady-state kinetic parameters kcat and Km for each β-lactamase/β-lactam pair were calculated.
Resistance frequency and stability determination.
Resistance frequencies were determined by growing P. aeruginosa PAO1 in LB broth to an OD600 of 0.5 (5.0 × 108 CFU/ml), concentrating 100-fold in fresh medium, and plating 100 μl on MHB plates (solidified with 1.5% agarose) containing a range of MC-1 concentrations. Serial dilutions of this suspension were also plated on nonselective medium to determine the total number of cells plated on each drug-containing plate. For comparative purposes, the frequency of resistance of this strain against aztreonam (ATM), an in-class comparator, was determined. Plates were incubated at 37°C for 40 h prior to colony counting, and resistance frequencies were calculated by dividing the total number of colonies on each plate by the original number of cells plated. To assess resistance stability, recovered colonies were first grown in the absence of drug pressure and frozen at −80°C. Frozen stocks were then used to inoculate fresh medium and grown overnight at 37°C. These overnight cultures were used to conduct MIC testing against the compounds from which they were raised. Strains that demonstrated a significant shift in the MIC (relative to the parental strain) were further characterized.
Construction of an isogenic panel of P. aeruginosa outer membrane siderophore receptors.
To evaluate the mechanism of entry of MC-1, a panel of isogenic TonB-dependent outer membrane siderophore receptor knockout mutants was constructed in P. aeruginosa PAO1. Below, Table 5 shows the open reading frame (ORF) designations and amino acids deleted in these mutants. The primers used to generate deletion mutants in each ORF are listed in Table S1 of the supplemental material. Knockout constructs were made either by splicing by overlap extension-PCR or by restriction digestion to remove a significant internal fragment of each gene and replacing the internal sequence with the GENr cassette from pPS856 (18). The mutated fragments were cloned into pEX100T (31) or pEX18ApGW (7) and delivered to PAO1 via conjugation or electroporation. GEN-resistant colonies recovered from PIA plates were screened for double-crossover recombination events by counterselecting on 5% sucrose-containing LB plates and further confirmed to be cured of the delivery plasmids by plating on LB containing 250 μg/ml carbenicillin. All mutants were verified by PCR and subsequent DNA sequencing of the resulting amplicons. When necessary, primary mutations were unmarked by conjugating pFLP2 (18) into each mutant strain to utilize the FLP recombination target sequences that flank the GENr cassette from pPS856. Exconjugants were confirmed to be GENs and verified by PCR, prior to introducing another knockout plasmid to delete an additional gene.
Table 5.
Genes, mutant deleted regions, and MICs of the P. aeruginosa PAO1 isogenic siderophore receptor mutant panel
| Gene(s) and ORF(s) | Protein size/deletion (aa) | MIC in MHB/MIC in low-iron medium (μg/ml) for strain: |
||
|---|---|---|---|---|
| MC-1 | BAL30072 | ATM | ||
| PAO1 | NAa | 0.25/0.25 | 4/2 | 8/4 |
| piuA/PA4514 | 753/4–750 | 8/0.25 | 16/2 | 4/4 |
| PA0470 | 803/233–643 | 0.25/0.25 | NDb | 8/4 |
| pirA/PA0931 | 743/28–606 | 0.5/0.25 | 4/2 | 4/4 |
| PA1322 | 733/53–531 | 0.25/0.125 | 4/2 | 8/4 |
| pfeA/PA2688 | 747/83–703 | 0.25/0.5 | 4/2 | 8/4 |
| cirA/PA1922 | 653/2–649 | 0.5/0.25 | ND | 8/2 |
| fecA/PA3901 | 784/1–782 | 0.06/0.25 | ND | 8/4 |
| fptA/PA4221 | 720/129–205 | 0.25/0.25 | 4/2 | 8/4 |
| foxA/PA2466 | 820/1–808 | 0.25/0.25 | ND | 8/4 |
| fpvA/PA2398 | 815/1–806 | 0.25/0.06 | 4/2 | 8/4 |
| fpvB/PA4168 | 802/1–796 | 0.25/0.25 | 4/2 | 8/4 |
| phuR/PA4710 | 765/95–475 | 0.25/0.25 | ND | 8/4 |
| hasR/PA3408 | 892/180–673 | 0.25/0.25 | ND | 8/4 |
| fpvA fpvB | 0.5/0.125 | 4/2 | 8/4 | |
| phuR hasR | 0.25/0.125 | ND | 8/4 | |
| piuA fpvA | 8/0.5 | 16/4 | 8/4 | |
| piuA fptA | 8/2 | 16/2 | 8/4 | |
| piuA cirA | 8/1 | ND | 8/4 | |
| piuA PA0470 | 8/1 | ND | 8/2 | |
| piuA PA0931 | 32/32 | 64/64 | 8/4 | |
| piuA PA1322 | 8/1 | 16/2 | 4/4 | |
| piuA PA2688 | 8/2 | 16/4 | 8/4 | |
| PA0931 PA2688 | 0.5/0.125 | 4/2 | 4/4 | |
| PA0931 PA1322 PA2688 | 1/0.125 | 4/2 | 8/4 | |
| piuA PA0931 PA2688 | 32/32 | 64/64 | 4/4 | |
| piuA PA0931 PA1322 | 32/16 | 64/64 | 4/4 | |
| piuA PA2688 PA1322 | 8/2 | 16/2 | 4/4 | |
| piuA PA0931 PA1322 PA2688 | 32/16 | 64/64 | 4/4 | |
| piuA PA0470 PA0931 PA1322 PA2688 | 32/32 | ND | 4/4 | |
NA, not applicable; ND, not determined.
Murine septicemia model of MC-1 efficacy.
Prior to each study, bacteria were grown overnight in brain heart infusion broth (Difco) with shaking at 150 rpm in an ambient air incubator at 37°C. The organisms were adjusted to an OD600 of 1.0 to 1.1 by using fresh medium. Once the inocula were normalized, 10−5 dilutions were made in sterile 3% brewer's yeast as the bacterial adjuvant (final inoculum of 104 CFU/ml). CF-1 female mice were infected intraperitoneally with 0.5 ml of the final inoculum preparation for each organism. MC-1 was prepared in 50 mM citrate buffer (pH 4.2), and 0.2 ml of each dose level was administered subcutaneously at 0.5 and 4.0 h postinfection. A 64-fold dose titration from the 50-mg/kg/dose to the 0.78-mg/kg/dose was evaluated in this study. Survivorship was recorded for 4 days postinfection (the day of infection was considered day zero). The 50% protective dose (PD50) values and their associated 95% confidence intervals were calculated based on the number of mice that survived on day 4. All procedures performed on these animals were in accordance with regulations and established guidelines and were reviewed and approved by an Institutional Animal Care and Use Committee.
RESULTS AND DISCUSSION
MC-1 is efficacious against porin-mutated strains of P. aeruginosa.
P. aeruginosa β-lactam resistance rates are increasing dramatically, partially due to the evolution and spread of ESBLs, as well as mutational events that decrease outer membrane permeability, thereby preventing entry of the drug into the cell. The latter has significantly affected the efficacy of the carbapenem class of β-lactam antibiotics due specifically to the loss of the OprD porin channel (34), which further emphasizes the need for new drug molecules that can circumvent this resistance mechanism. To demonstrate that changes in outer membrane permeability would not affect the potency of MC-1, we constructed both an isogenic oprD mutant of P. aeruginosa PAO1 and an in trans-complemented oprD mutant, and we conducted MICs with MC-1 and meropenem (MEM), a carbapenem known to be affected by porin downregulation. In addition, several recent clinical isolates with OprD deficiencies (as assessed by Western blot analysis) were identified, and the efficacies of this same set of antibiotics were compared. As shown in Table 1, MC-1 MICs against ΔoprD and the oprD-downregulated clinical isolates were indistinguishable from PAO1, while the MICs of MEM increased 16- to 128-fold in these engineered or clinical strains. This demonstrated that MC-1 resistance, unlike carbapenem antibiotics, cannot be achieved by OprD-related decreases in outer membrane permeability and that this novel molecule is efficacious against carbapenem-resistant clinical isolates of P. aeruginosa that have evolved this resistance mechanism.
Table 1.
Downregulation of the major P. aeruginosa outer membrane porin OprD did not affect the activity of MC-1, as indicated by the lack of shift in MICs for isogenic mutants and clinical isolates
| Strain | OprD expression | MIC (μg/ml) |
|
|---|---|---|---|
| MC-1 | MEM | ||
| PAO1 | + | 0.5 | 0.5 |
| PAO1 ΔoprD | − | 0.5 | 8 |
| PAO1 ΔoprD/pUCP26 | − | 0.5 | 8 |
| PAO1 ΔoprD/pUCP26-oprD | + | 0.5 | 0.5 |
| PA4992 | − | 0.06 | 64 |
| PA4993 | − | 0.06 | 64 |
| PA4996 | − | 0.25 | 16 |
| PA4997 | − | 0.25 | 16 |
| PA4999 | − | 0.125 | 64 |
| PA5000 | − | 0.5 | 64 |
| PA1561-08 | − | 0.25 | 32 |
MC-1 is not resisted by the activity of various RND efflux pumps in P. aeruginosa.
Efflux pump expression is a clinically relevant resistance mechanism that many Gram-negative pathogens use to resist the action of multiple classes of antibiotics, including β-lactams (34). We therefore examined whether MC-1 resistance in P. aeruginosa could be achieved by efflux of the compound. To address this, we utilized an extensive panel of genetically engineered P. aeruginosa strains derived from PAO1 and measured the MIC values of MC-1 and two comparator agents known to be efflux pump substrates. Table 2 shows the activity and compound spectrum of several resistance-nodulation-cell division (RND) efflux pumps expressed by P. aeruginosa represented by different strains in this panel. The ability of the MexAB-OprM RND efflux pump to aid in resistance to both ATM and ciprofloxacin (CIP) was demonstrated by the elevation in each MIC (relative to the parent strain) when the activity of MexR, the repressor for this particular efflux pump, was abolished by disruption of the mexR gene in strain PA142. This was further supported by the MIC results of PAO200, which cannot express MexAB-OprM due to a mutation, for which the ATM and CIP MICs were reduced 16- and 8-fold, respectively, relative to the PAO1 parental strain. Importantly, the activity of MC-1 was not compromised by the expression of any of the P. aeruginosa RND efflux pump systems represented in this panel, as evidenced by the lack of a MIC increase in any of the strains evaluated. Further confirmation that efflux pump expression/upregulation does not contribute to MC-1 resistance was obtained when various clinical isolates of P. aeruginosa that have been characterized to have hyperactive efflux pump systems did not show elevated MICs relative to susceptible strains, like PAO1 (data not shown). We speculated that this lack of efflux potential as a viable resistance mechanism is at least partially due to the siderophore component of this compound, since ATM, which is structurally similar to MC-1, was shown to be a substrate for the MexAB-OprM efflux pump (Table 2). With that in mind, it can also be inferred that the physical positioning of the siderophore's conjugation to the β-lactam is important in achieving this efflux-resistant phenotype, since BAL30072, which was tested using a similar panel of isogenic efflux pump up- and downregulated strains of PAO1, appeared to have a liability for this clinically relevant resistance mechanism (27).
Table 2.
MICs for isogenic efflux pump-expressing mutants demonstrated that efflux does not contribute to MC-1 resistance in P. aeruginosa
| Strain | Genotype | MIC (μg/ml) |
||
|---|---|---|---|---|
| MC-1 | ATM | CIP | ||
| PAO1 | WT (MexAB-OprM basal) | 0.25 | 4 | 0.125 |
| PA142 | ΔmexR (MexAB-OprM overproduced) | 0.5 | 16 | 0.5 |
| PAO200 | ΔmexAB-oprM | 0.125 | 0.25 | 0.016 |
| PAO200-2 | ΔmexAB-oprM nfxB (MexCD-OprJ+) | 0.125 | 0.25 | 0.25 |
| PAO238 | ΔmexAB-oprM ΔmexCD-oprJ | 0.06 | 0.5 | <0.008 |
| PAO253 | ΔmexAB-oprM mexS/T (MexEF-OprN+) | 0.25 | 0.125 | 2 |
| PAO255 | ΔmexAB-oprM ΔmexEF-oprN | 0.125 | 0.25 | 0.5 |
| PAO267 | ΔmexAB-oprM mexZ (MexXY+) | 0.06 | 0.25 | 0.016 |
| PAO280 | ΔmexAB-oprM ΔmexXY | 0.25 | 0.25 | 0.016 |
| PAO238-1 | ΔmexAB-oprM ΔmexCD-oprJ ΔmexL (MexJK+) | 0.125 | 0.5 | <0.008 |
| PAO314 | ΔmexAB-oprM ΔmexCD-oprJ ΔmexJK | 0.125 | 0.5 | <0.008 |
| PAO325 | ΔmexAB-oprM ΔmexCD-oprJ ΔmexJK ΔmexXY | 0.06 | 0.5 | <0.008 |
| PAO397 | ΔmexAB-oprM ΔmexCD-oprJ ΔmexEF-oprN ΔmexJK ΔmexXY | 0.06 | 0.5 | <0.008 |
Genetic and biophysical assays demonstrated that MC-1 is not a good substrate for multiple classes of β-lactamases.
Given that different classes of β-lactamases (designated Ambler classes A to D) have been thoroughly studied and described to inactivate multiple classes of β-lactam antibiotics (5, 19), it was crucial for us to determine whether any β-lactamases would be capable of hydrolyzing MC-1. To address this, we took both genetic and biophysical approaches to study resistance development and substrate-enzyme interactions. First, a comprehensive library of isogenic β-lactamase-producing strains was constructed in the fully susceptible strain E. coli DH5α. This approach has been utilized before to assess substrate specificity differences and propensities for resistance development (2, 10), but one significant difference between those previous studies and our approach was the isogenic nature of our library. We synthesized several variants/subtypes of each class of β-lactamase, cloned them into the same vector under the control of the same promoter, and expressed them in the same parental strain. This strategy was employed to normalize, as much as was reasonably achievable, the relative amounts of enzyme produced when comparing one enzyme variant to another, so that differences seen in MIC values were less likely to be attributable to differences in amounts of enzyme produced. Table 3 shows the MIC50 and MIC90 values of the various classes of β-lactamases examined in our study. The substrates used for these studies were chosen to demonstrate the broad spectrum of activity that many of these enzymes have on different classes of β-lactam antibiotics. These data showed that while all classes of β-lactamases tested are capable of cleaving several types of marketed β-lactam antibiotics, MC-1 does not appear to be a substrate for any of them. The MIC data for each individual enzyme variant can be found in Table S2 of the supplemental material.
Table 3.
An isogenic β-lactamase library demonstrated that MC-1 is not a good substrate for various types of clinically relevant β-lactamases
| β-Lactamase | No. of strains tested | MIC50/MIC90 in μg/ml (range) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MC-1 | MC-1 + 4 μg/ml tazobactam | BAL30072 | Aztreonam | Meropenem | Cefepime | Cefotaxime | Cefotaxime + clavulanic acid at 4 μg/ml | Ceftazidime | Ceftazidime + clavulanic acid at 4 μg/ml | ||
| SHV | 39 | 0.125/0.25 (0.06–0.5) | 0.125/0.125 (0.03–0.25) | 0.25/>64 (0.06 to >64) | 0.5/>64 (0.125 to >64) | 0.015/0.03 (0.015–0.03) | 0.125/1 (0.03–2) | 0.06/8 (0.03–16) | 0.03/0.06 (0.03–0.125) | 1/64 (0.25 to >64) | 0.25/0.25 (0.125–0.25) |
| TEM | 38 | 0.03/0.06 (0.03–0.25) | 0.03/0.06 (0.015–0.125) | 0.06/16 (0.015 to >64) | 0.125/32 (0.03–64) | 0.015/0.03 (0.015–0.03) | 0.06/1 (0.015–2) | 0.03/2 (0.015–4) | 0.03/0.06 (0.015–0.06) | 0.25/32 (0.06 to >64) | 0.125/0.25 (0.06–0.5) |
| KPC* | 5 | 0.06/0.06 (0.03–0.06) | 0.06/0.06 (0.03–0.06) | 0.06/1 (0.06–1) | 4/32 (2–32) | 0.06/0.06 (0.015–0.06) | 0.25/0.25 (0.125–0.25) | 0.5/0.5 (0.25–0.5) | 0.03/0.03 (0.03–0.03) | 2/8 (0.5–8) | 0.125/0.25 (0.125–0.25 |
| CTX-M* | 5 | 0.125/0.25 (0.06–0.25) | 0.06/0.06 (0.03–0.06) | 1/1 (0.125–1) | 32/>64 (8 to >64) | 0.25/0.25 (0.25–0.25) | 8/8 (1–8) | >64/>64 (32 to >64) | 0.06/0.06 (0.03–0.06) | 2/32 (1–32) | 0.125/0.25 (0.125–0.25) |
| GES* | 3 | 0.06/0.06 (0.06) | 0.06/0.06 (0.06–0.06) | 0.25/1 (0.125–1) | 0.5/0.5 (0.25–0.5) | 0.25/0.25 (0.125–0.25) | 0.125/0.5 (0.03–0.5) | 2/4 (0.25–4) | 0.06/2 (0.03–2) | 16/16 (1–16) | 0.25/16 (0.125–16) |
| VEB* | 4 | 0.125/0.5 (0.06–0.5) | 0.06/0.5 (0.06–0.5) | 2/16 (2–16) | 16/32 (2–32) | 0.125/0.25 (0.125–0.25) | 0.5/0.5 (0.015–0.5) | 4/16 (0.125–16) | 0.03/0.06 (0.03–0.06) | >64/>64 (0.5 to >64) | 0.125/0.2 (0.03–0.25) |
| MβL* | 7 | 0.03/0.125 (0.03–0.125) | 0.03/0.125 (0.03–0.125) | 0.125/0.25 (0.03–0.25) | 0.125/0.5 (0.06–0.5) | 4/16 (0.5–16) | 2/4 (0.06–4) | 32/64 (8–64) | 16/64 (4–64) | 64/>64 (1 to >64) | 64/>64 (0.5 to >64) |
| OXA | 12 | 0.25/0.25 (0.125–32) | 0.125/0.25 (0.06–1) | 0.125/0.25 (0.06–16) | 0.125/1 (0.125–64) | 0.015/0.03 (0.015–0.03) | 0.06/0.125 (0.03–4) | 0.06/0.25 (0.03–32) | 0.03/0.06 (0.03–0.06) | 0.25/0.5 (0.25–32) | 0.25/0.25 (0.125–0.25) |
Of particular significance was the inclusion and testing of the New Delhi metallo-β-lactamase (NDM-1) (22), as well as representatives from the VIM and IMP families of class B metallo-β-lactamases (MβLs), none of which showed any hydrolysis-mediated increases in the MIC (Table 3). Other β-lactamases known to be closely associated with P. aeruginosa, such as variants of VEB (29) and GES (6), also did not demonstrate any detectable MC-1 cleavage activity. A small sampling of class C enzymes were also tested, none of which showed elevated MICs relative to the empty vector control (see Table S2 in the supplemental material). Although not shown, the MIC of a site-directed ampR deletion mutant in P. aeruginosa PAO1 remained unchanged relative to the parent strain which, taken together with susceptible MICs seen with P. aeruginosa clinical isolates known to have hyperproduction of the chromosomal AmpC β-lactamase, suggest that this resistance mechanism does not play a role in MC-1 resistance. In our analysis we also noted that the KPC variant MICs against meropenem did not indicate that these enzymes cleaved this carbapenem antibiotic, contrary to the phenotype that their name would indicate. After confirming the construct integrity in these clones and seeing elevated MICs with other non-carbapenem β-lactams (seeTable S2), we speculated that the level of expression the lac promoter provides from pUCP26 in the absence of isopropyl-β-d-thiogalactopyranoside induction is insufficient for providing carbapenem resistance in E. coli.
We postulated that MC-1 resistance to β-lactamase activity is due both to the presence and positioning of the siderophore moiety as well as to the linker that joins it to the monocyclic β-lactam ring. Preliminary computational modeling of MC-1 with various ESBL-type β-lactamases showed that these enzymes cannot effectively dock with this compound, indicating that cleavage of the β-lactam ring is sterically hindered by the presence of the linker and siderophore components of the molecule (V. Shanmugasundaram, personal communication). As was the case for resisting the activity of efflux pumps, these data support the notion that the strategic positioning of the siderophore on MC-1 provides added benefit beyond facilitating transport across the outer membrane. There are clear differences in the β-lactamase hydrolysis potentials between MC-1 and BAL30072 (Table 3), the latter of which has been described to have some ESBL-mediated resistance liabilities (27). Additionally, the MIC profile that this library showed against ATM, which is more closely related structurally to MC-1 than BAL30072, further reinforced this important difference.
To confirm our genetic data by using an alternate approach, we purified four clinically relevant representative β-lactamases (KPC-2, SHV-12, TEM-1, and CTX-M-15) and evaluated their ability to bind and subsequently hydrolyze the β-lactam ring of MC-1. We first attempted to apply traditional spectrophotometric methods to detect β-lactamase-mediated MC-1 hydrolysis, but we quickly found that we could not detect significant, reliable absorbance differences between the parent molecule and its ring-open derivative (data not shown). As a result, we explored the utility of ITC, which is used to measure the amount of compensatory power needed to maintain constant temperature, relative to an adjacent reference cell, as a result of changes in heat generated by substrate recognition and subsequent cleavage (1). These changes in heat are then used to calculate both the Km and kcat values, providing a catalytic efficiency value for each enzyme with each substrate. As this method demonstrated that all β-lactamases tested bind to and cleave ampicillin extremely efficiently, we compared the relative rates of hydrolysis of MC-1 to those of ampicillin and expressed these values as the percentage of ampicillin catalytic efficiency (Table 4). These data demonstrated that each β-lactamase, although capable of cleaving ampicillin readily, did not exhibit similar hydrolytic properties when MC-1 was used as a substrate. In fact, the MC-1 reactivity of TEM-1 was so miniscule that accurate Km and kcat calculations were not possible. To further validate our ITC data, we attempted to measure MC-1 hydrolysis by using mass spectrometry-based methods and these same enzyme preparations, but we were unable to detect any (<0.1%) ring-open product, even after several hours of coincubation of the various β-lactamases and MC-1 (data not shown). Therefore, we are confident that this novel ITC approach effectively complemented our genetic approach for understanding the susceptibilities, if any, of our novel siderophore-conjugated monocarbam to many types of clinically relevant β-lactamases.
Table 4.
ITC indicated that MC-1 is not significantly hydrolyzed by clinically-relevant β-lactamases
| Compound | SHV-12 |
KPC-2 |
TEM-1 |
CTX-M-15 |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| kcat (s−1) | Km (μM) | kcat/Km (μM−1 s−1) | Relative kcat/Km (μM−1 s−1) | kcat (s−1) | Km (μM) | kcat/Km (μM−1 s−1) | Relative kcat/Km (μM−1 s−1) | kcat (s−1) | Km (μM) | kcat/Km (μM−1 s−1) | Relative kcat/Km (μM−1 s−1) | kcat (s−1) | Km (μM) | kcat/Km (μM−1 s−1) | Relative kcat/Km (μM−1 s−1) | |
| Ampicillin | 116 | 13.3 | 8.72 | 100 | 128 | 145 | 0.883 | 100 | 2,170 | 60 | 36 | 100 | 545 | 39 | 14 | 100 |
| MC-1 | 0.39 | 25 | 0.016 | 0.183 | 0.074 | 705 | 0.0001 | 0.011 | <0.01 | NDa | ND | 0.48 | 202 | 0.002 | 0.014 | |
ND, not determined.
The frequency, but not mechanism, of resistance to MC-1 is consistent with other marketed β-lactam antibiotics.
Given that MC-1 is one of the first examples of an antimicrobial compound that exploits iron acquisition for efficacy and that it appears to be unaffected by resistance mechanisms commonly associated with clinical isolates of P. aeruginosa, we sought to understand what resistance mechanisms could be employed by Gram-negative pathogens to resist its activity. We used standard population analysis methods to determine the frequency of resistance of P. aeruginosa PAO1 against MC-1 and ATM, which was included as an in-class comparator. From these experiments, we found the frequency of resistance to MC-1 to be 9.3 × 10−7 at 4× MIC (2 μg/ml), with no recoverable colonies at concentrations of >16× MIC. By comparison, the frequency of resistance to ATM was 8.6 × 10−7 at 4× MIC (8 μg/ml), again with no colonies detected at concentrations of>16× MIC. While these attributes are not significantly different, the mechanisms of resistance contributing to the resistance frequencies against these two compounds are quite distinct. While recovered ATM-resistant mutants were found to be cross-resistant to cephalosporins, likely indicative of porin mutations, derepressed ampC, and/or upregulated efflux, such a phenotype was not seen with any MC-1r mutants (data not shown). To identify potential MC-1 resistance mechanisms, we performed whole-genome sequencing on a stable resistant isolate (MIC, 8 μg/ml) of PAO1. From this, we identified a single cytosine base pair insertion within the piuC (PA4515) open reading frame, which resulted in a frameshift mutation that ultimately led to the premature termination of translation of the PiuC protein. While the function of this gene is still unknown, it has been shown to be iron regulated (25), and the gene immediately preceding it is predicted to encode a TonB-dependent outer membrane siderophore receptor. The expression of this adjacent gene, piuA (PA4514), was determined to be significantly diminished in the piuC frameshift mutant by reverse transcription-PCR (data not shown), and a site-directed deletion mutant of piuA was constructed. Consistent with the original whole-genome-sequenced mutant, the piuA deletion mutant showed a 32-fold increase in the MIC over the parent strain when assayed under standard conditions (Table 5). Using directed Sanger sequencing, we characterized 9 other MC-1r isolates generated via this method and found the piuAC locus to be mutated in 5 of them. Most importantly, our mutant characterization efforts did not identify any established, clinically relevant antibiotic resistance determinants as mechanisms by which P. aeruginosa resists MC-1, including those that have been described to be involved in ATM resistance (23, 30).
The mechanism of entry of MC-1 is dependent on specific outer membrane receptors in P. aeruginosa.
Based on the preliminary results obtained with the piuAC mutants, we wanted to identify which of the other putative siderophore receptors could be used for MC-1 uptake. The P. aeruginosa genome includes at least 25 annotated TonB-dependent outer membrane siderophore receptors (37), and the potential involvement of many of them in MC-1 uptake was determined by constructing an extensive panel of isogenic mutants from the parent strain PAO1. As shown in Table 5, mutations in 13 separate genes were created, either alone or in combination with other receptor mutations, to conduct MIC experiments to identify the receptors (or combination of receptors) that led to an increased MIC relative to the parent strain. Receptor mutants were subjected to MIC testing against MC-1, BAL30072, and ATM, which was included to demonstrate specific changes in the efficacies of MC-1 and/or BAL30072 relative to each mutation. Additionally, MICs were tested in both iron-replete Mueller-Hinton broth (MHB) and an iron-chelated version of MHB (see Materials and Methods) to determine whether the efficacy of MC-1 was dependent upon the amount of free iron in the medium. This analysis indicated that the mutant lacking PiuA (encoded by PA4514) was the only strain with a single receptor deletion that had an increased MIC relative to PAO1 (32-fold). This also seemed to be the case for BAL30072, despite starting with slightly elevated MICs against the wild-type PAO1 strain. It was previously reported that BAL30072 activity is unaffected by the loss of various siderophore receptors (27); however, a piuA mutant was not generated or tested for MIC changes relative to the wild-type PAO1 strain in that earlier study. In addition, bipyridyl was used as an iron chelator in both MIC tests as well as resistance frequency determination studies, which corresponded to the low-iron conditions described in this report. Based on the data presented here, a PiuA-deficient strain would have been unlikely to be identified, given the fully susceptible nature of the ΔpiuA mutant in iron-depleted medium.
It was interesting that while the ΔpiuA MIC increased in the iron-replete medium, the corresponding MICs were indistinguishable from the parent strain when tested in the low-iron medium. This suggested the involvement of another siderophore receptor that mediates MC-1 entry in the absence of a functional PiuA. The introduction of additional deletion mutations in the ΔpiuA background revealed that the mutation of a second siderophore receptor, PirA (PA0931), caused an additional 4-fold increase in the MC-1 MIC relative to the ΔpiuA strain (Table 5). This phenotype was confirmed by random transposon mutagenesis of the ΔpiuA strain, in which mutants with increased MC-1 resistance levels were selected, and independently demonstrated that PirA is involved in MC-1 transport (data not shown). Unlike the ΔpiuA mutant, the MIC increase seen in the ΔpiuA ΔpirA double mutant was evident in both iron-replete and iron-limiting media, suggesting that these are the major receptors responsible for MC-1 uptake. This determination was further supported by the lack of MIC increase when any other receptors (or combination of receptors) were deleted in the ΔpiuA background. Again, consistent with the results obtained with MC-1, the activity of BAL30072 was further diminished in iron-replete medium when both piuA and pirA were deleted, and this MIC shift was also seen in the low-iron medium (Table 5). These uptake phenotype similarities shared by MC-1 and BAL30072 are not surprising, however, given the similar catechol-based nature of the siderophore moieties that they possess.
It should be noted that single mutants with deletions in the receptors responsible for uptake of the major siderophores in P. aeruginosa, pyoverdine (FpvA) and pyochelin (FptA), did not show an increase in the MIC relative to PAO1 (Table 5). Further supporting the lack of FpvA or FptA involvement in MC-1 uptake, MIC shifts were not detected when ΔpiuA ΔfpvA or ΔpiuA ΔfptA mutants were compared to ΔpiuA. This result was not surprising, since the siderophore on MC-1 does not resemble either pyoverdine or pyochelin. Instead, the receptors that promote the uptake of the major E. coli siderophore enterobactin are those that appear to be involved in the import of this compound into P. aeruginosa cells. PiuA is part of the Fiu family of TonB-dependent outer membrane siderophore receptors (COG4774), which has been described to import similar types of molecules previously (24). PirA and its highly conserved homolog, PfeA (PA2688), have been extensively described for their involvement in enterobactin uptake in E. coli (15). Interestingly, while the ΔpiuA ΔpirA double mutant showed elevated MICs relative to the ΔpiuA single mutant, the ΔpirA single mutant did not demonstrate any increase in the MIC relative to PAO1 (Table 5). This suggested a hierarchy for MC-1 uptake in P. aeruginosa, with PiuA being the primary uptake mechanism and PirA being utilized secondarily in the absence of a functional PiuA. It is also interesting that the ΔpiuA ΔpfeA mutant did not exhibit the same elevation in MIC as ΔpiuA ΔpirA, despite the high degree of similarity shared between PfeA and PirA. The possibility of PfeA acting as a tertiary receptor was refuted by the lack of MIC shift (relative to the ΔpiuA ΔpirA double mutant) when all three receptors were deleted (Table 5).
The primary in vitro MC-1 resistance mechanism in P. aeruginosa does not translate in vivo.
Given the frequency of resistance and corresponding MIC shift associated with a single mutation resulting in downregulation of piuA, it was concerning that clinical resistance to MC-1 could be achieved from a single mutational event that resulted in the lack of production of the PiuA receptor. In order to address this possibility, the in vivo efficacy of MC-1 was tested against PAO1, ΔpiuA, and ΔpiuA ΔpirA in a murine septicemia model of infection. The results showed that MC-1 retained significant activity against the ΔpiuA mutant in vivo (Table 6), despite having an elevated MIC in MHB in vitro. This indicated that infections with PiuA-deficient mutants could still be effectively treated with MC-1 and was further supported by the pharmacokinetics of this compound in vivo, as concentrations needed to effectively treat the ΔpiuA mutant are reasonably achievable (J. Hardink, personal communication). This in vivo result strongly correlated with the iron-depleted MHB MIC data obtained in vitro, where no elevation in the MIC was seen compared to the parent strain. With this correlation in mind, it was not surprising to find that the ΔpiuA ΔpirA double mutant had a significantly elevated PD50 compared to both PAO1 and ΔpiuA (Table 6). However, in order to achieve this high-level resistance in vivo, two separate mutational events would have to occur within the same bacterial cell to prevent the production of both PiuA and PirA. The probability of such a multistep event occurring is extremely low and is supported by our inability to isolate such a mutant in studies evaluating frequency of resistance with MC-1. For this reason, we feel confident that clinical resistance to this siderophore-conjugated monocarbam will not occur as a result of decreased or abolished drug entry.
Table 6.
In vivo resistance to MC-1 requires inactivation of two distinct outer membrane siderophore receptors
| Strain | PD50 (mg/kg/dose) | 95% CI |
|---|---|---|
| PAO1 | <0.78 | 0.9–0.9 |
| ΔpiuA | 2.9 | 0.8–4.9 |
| ΔpiuA ΔpirA | >50 | NDa |
ND, not determined.
In summary, we have developed and characterized a novel β-lactam with potent Gram-negative activity that circumvents the most common, clinically relevant antibiotic resistance mechanisms identified in clinical isolates to date. We have demonstrated that the inclusion of the siderophore moiety affords MC-1 a substantial advantage over in-class comparators, like ATM, which can be resisted by several different mechanisms, including lowering of outer membrane permeability, the activity of efflux pumps, and the enzymatic activity of a wide spectrum of β-lactamases. Unlike ATM, however, we identified the primary first-step resistance mechanism against MC-1 to be related to compound transport across the outer membrane, which was not surprising, given the dependence on siderophore-mediated iron acquisition for this compound to be efficacious. The data generated in this study also provide evidence that the siderophore location on MC-1 affords it a significant advantage over other described siderophore-linked β-lactams, like BAL30072, as its resistance potential via efflux and the action of β-lactamases was not observed. Encouragingly, the in vitro-generated MC-1r mutants were not equally resistant to the action of this compound in vivo unless multiple inactivating mutations were identified in the same cell. Such a genotype has an extremely low probability of preexisting in successful clinical isolates or of occurring spontaneously in the future. Taken together, these data support the concept of incorporating features into antibacterial compounds that allow for the hijacking of normal cellular processes in pathogens of interest. MC-1 may potentially represent a best-in-class molecule that will withstand the action of multiple antibiotic resistance mechanisms and provide clinicians with a weapon in the constant battle with MDR Gram-negative pathogens like P. aeruginosa.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Herbert Schweizer for providing us with the strains comprising the P. aeruginosa efflux panel and to Mike Vasil for supplying the PAO1 and ΔfptA strains. We thank Richard Miller for assisting in the design of the β-lactamase library. We also thank Chris Zook and Steve Dunham for coordinating whole-genome sequencing efforts and Alita Miller for critically reading the manuscript.
Footnotes
Published ahead of print 1 October 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Bains G, Freire E. 1991. Calorimetric determination of cooperative interactions in high affinity binding processes. Anal. Biochem. 192:203–206 [DOI] [PubMed] [Google Scholar]
- 2. Barlow M, Hall BG. 2002. Predicting evolutionary potential: in vitro evolution accurately reproduces natural evolution of the TEM beta-lactamase. Genetics 160:823–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bebrone C, et al. 2001. CENTA as a chromogenic substrate for studying β-lactamases. Antimicrob. Agents Chemother. 45:1868–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Best DJ, et al. 1990. Structure-activity relationships of some arylglycine analogues and catechol isosteres of BRL 36650, a 6 alpha-formamido penicillin. J. Antibiot. (Tokyo) 43:574–577 [DOI] [PubMed] [Google Scholar]
- 5. Bush K, Jacoby GA. 2010. Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 54:969–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Castanheira M, Mendes RE, Walsh TR, Gales AC, Jones RN. 2004. Emergence of the extended-spectrum beta-lactamase GES-1 in a Pseudomonas aeruginosa strain from Brazil: report from the SENTRY antimicrobial surveillance program. Antimicrob. Agents Chemother. 48:2344–2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Choi K-H, Schweizer H. 2005. An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol. 5:30 doi:10.1186/1471-2180-5-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. CLSI 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically: approved standard, M07. Clinical Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 9. CLSI 2009. Performance standards for antimicrobial susceptibility testing: 19th informational supplement. M100 Clinical Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 10. Ehrhardt AF, Sanders CC, Moland ES. 1999. Use of an isogenic Escherichia coli panel to design tests for discrimination of beta-lactamase functional groups of Enterobacteriaceae. Antimicrob. Agents Chemother. 43:630–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Farra A, Islam S, Strålfors A, Sörberg M, Wretlind B. 2008. Role of outer membrane protein OprD and penicillin-binding proteins in resistance of Pseudomonas aeruginosa to imipenem and meropenem. Int. J. Antimicrob. Agents 31:427–433 [DOI] [PubMed] [Google Scholar]
- 12. Figurski DH, Helinski DR. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. U. S. A. 76:1648–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Flanagan ME, et al. 2011. Preparation, Gram-negative antibacterial activity, and hydrolytic stability of novel siderophore-conjugated monocarbam diols. ACS Med. Chem. Lett. 2:385–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fung-Tomc J, et al. 1997. Antibacterial activity of BMS-180680, a new catechol-containing monobactam. Antimicrob. Agents Chemother. 41:1010–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ghysels B, et al. 2005. The Pseudomonas aeruginosa pirA gene encodes a second receptor for ferrienterobactin and synthetic catecholate analogues. FEMS Microbiol. Lett. 246:167–174 [DOI] [PubMed] [Google Scholar]
- 16. Han S, et al. 2010. Structural basis for effectiveness of siderophore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 107:22002–22007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Helfand MS, Bonomo RA. 2005. Current challenges in antimicrobial chemotherapy: the impact of extended-spectrum β-lactamases and metallo-β-lactamases on the treatment of resistant Gram-negative pathogens. Curr. Opin. Pharmacol. 5:452–458 [DOI] [PubMed] [Google Scholar]
- 18. Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomal located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86 [DOI] [PubMed] [Google Scholar]
- 19. Lee JH, Bae IK, Hee Lee S. 2012. New definitions of extended-spectrum β-lactamase conferring worldwide emerging antibiotic resistance. Med. Res. Rev. 32:1–17 [DOI] [PubMed] [Google Scholar]
- 20. Luo L, et al. 2011. Efflux pump overexpression in conjunction with alternation of outer membrane protein may induce Acinetobacter baumannii resistant to imipenem. Chemotherapy 57:77–84 [DOI] [PubMed] [Google Scholar]
- 21. Livermore DM. 2002. Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: our worst nightmare? Clin. Infect. Dis. 34:634–640 [DOI] [PubMed] [Google Scholar]
- 22. Marra A. 2011. NDM-1: a local clone emerges with worldwide aspirations. Future Microbiol. 6:137–141 [DOI] [PubMed] [Google Scholar]
- 23. Masuda N, et al. 2000. Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-oprM efflux pumps in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44:3322–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nikaido H, Rosenberg EY. 1990. Cir and Fiu proteins in the outer membrane of Escherichia coli catalyze transport of monomeric catechols: study with beta-lactam antibiotics containing catechol and analogous groups. J. Bacteriol. 172:1361–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ochsner UA, Vasil ML. 1996. Gene repression by the ferric uptake regulator in Pseudomonas aeruginosa: cycle selection of iron-regulated genes. Proc. Natl. Acad. Sci. U. S. A. 93:4409–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ohman DE, Sadoff JC, Iglewski BH. 1980. Toxin A-deficient mutants of Pseudomonas aeruginosa PA103: isolation and characterization. Infect. Immun. 28:899–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Page MG, Dantier C, Desarbre E. 2010. In vitro properties of BAL30072, a novel siderophore sulfactam with activity against multiresistant gram-negative bacilli. Antimicrob. Agents Chemother. 54:2291–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patel G, Bonomo RA. 2011. Status report on carbapenemases: challenges and prospects. Expert Rev. Anti Infect. Ther. 9:555–570 [DOI] [PubMed] [Google Scholar]
- 29. Poirel L, Rotimi VO, Mokaddas EM, Karim A, Nordmann P. 2001. VEB-1-like extended-spectrum beta-lactamases in Pseudomonas aeruginosa, Kuwait. Emerg. Infect. Dis. 7:468–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Quale J, Bratu S, Gupta J, Landman D. 2006. Interplay of efflux system, ampC, and oprD expression in carbapenem resistance of Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 50:1633–1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schweizer HP, Hoang TT. 1995. An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene 158:15–22 [DOI] [PubMed] [Google Scholar]
- 32. Silley P, Griffiths JW, Monsey D, Harris AM. 1990. Mode of action of GR69153, a novel catechol-substituted cephalosporin, and its interaction with the tonB-dependent iron transport system. Antimicrob. Agents Chemother. 34:1806–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Spellberg B, et al. 2008. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin. Infect. Dis. 46:155–164 [DOI] [PubMed] [Google Scholar]
- 34. Tomas M, et al. 2010. Efflux pumps, OprD porin, AmpC β-lactamase, and multiresistance in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Antimicrob. Agents Chemother. 54:2219–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsai Y-K, et al. 2011. Klebsiella pneumoniae outer membrane porins OmpK35 and OmpK36 play roles in both antimicrobial resistance and virulence. Antimicrob. Agents Chemother. 55:1485–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. West SEH, Schweizer HP, Dall C, Sample AK, Runyen-Janecky LJ. 1994. Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa. Gene 148:81–86 [DOI] [PubMed] [Google Scholar]
- 37. Winsor GL, et al. 2011. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 39:D596–D600 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.