

Abstract
The combination of ceftazidime and avibactam possesses potent activity against resistant Gram-negative pathogens, including Pseudomonas aeruginosa. We compared the efficacies of human simulated doses of ceftazidime and ceftazidime-avibactam using a hollow-fiber system and neutropenic and immunocompetent murine thigh infection models. Twenty-seven clinical P. aeruginosa isolates with ceftazidime MICs of 8 to 128 mg/liter and ceftazidime-avibactam MICs of 4 to 32 mg/liter were utilized in neutropenic mouse studies; 15 of the isolates were also evaluated in immunocompetent mice. Six isolates were studied in both the hollow-fiber system and the neutropenic mouse. In both systems, the free drug concentration-time profile seen in humans given 2 g of ceftazidime every 8 h (2-h infusion), with or without avibactam at 500 mg every 8 h (2-h infusion), was evaluated. In vivo activity was pharmacodynamically predictable based on the MIC. Ceftazidime decreased bacterial densities by ≥0.5 log unit for 10/27 isolates, while ceftazidime-avibactam did so for 22/27 isolates. In immunocompetent animals, enhancements in activity were seen for both drugs, with ceftazidime achieving reductions of ≥0.3 log unit for 10/15 isolates, whereas ceftazidime-avibactam did so against all 15 isolates. In vitro, ceftazidime resulted in regrowth by 24 h against all isolates, while ceftazidime-avibactam achieved stasis or better against 4/7 isolates. Mutants with elevated ceftazidime-avibactam MICs appeared after 24 h from 3/7 isolates studied in vitro; however, no resistant mutants were detected in vivo. Against this highly ceftazidime-nonsusceptible population of P. aeruginosa, treatment with human simulated doses of ceftazidime-avibactam resulted in pharmacodynamically predictable activity, particularly in vivo, against isolates with MICs of ≤16 mg/liter, and this represents a potential new option to combat these difficult-to-treat pathogens.
INTRODUCTION
Avibactam (formerly NXL104) is a novel non-β-lactam β-lactamase inhibitor with activity against a wide variety of enzyme-mediated resistance mechanisms, including both class A and class C enzymes (25). Given the increasing prevalence of resistant Gram-negative pathogens and the high likelihood that portions of these resistance mechanisms are enzyme based, β-lactam combinations with avibactam represent an excellent opportunity to increase potency against these organisms, where so few options are currently available (12).
One such combination that has received considerable interest is avibactam with ceftazidime. Recent in vitro studies evaluating this combination have shown significant potency increases compared with ceftazidime alone against a wide variety of Gram-negative pathogens (7, 15, 19, 29). Endimiani and colleagues used this combination in mice to evaluate its efficacy against KPC-producing Klebsiella pneumoniae compared with ceftazidime alone (10). Using a set 4:1 ceftazidime-to-avibactam ratio, they found that while a number of ceftazidime-avibactam regimens within the dose range resulted in efficacy, ceftazidime monotherapy showed no activity.
From a clinical standpoint, the combination of ceftazidime-avibactam has shown favorable results in the recently completed phase II clinical trials for the treatment of complicated urinary tract and complicated intra-abdominal infections (NCT00690378 and NCT00752219, respectively) (17, 28). Given the high propensity of Pseudomonas aeruginosa to be the causative pathogen for these and other infection types (11, 22, 26), understanding the pharmacodynamics of ceftazidime-avibactam against this pathogen is critical. The goal of the work described here was to evaluate the activity of human simulated free concentration-time profiles of ceftazidime and ceftazidime-avibactam against several resistant clinical isolates of P. aeruginosa in an in vitro hollow-fiber model and in neutropenic and immunocompetent murine thigh infection models. Moreover, several isolates were studied in all three models, allowing for direct comparisons between the models.
MATERIALS AND METHODS
Antimicrobial test agent.
Commercially available ceftazidime (Fortaz; GlaxoSmithKline, Philadelphia, PA) was obtained from the Hartford Hospital Pharmacy Department and utilized for all in vivo studies. Fortaz from GlaxoSmithKline, United Kingdom, was used for the hollow-fiber studies, while analytical grade ceftazidime (Sigma-Aldrich, St. Louis, MO) was utilized for MIC measurements. Analytical grade avibactam was supplied by AstraZeneca Pharmaceuticals (Waltham, MA) and utilized for both in vitro and in vivo analyses. Clinical vials of ceftazidime were reconstituted as described in the prescribing information and diluted as appropriate to achieve the desired concentrations; analytical ceftazidime and avibactam powders were weighed in a quantity sufficient to achieve the required concentrations and reconstituted immediately prior to use.
Bacterial isolates.
A total of 91 clinical P. aeruginosa isolates were screened in vitro (MIC assay) and/or in vivo for inclusion in this analysis. Isolates were maintained in double-strength skim milk (BD Biosciences, Sparks, MD) at −80°C. Each isolate was subcultured twice on Trypticase soy agar with 5% sheep blood (BD Biosciences) prior to use in the experiments.
A subset of the screened P. aeruginosa isolates (3607, 1382, 1383, 1384, 1386, 1387, and 1388) were tested in vitro in the hollow-fiber system. These strains were obtained from a ceftazidime-avibactam surveillance study (24) and were kindly provided by JMI Laboratories (North Liberty, IA).
Susceptibility testing.
The MICs of ceftazidime and ceftazidime-avibactam were determined for each isolate using the broth microdilution methodology as outlined by the Clinical and Laboratory Standards Institute (CLSI) (5). For ceftazidime-avibactam, doubling dilutions of ceftazidime were utilized in combination with a fixed 4-mg/liter concentration of avibactam. Staphylococcus aureus ATCC 29213 and extended-spectrum-β-lactamase-producing K. pneumoniae ATCC 700603 were used as quality control strains for ceftazidime-avibactam studies; P. aeruginosa ATCC 27853 served as the quality control strain for ceftazidime. MIC studies were conducted in at least triplicate, and the modal MIC was reported.
Hollow-fiber system.
The pharmacological activity of the selected human dose of 2 g/0.5 g of ceftazidime-avibactam was tested in the hollow-fiber system against seven strains of P. aeruginosa with ceftazidime-avibactam MICs of 4 (n = 3) and 8 (n = 4) mg/liter. The schematic of the hollow-fiber system has been described in detail elsewhere (2, 18, 21). For these studies, cellulosic cartridges from FiberCell Systems, Inc. (Frederick, MD), with a molecular mass cutoff of 20 kDa, were inoculated with log-phase cultures of the test microorganism. The inoculum was prepared by inoculating 20 ml of fresh cation-adjusted Mueller-Hinton broth with a swab from a plate that had been streaked with the microorganism and incubated overnight. The suspension was incubated with shaking at 37°C for 1 h until log-phase growth was attained. The culture was diluted in 150 ml of fresh medium according to absorbance at 600 nm in order to reach an inoculum size of 106 CFU/ml. Fifteen milliliters of this final culture was injected into the extracapillary compartment of the hollow-fiber cartridge. For every microorganism tested, the experimental setup included 1 control cartridge dosed with ceftazidime alone, 1 control cartridge dosed with avibactam alone, and 2 cartridges dosed with ceftazidime-avibactam, all at human target doses.
The system was perfused with cation-adjusted Mueller-Hinton broth continuously in order to dilute the drug and simulate a 2-h half-life. One hour after inoculation of the cartridge, ceftazidime-avibactam was dosed into the central reservoir in order to reach target peak concentrations for a 2- to 0.5-g dose of 46.5 and 7 mg/liter of ceftazidime and avibactam, respectively. Ceftazidime-avibactam was administered by a 2-h infusion every 8 h in order to mimic the target human profile and dose regimen (i.e., 2 g/0.5 g every 8 h). The free plasma concentration time courses of ceftazidime and avibactam intended to be representative of patients were based on pharmacokinetic parameters derived from patients studied in a phase II clinical trial of complicated intra-abdominal infections (17) combined with phase I data from healthy volunteers (J. Li, AstraZeneca Pharmaceuticals, personal communication). After dosing, samples were collected at specific time points from the central and bacterial compartments for determination of drug concentrations. The samples were frozen and were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) at the end of the experiment. Samples were also collected from the bacterial compartment for CFU determination. These samples were diluted by serial 10-fold dilutions, and each dilution was plated onto drug-free blood agar plates and incubated at 37°C for at least 16 h prior to counting. The samples collected at 24 h were also plated on blood agar plates containing 32 mg/liter of ceftazidime and 4 mg/liter of avibactam for phenotypic assessment of resistance.
Neutropenic mouse thigh infection model.
Pathogen-free, female ICR mice weighing approximately 25 g were acquired from Harlan Sprague Dawley, Inc. (Indianapolis, IN) and utilized throughout these experiments. Animals were maintained and used in accordance with National Research Council recommendations and were provided food and water ad libitum. Mice were rendered neutropenic with 100- and 150-mg/kg intraperitoneal injections of cyclophosphamide (Cytoxan; Bristol-Myers Squibb, Princeton, NJ) given 1 and 4 days prior to inoculation, respectively. Three days prior to inoculation, mice were also given a single 5-mg/kg intraperitoneal injection of uranyl nitrate. This produces a predictable degree of renal impairment to slow drug clearance (1). Two hours prior to the initiation of antimicrobial therapy, each thigh was inoculated intramuscularly with a 0.1-ml solution containing approximately 107 CFU/ml of the test isolate (i.e., inocula of ca. 1 × 106 CFU).
Immunocompetent mouse thigh infection model.
Mice utilized in the immunocompetent mouse studies underwent the same procedures as outlined above, except that cyclophosphamide was not given and an inoculum of 108 CFU/ml was used to produce thigh infection (i.e., inocula of ca. 1 × 107 CFU).
Determination of the in vivo dosing regimen.
In these studies, we determined a dosing regimen that, in mice, simulated the percentage of the dosing interval in which free drug concentrations remained above the MIC (fT>MIC) observed in humans given 2 g ceftazidime plus 500 mg avibactam every 8 h as a 2-h infusion and the same dose of ceftazidime as monotherapy across a range of MICs. The target human exposures were identical to those used in the hollow-fiber system, as noted above. For free drug calculations, protein binding values of 26% and 15% were used for ceftazidime in mice and humans, respectively. These values for avibactam were 10% and 8%, respectively.
First, single-dose studies with ceftazidime-avibactam and ceftazidime alone were undertaken in thigh-infected neutropenic mice. For these analyses, animals were dosed with a single weight-based, 0.2-ml subcutaneous injection of the study drug(s), and groups of 6 mice were euthanized at 8 time points over the following 12 h. Blood samples were taken via cardiac puncture, and plasma was stored at −80°C until analysis. Pharmacokinetic parameters for single doses of ceftazidime-avibactam and ceftazidime alone were calculated using first-order input and elimination, by nonlinear least-squares techniques (WinNonlin version 5.0.1; Pharsight, Mountain View, CA). Compartment model selection and weighting schemes were based on visual inspection of the fit and use of the correlation between the observed and calculated concentrations.
After pharmacokinetic characterization of single doses, these data were used to determine regimens in mice that simulated the free drug exposure profile seen in humans for ceftazidime-avibactam and ceftazidime alone as noted above. Confirmatory pharmacokinetics studies in infected mice were undertaken prior to the use of these regimens in the pharmacodynamic analyses. For these studies, infected neutropenic mice were dosed with the regimens calculated as described above, and groups of 6 mice were euthanized at 6 to 8 time points throughout the dosing interval to confirm target exposures. These studies were conducted over the first interval (i.e., 8 h) for ceftazidime, while ceftazidime-avibactam studies were conducted for all 3 treatment intervals (i.e., 24 h). Confirmatory studies in immunocompetent animals were also undertaken to ensure similar exposures.
In vivo efficacy.
For each of the 27 P. aeruginosa isolates, groups of 3 mice were administered human simulated regimens of ceftazidime or ceftazidime-avibactam beginning 2 h after inoculation. All doses were administered as 0.2-ml subcutaneous injections and consisted of 3 8-h dosing intervals (i.e., 24 h). To serve as control animals, an additional group of mice were administered normal saline with the same volume, route, and frequency as the most frequent treatment regimen. All animals were harvested at 24 h after the initiation of therapy; mice that failed to survive for 24 h were harvested at the time of expiration (13). The harvesting procedure for all study mice began with euthanization by CO2 exposure followed by cervical dislocation. After sacrifice, thighs were removed and individually homogenized in normal saline. Serial dilutions of the thigh homogenate were plated on Trypticase soy agar with 5% sheep blood for CFU determination. In addition to the above-mentioned treatment and control groups, another group of 3 infected, untreated mice were harvested at the initiation of dosing and served as 0-h controls. Efficacy was calculated as the change in log10 bacterial CFU obtained for treated mice after 24 h from the starting densities observed in 0-h control animals.
During portions of the neutropenic mouse studies, a phenotypic assessment of resistance was conducted against the 7 isolates evaluated in vitro using a methodology similar to that described above.
Bioanalytical studies.
(i) Hollow-fiber samples. Samples from the hollow-fiber system were first diluted at least 10-fold in a 1:1 mixture of rat plasma and cation-adjusted Mueller-Hinton broth. Proteins were precipitated by adding 400 μl of 80:20 acetonitrile-methanol containing carbutamide (250 ng/ml) and 100-μl aliquots of a 500-ng/ml avibactam analog (AstraZeneca Pharmaceuticals, Waltham, MA) as an internal standard. Mixtures were vortex mixed for 15 s and precipitates sedimented by centrifugation at 2,500 × g for 5 min. Two 200-μl samples of the supernatant solution were processed further as follows. The first was evaporated to dryness under nitrogen and then reconstituted with 200 μl of water for the analysis of avibactam by LC-MS/MS with selective reaction monitoring. The second was diluted with 200 μl of water for the analysis of ceftazidime by LC-MS/MS with selective reaction monitoring. Calibration curves were prepared by dissolving avibactam and ceftazidime in dimethyl sulfoxide at 2 mg/ml and diluting to various concentrations with a mixture of 50:50 rat plasma–cation-adjusted Mueller-Hinton broth.
Avibactam was measured as follows. Chromatographic separation was performed using an Aqua C18 2.0- by 50-mm, 3-μm-particle-size LC column (Phenomenex, Torrance, CA) held at ambient temperature, connected to a Shimadzu high-performance liquid chromatography (HPLC) system with an LC10ADvP pump, CBM20A controller (Shimadzu Scientific Instruments, Columbia, MD), and CTC PAL autosampler (CTC Analytics, Zwingen, Switzerland) held at 10°C and performing 10-μl injections. Samples were eluted at 500 μl/min with a 3.2-min gradient elution method. Mobile phase A consisted of 0.1% (vol/vol) formic acid in water. Mobile phase B consisted of 0.1% (vol/vol) formic acid in acetonitrile. The elution method was as follows: 0% B for 0.5 min, then a linear increase from 0 to 90% in 1 min, a hold at 90% B for 0.5 min, and then a return to 0% B for 1.2 min. Mass spectrometer measurements used a Sciex API4000 mass spectrometer (Applied Biosystems, Framingham, MA) fitted with a TurboIonSpray sample sprayer operated in negative-ion mode with selected reaction monitoring of either m/z 263.8 to 96.0 with a collision energy of −34 V (avibactam) or m/z 303.0 to 96.0 with a collision energy of −34 V (internal standard) and using a 75-ms dwell time. Data were collected and analyzed using Analyst software v1.4.1 (Applied Biosystems).
Ceftazidime was measured as follows. Chromatographic separation was performed using an Atlantis T3 3.0- by 20-mm, 3-μm-particle-size LC column (Waters Corp., Milford, MA) held at ambient temperature, connected to a Shimadzu HPLC system with an LC10ADvP pump, CBM20A controller (Shimadzu Scientific Instruments, Columbia, MD), and CTC PAL autosampler (CTC Analytics, Zwingen, Switzerland) held at 10°C and performing 10-μl injections. Samples were eluted at 500 μl/min with a 2.5-min gradient elution method. Mobile phase A consisted of 10 mM ammonium formate plus 0.1% (vol/vol) formic acid. Mobile phase B consisted of 0.1% (vol/vol) formic acid in acetonitrile. The elution method was as follows: 5% B for 0.5 min, then a linear increase from 5 to 95% in 1 min, a hold at 95% B for 0.5 min, and then a return to 5% B for 0.5 min. Mass spectrometer measurements used a Sciex API4000 mass spectrometer (Applied Biosystems, Framingham, MA) fitted with a TurboIonSpray sample sprayer operated in positive-ion mode with selected reaction monitoring of either m/z 547.3 to 467.9 with a collision energy of 17 V (ceftazidime) or m/z 272.1 to 156.1 with a collision energy of 25 V (internal standard) and using a 75-ms dwell time. Data were collected and analyzed using Analyst software v1.4.1 (Applied Biosystems).
(ii) In vivo samples.
Ceftazidime concentrations in serum samples were analyzed at the Center for Anti-Infective Research and Development (Hartford, CT) using a previously described high-performance liquid chromatography assay (20), while avibactam concentrations were determined by Eurofins Medinet, Inc (Chantilly, VA) using a previously described LC-MS/MS assay (30).
RESULTS
Bacterial isolates.
The ceftazidime and ceftazidime-avibactam MIC distributions for 91 isolates screened for inclusion are shown in Fig. 1. The ceftazidime MIC50 and MIC90 were 64 and ≥128 mg/liter, respectively, whereas these values for ceftazidime-avibactam were 8 and 32 mg/liter, respectively. The collection represented a highly resistant sample of P. aeruginosa. This can be inferred by comparing the ceftazidime-avibactam MIC90 of 32 mg/liter for these isolates with that of 8 mg/liter as observed in recent surveillance data from Canada (29), the United States (24), and Europe (14).
Fig 1.

Ceftazidime and ceftazidime-avibactam MIC distributions for the 91 P. aeruginosa isolates screened for inclusion in efficacy studies.
Based on the desired phenotypic profiles, coupled with the ability to produce infection in vivo, 27 P. aeruginosa isolates were utilized for the in vivo portions of this study (Table 1). The majority of these 27 isolates were collected from the respiratory tract (44%), followed by equal numbers (19%) from blood and skin. Ceftazidime-avibactam MICs ranged from 4 to 32 mg/liter; all but 1 isolate were nonsusceptible to ceftazidime, with MICs ranging from 8 to >128 mg/liter.
Table 1.
Demographic data and phenotypic profile of the P. aeruginosa isolates included in the in vivo and in vitro efficacy studiesa
| Isolate no. | Source | Location | MIC (mg/liter) |
|||||
|---|---|---|---|---|---|---|---|---|
| CAZ-AVI | CAZ | MER | TZP | CIP | TOB | |||
| 971 | Respiratory | ND | 4 | 16 | >32 | >256 | >32 | 1.5 |
| 22 | Respiratory | ND | 4 | 64 | 16 | 32 | 0.25 | 0.5 |
| AZ37-8 | Respiratory | ICU | 4 | 64 | 8 | 128 | 16 | 1 |
| 3607 | Burn | Non-ICU | 4 | 64 | 8 | >64 | ND | ND |
| 1383 | Respiratory | ICU | 4 | 64 | 4 | >64 | ND | ND |
| 1384 | Respiratory | Non-ICU | 4 | 64 | 8 | >64 | ND | ND |
| 856 | Respiratory | ND | 8 | 8 | >32 | >256 | >32 | 32 |
| AZ24-2 | Respiratory | ICU | 8 | 32 | 64 | 64 | 16 | 64 |
| JJ4-32 | Respiratory | ICU | 8 | 32 | 64 | 16 | 64 | 128 |
| JJ4-39 | Skin | Non-ICU | 8 | 32 | 4 | 128 | 64 | 64 |
| 1387 | Blood | Non-ICU | 8 | 32 | >8 | >64 | ND | ND |
| JJ2-69 | Blood | Non-ICU | 8 | 64 | 1 | 256 | 32 | 64 |
| JJ8-16 | Respiratory | ICU | 8 | 64 | 2 | 256 | 0.5 | 0.25 |
| 1389 | Urine | Non-ICU | 8 | 64 | >8 | >64 | ND | ND |
| AZ28-19 | Skin | ICU | 8 | 128 | 4 | 256 | 16 | 1 |
| 1382 | Blood | ND | 8 | 128 | >8 | >64 | ND | ND |
| 1386 | ND | Non-ICU | 8 | 128 | >8 | >64 | ND | ND |
| 1388 | Respiratory | ICU | 8 | 128 | >8 | >64 | ND | ND |
| 968 | Respiratory | ND | 16 | 32 | >32 | 16 | >32 | 96 |
| JJ1-29 | ND | ND | 16 | 32 | >64 | 32 | 4 | 0.5 |
| JJ4-36 | Skin | Non-ICU | 16 | 64 | 32 | 128 | 8 | 0.5 |
| JJ4-84 | Skin | Non-ICU | 16 | 64 | 32 | 256 | 16 | >128 |
| 1394 | Respiratory | ICU | 16 | 64 | >8 | >64 | ND | ND |
| JJ1-25 | ND | ND | 16 | 128 | 32 | 256 | 0.5 | 1 |
| 1391 | Blood | Non-ICU | 16 | 128 | >8 | >64 | ND | ND |
| JJ3-9 | Blood | Non-ICU | 16 | >128 | 32 | 64 | 16 | 1 |
| JJ11-54 | Skin | Non-ICU | 32 | 128 | 8 | 512 | 4 | 0.5 |
| JJ7-6 | Other | Non-ICU | 32 | 128 | 1 | 4 | 64 | 128 |
CAZ-AVI, ceftazidime-avibactam; CAZ, ceftazidime; MER, meropenem; TZP, piperacillin-tazobactam; CIP, ciprofloxacin; TOB, tobramycin; ND, not determined; ICU, intensive care unit.
In vitro hollow-fiber efficacy.
The average concentration-time profiles for ceftazidime and avibactam achieved in the hollow-fiber system when dosed together are shown in Fig. 2. There was good consistency across cartridges, as demonstrated by the small standard deviations (SD). In addition, avibactam and ceftazidime, when dosed in combination, in the bacterial compartment of the system reached similar concentrations, which overlapped with the concentration in the central compartment (data not shown). On average, the ceftazidime peak concentration (±SD) was 32.1 ± 2.0 mg/liter, and the avibactam average peak concentration was 5.1 ± 1.1 mg/liter. The average trough concentrations (±SD) were 7.1 ± 1.1 and 1.2 ± 0.4 mg/liter for ceftazidime and avibactam, respectively. Degradation of ceftazidime was observed in the bacterial compartment when the compound was dosed alone (Fig. 3). Such degradation was not observed when ceftazidime was dosed in combination with avibactam, which demonstrated the protective effect of avibactam on ceftazidime at the concentrations tested. Similar observations were reported for ceftaroline and avibactam studies in the hollow-fiber model (16).
Fig 2.
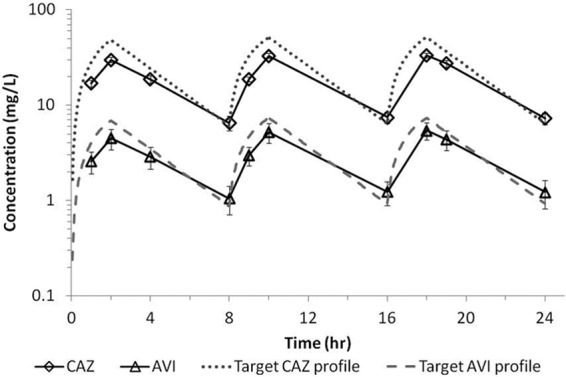
Average ceftazidime (CAZ) and avibactam (AVI) concentration-time profiles in the hollow-fiber central compartment when dosed in combination. Profiles represent averages of the concentrations in different cartridges infected with different strains of P. aeruginosa. Error bars represent standard deviations.
Fig 3.
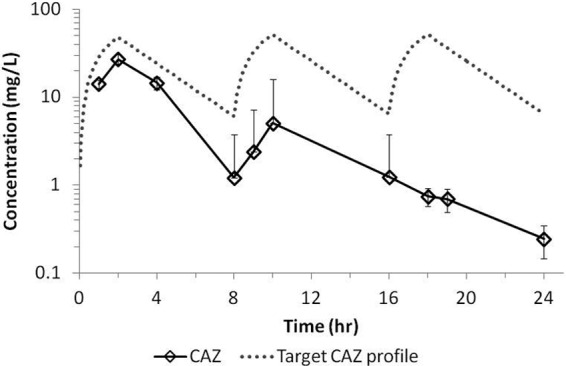
Ceftazidime (CAZ) average concentration-time profile in the bacterial compartment of the hollow-fiber system when dosed as monotherapy. The profile represents averages of the concentrations in 6 cartridges infected with different strains of P. aeruginosa. Error bars represent standard deviations.
Ceftazidime-avibactam was efficacious against P. aeruginosa in the hollow-fiber system (Fig. 4). Ceftazidime demonstrated minimal to no effect on the 24-h growth of the strains tested in the hollow-fiber system when dosed alone at the target dose, as shown by the control curves. This result was consistent with the high MICs of ceftazidime (Table 1). A similar observation was made for avibactam alone, which also had no impact on the growth curves at the target dose. A maximum effect was observed for all 3 P. aeruginosa strains tested with a ceftazidime-avibactam MIC of 4 mg/liter, demonstrating a clear effect of the dose on these strains. The response against the P. aeruginosa strains with a ceftazidime-avibactam MIC of 8 mg/liter was more variable. Significant regrowth was observed in 24 h for 2 of the 4 strains with a ceftazidime-avibactam MIC of 8 mg/liter. One of the other 2 strains, 1382, showed a static response by the end of treatment. The fourth strain (1387) showed the maximum response in the hollow-fiber system, but the MIC of ceftazidime against that strain (32 mg/liter) was low compared with those against the other strains tested (Table 1). For all cartridges that showed regrowth in 24 h (Fig. 4), bacterial colonies equating to the entire bacterial density were observed on drug-containing plates. Upon further profiling, these colonies showed a stable shift in the ceftazidime-avibactam MIC of 3 to 5 doubling dilutions (i.e., 64 to 128 mg/liter). Based on the ability to produce an infection, 6 of the isolates studied in the hollow-fiber system were also examined in the neutropenic mouse thigh model.
Fig 4.

Effect of ceftazidime (CAZ) squares and dashed lines), avibactam (AVI) (triangles and dotted lines), and ceftazidime-avibactam (CAZ-AVI) (diamonds with solid lines) against P. aeruginosa in the hollow-fiber system. Curves represent the average CFU/ml across cartridges infected with the same P. aeruginosa strain.
Determination of dosing regimen for in vivo studies.
The free drug pharmacokinetic profiles determined in vivo for ceftazidime-avibactam are shown in Fig. 5, and the profile for ceftazidime alone is shown in Fig. 6. While it is clear from these figures that the murine exposure profiles are similar to those observed in humans, importantly, the fT>MICs attained for these regimens across the range of MICs tested were comparable (Table 2). Also of note, target concentrations were similar in immunocompetent and neutropenic animals (data not shown); therefore, these same regimens were used in both models.
Fig 5.

Free concentration-time profiles for 2-h infusions of ceftazidime (2 g) and avibactam (500 mg) in humans (solid line and dotted line, respectively) and mice (circles and triangles, respectively). Murine data represent the mean ± standard deviation.
Fig 6.

Free concentration-time profiles for 2-h infusions of ceftazidime 2 g in humans (solid line) and mice (circles). Murine data represent the mean ± standard deviation.
Table 2.
Free drug T>MIC profile for ceftazidime at 2 g every 8 h (2-h infusion) as monotherapy and in combination with avibactam in mice and humans
| MIC (mg/liter) | Ceftazidime fT>MIC (%) in: |
||
|---|---|---|---|
| Humans | Mice | Mice as CAZ-AVIa | |
| 4 | 100 | 100 | 99 |
| 8 | 96 | 87 | 88 |
| 16 | 63 | 58 | 62 |
| 32 | 24 | 28 | 34 |
| 64 | 0 | 0 | 0 |
CAZ-AVI, ceftazidime-avibactam.
In vivo efficacy.
During the neutropenic mouse studies, 0-h control mice (for the bacterial load at start of treatment) displayed a mean bacterial density of 5.03 ± 0.31 log10 CFU per thigh, which increased by an average of 3.25 ± 0.53 log units in untreated mice after 24 h. The mean bacterial density in control animals during immunocompetent mouse studies was 6.59 ± 0.24 log10 CFU at 0 h and increased by 1.52 ± 0.72 log units after 24 h. The results of the neutropenic mouse efficacy studies are shown in Fig. 7. Human simulated doses of ceftazidime-avibactam resulted in predictable efficacy (based on the MIC), with bacterial killing against 16 of 17 isolates with ceftazidime-avibactam MICs of ≤8 mg/liter (fT>MIC of ≥96%) and 5 of 8 isolates with ceftazidime-avibactam MICs of 16 mg/liter (fT>MIC of 63%). After the 24-h treatment period with ceftazidime-avibactam, no bacterial colonies were observed from thigh homogenates plated on drug-containing plates (2 to 3 doublings above the MICs of 4 to 8 mg/liter), suggesting that there was no development of resistance. This was in contrast to the findings in the hollow-fiber experiments, in which variants with elevated MICs were detected in 24 h for a number of the analyzed isolates. In the neutropenic mice, efficacy was noted for ceftazidime monotherapy against 7 of 8 isolates with MICs of ≤32 mg/liter; minimal activity was seen against the remaining 19 strains (MIC range, 64 to >128 mg/liter).
Fig 7.

Comparative efficacies of ceftazidime-avibactam (CAZ-AVI) and ceftazidime alone against a distribution of P. aeruginosa in neutropenic mice. Error bars represent standard deviations.
When tested in immunocompetent animals, treatment with ceftazidime-avibactam resulted in efficacy against all isolates (range of bacterial reductions, 0.3 to 1.95 log10 CFU), with 13 of 15 achieving reductions of 0.75 log unit or greater (Fig. 8). Of note, this group of isolates included 3 organisms with ceftazidime-avibactam MICs of ≤16 mg/liter against which ceftazidime-avibactam treatment yielded increases in bacterial density in the neutropenic animals (Fig. 4), clearly highlighting the enhancement of activity in the presence of a functioning immune system.
Fig 8.

Comparative efficacies of ceftazidime-avibactam and ceftazidime alone against a distribution of P. aeruginosa in immunocompetent animals. Error bars represent standard deviations.
DISCUSSION
Over the last few years, discussions pertaining to the lack of available agents to treat resistant Gram-negative pathogens have appeared at nearly every infectious disease-centered scientific meeting, and they have even begun to emerge regularly in the general media. In light of this, companies worldwide have invested time and resources into searching for new drug targets for Gram-negative organisms. The addition of a novel β-lactamase inhibitor such as avibactam to a clinically available compound offers an excellent opportunity to combat these organisms without the need to identify a novel target. In this study, we evaluated the pharmacodynamics of a humanized profile of ceftazidime combined with avibactam in comparison to the efficacy of ceftazidime alone against a wide distribution of clinical P. aeruginosa isolates. Under the simulated dynamic conditions, the addition of avibactam potentiated both the in vitro and in vivo activities of ceftazidime against this highly ceftazidime-resistant population of P. aeruginosa.
Studying the pharmacodynamics of a simulated human pharmacokinetic exposure of an antibacterial agent or combination of agents in the in vitro hollow-fiber model, as described here, eliminates the cellular and humoral immune systems and animal-to-animal pharmacokinetic variability intrinsic to animal models (6). The advantages of this are 2-fold. First, the use of animals can be minimized while still allowing the interaction between pharmacokinetics and pharmacodynamics to be studied, and second, the biological variation is confined to the bacterial response. On the other hand, it exposes the antibacterial agent or combination of agents to a comparatively harsh test of efficacy, by allowing outgrowth of any spontaneous higher-MIC variants that might arise (3, 16). In that respect, exposure of bacteria to antibacterial agents in the hollow-fiber system is similar to “serial-passage” exposure (23). Indeed, one way to use a hollow-fiber model has been to define a pharmacokinetic profile that can prevent the outgrowth of resistant mutants selected from test bacterial strains in the absence of cellular or humoral immune systems (16). The hollow-fiber system in the present work was used to examine the response of P. aeruginosa strains that displayed ceftazidime-avibactam MICs of 8 mg/liter, which has been identified as the MIC90 of the combination in surveillance studies (14, 24, 29). Against isolates with ceftazidime-avibactam MICs of 8 mg/liter, the response to ceftazidime-avibactam in the hollow-fiber system was variable, with 2 of the 4 strains showing regrowth, 1 showing stasis, and the fourth showing maximum bactericidal response at 24 h (Fig. 4). Of note, 3 of these 4 strains exhibited development of isolates with elevated MICs by the 24-h time of sampling, including the strain with the 24-h response of stasis. As a result of the variable response, strains with ceftazidime-avibactam MICs of 4 mg/liter were also tested. A consistent maximum bactericidal response was observed at 24 h for all 3 of these isolates (Fig. 4), and, in contrast to the findings described above, resistant mutants were not isolated. As yet, biochemical mechanisms of resistance have not been elucidated. In this context, it is worth noting that in the control experiment in which the same pharmacokinetic profile of ceftazidime was studied in the same hollow-fiber system against isolates that displayed a ceftazidime-alone MIC of 8 mg/liter, spontaneous ceftazidime-resistant mutants also arose (data not shown).
Using simulated human pharmacokinetics in neutropenic mice, it can be seen from Fig. 7 that isolates 1382, 1386, and 1388, from which resistant variants grew in the hollow-fiber system, were killed by ceftazidime-avibactam and, importantly, that no resistant colonies were recovered from the thighs of mice inoculated with these strains. Studying the same isolates in the hollow-fiber and neutropenic mouse models provided an understanding of the “translatability” between these models, which is expected to be helpful in future studies of novel antibacterial agents in the hollow-fiber system, particularly when optimizing novel compounds that might not be appropriate for animal efficacy studies and/or in an effort to reduce animal usage. As would be expected (3, 4, 16), the hollow-fiber model was a conservative model, in the sense that it was a severe test of the ability of the combination to prevent selection of resistant mutants. By comparing results for isolates studied in both the hollow-fiber and the neutropenic mouse models, as well as strains that were studied in both the neutropenic and immunocompetent mouse models, one can infer that a hierarchy exists between the different models; that is, in terms of the severity of the model to evaluate pharmacodynamics, hollow-fiber model > neutropenic mouse thigh model > immunocompetent mouse thigh model. This is likely to be useful for understanding the place of the hollow-fiber model in future studies of novel preclinical compounds.
In contrast to the hollow-fiber system, animal models used to study pharmacodynamic responses to simulated pharmacokinetic profiles offer the advantage of showing responses likely to be closer to those that would be expected in humans, owing to host immunity (6). Moreover, there are other advantages of animal models that might not be appreciated at first sight. One such example is that the animal model inherently possesses variation in the pharmacokinetic profile between individual animals, offering insight into patient-to-patient variation, usually understood through population pharmacokinetic analysis and Monte-Carlo simulation (9). In addition to alterations in drug kinetics, this in vivo system may better mimic clearance or degradation of organism-produced enzymes as would be anticipated in patients. This concept is supported by the observation that, unlike in the in vitro model (Fig. 3), there was no enzyme-mediated loss in ceftazidime exposure in vivo. Lastly, the in vivo model displays variable immunity, which can be further modulated to understand the relationship between “host, bug, and drug,” namely, the required exposure for eradication of the organism, as well as the prevention of resistance. Animal-to-animal variation with respect to the magnitudes of pharmacodynamic indices in the present study is discussed below.
When one considers the relative potency of ceftazidime compared with ceftazdime-avibactam (Table 1), the addition of avibactam to ceftazidime resulted in a median 8-fold increase in potency against the 27 isolates studied in vivo. Not unexpectedly given the ceftazidime resistance of the population of P. aeruginosa isolates specifically selected, this shift is greater than was noted in a study of 470 P. aeruginosa isolates collected from Canadian hospitals, where the MIC50 and MIC90 for ceftazidime were 4 and 32 mg/liter, respectively, while the respective values for ceftazidime-avibactam were 2 and 8 mg/liter (29). Importantly, these data highlight the fact that the population of isolates included in the current in vivo analysis represents the least susceptible organisms within the clinical distribution. While genetic information is not available to explain resistance mechanisms for these clinical isolates, based on a study conducted by Mushtaq and colleagues evaluating the potency shift observed upon the addition of avibactam to ceftazidime against a population of P. aeruginosa with known genotypes (19), it is reasonable to assume that upregulation of AmpC β-lactamases played a significant role in resistance to ceftazidime for the included organisms.
Given the ceftazidime MICs of the P. aeruginosa isolates studied, it is not unexpected that human simulated doses of ceftazidime monotherapy resulted in some efficacy, as assessed by a reduction in bacterial densities below baseline, for only a small percentage (14/27; 52%) of the isolates evaluated in neutropenic animals. The fT>MIC of ceftazidime alone achieved for the simulated dose of 2 g every 8 h (2-h infusion) against 26 of 27 isolates was ≤34%, compared with the 60 to 70% fT>MIC target identified for maximal efficacy of cephalosporins (6, 8, 27). In light of the increased potency noted after the addition of avibactam, the fT>MIC achieved by the ceftazidime portion of the combination ranged from 34 to 99% and correlated well with the observed efficacy in vivo. Specifically, within neutropenic mouse studies, pharmacodynamically predictable efficacy against a majority of isolates with MICs of ≤16 mg/liter (≥64% fT>MIC) was noted, and efficacy was seen for 1 of 2 isolates with an MIC of 32 mg/liter (34% fT>MIC). The observation that pharmacodynamic targets derived from cephalosporin monotherapy translate in vivo to this combination of β-lactam and β-lactamase inhibitor is certainly worth noting. Namely, MIC testing of ceftazidime-avibactam utilized a constant 4-mg/liter concentration of avibactam while altering the ceftazidime concentration; this is different from what happens to the drugs in vivo, where both ceftazidime and avibactam concentrations change over time. Given that the function of avibactam is to protect ceftazidime from inactivation by β-lactamases, it can be theorized that once concentrations fall below a certain threshold, MICs could increase above what is predicted in vitro. However, in the current study, which mimicked human exposure profiles of both compounds, this was seemingly not the case, and it is reasonable to assume that the pharmacodynamic findings in mice will readily translate to humans.
In light of the fact that a large proportion of patients treated in the clinic have a functioning immune system, translatability to humans was furthered in this study by evaluating efficacy in immunocompetent animals. In doing so, the activity of ceftazidime-avibactam was enhanced, such that reductions in bacterial density were noted for all isolates tested (MIC range, 4 to 32 mg/liter), with a large proportion being greater than 1 log unit. This included isolates JJ2-69 (ceftazidime-avibactam MIC of 8 mg/liter) and JJ4-36 and JJ4-84 (both with ceftazidime-avibactam MICs of 16 mg/liter), which had resulted in modestly increased bacterial densities in neutropenic animals. Slight enhancements were seen with ceftazidime monotherapy against a number of isolates, but profound increases were likely repressed secondary to the extraordinarily high MICs and, likewise, minimal fT>MICs.
With any human simulated study in mice, one must choose a human profile to mimic in animals. While we used the best available data to choose this regimen, clearly there were patients in the phase II study who received exposures both above and below this target regimen. Importantly, if one considers the variability around the mean for the composite pharmacokinetic curves, as shown in Fig. 5 and 6, it is evident that the individual animal exposures attained during efficacy studies help to capture the likely range in exposures that could be anticipated in humans.
It is also important to note that in this analysis, we evaluated ceftazidime alone at a dose of 2 g every 8 h (2-h infusion) so that we could assess the benefits of avibactam. This ceftazidime dosing regimen provided a greater fT>MIC than would be expected from this same dose given as a standard 0.5-h infusion. Namely, the fT>MICs provided by a 0.5-h infusion would be 80%, 55%, and 26% at MICs of 8, 16, and 32 mg/liter, respectively. Thus, if one compares results for ceftazidime-avibactam to those for conventional ceftazidime at approved dosing regimens, the activity of the combination would be further differentiated. These pharmacodynamic differences resulting from prolonging the infusion time will also be of importance when considering potential breakpoints for ceftazidime-avibactam. Namely, the current ceftazidime breakpoint of 8 mg/liter recommended by CLSI and the European Committee on Antimicrobial Susceptibility Testing is based on a standard 0.5-h infusion, whereas the data contained here, secondary to the pharmacodynamic enhancement achieved by prolonging the infusion, show that ceftazidime-avibactam pharmacokinetics can be adequate to provide a substantial fT>MIC at an MIC of 16 mg/liter (i.e., 62% fT>MIC) (Table 2).
The ability to simulate human exposures in an in vitro hollow-fiber system and in mice allowed us the opportunity to compare the pharmacodynamics of ceftazidime and ceftazidime-avibactam against a highly ceftazidime-resistant population of P. aeruginosa. While ceftazidime alone was unable to achieve efficacy against a large number of the isolates in vivo, treatment with ceftazidime-avibactam in vivo resulted in activity against all but those at the upper end of the MIC range and was predictable based on the fT>MIC profile. With the increased prevalence of multidrug-resistant Gram-negative pathogens, the use of ceftazidime plus avibactam at doses of 2 g and 0.5 g, respectively, given every 8 h as a 2-h infusion represents an excellent opportunity to combat a vast majority of organisms within the clinical distribution, particularly in the presence of a functioning immune system.
ACKNOWLEDGMENTS
Thanks go to Jianguo Li (AstraZeneca Pharmaceuticals) for providing the pharmacokinetic time course of ceftazidime and avibactam modeled for patients. Karthick Vishwanathan (AstraZeneca Pharmaceuticals) advised on the logistics of handling samples from the hollow-fiber system for determination of analyte concentrations. We also thank Amira Bhalodi, Henry Christenson, Mao Hagihara, Seth Housman, Jennifer Hull, Debora Santini, Christina Sutherland, Pam Tessier, Lindsey Tuttle, and Dora Wiskirchen (Center for Anti-Infective Research and Development) for their assistance with the in vivo studies.
Footnotes
Published ahead of print 17 September 2012
REFERENCES
- 1. Andes D, Craig WA. 1998. In vivo activities of amoxicillin and amoxicillin-clavulanate against Streptococcus pneumoniae: application to breakpoint determinations. Antimicrob. Agents Chemother. 42:2375–2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bilello JA, Bauer G, Dudley MN, Cole GA, Drusano GL. 1994. Effect of 2′,3′-didehydro-3′-deoxythymidine in an in vitro hollow-fiber pharmacodynamic model system correlates with results of dose-ranging clinical studies. Antimicrob. Agents Chemother. 38:1386–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blaser J, Stone BB, Groner MC, Zinner SH. 1987. Comparative study with enoxacin and netilmicin in a pharmacodynamic model to determine importance of ratio of antibiotic peak concentration to MIC for bactericidal activity and emergence of resistance. Antimicrob. Agents Chemother. 31:1054–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blaser J, Stone BB, Zinner SH. 1985. Two compartment kinetic model with multiple artificial capillary units. J. Antimicrob. Chemother. 15(Suppl A):131–137 [DOI] [PubMed] [Google Scholar]
- 5. Clinical Laboratory Standard Institute 2011. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 8th ed CLSI publication M07-A8. Clinical Laboratory Standard Institute, Wayne, PA [Google Scholar]
- 6. Crandon JL, Nicolau DP. 2012. In vivo pharmacodynamic modeling for drug discovery, p 1035–1054 In Dougherty TJ, Pucci MJ. (ed), Antibiotic discovery and development, vol 2 Springer, Berlin, Germany [Google Scholar]
- 7. Curcio D. 2011. Activity of a novel combination against multidrug-resistant nonfermenters: ceftazidime plus NXL104. Expert Rev. Anti Infect. Ther. 9:173–176 [DOI] [PubMed] [Google Scholar]
- 8. Drusano GL. 2004. Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug’. Nat. Rev. Microbiol. 2:289–300 [DOI] [PubMed] [Google Scholar]
- 9. Drusano GL, et al. 2001. Use of preclinical data for selection of a phase II/III dose for evernimicin and identification of a preclinical MIC breakpoint. Antimicrob. Agents Chemother. 45:13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Endimiani A, et al. 2011. Evaluation of ceftazidime and NXL104 in two murine models of infection due to KPC-producing Klebsiella pneumoniae. Antimicrob. Agents Chemother. 55:82–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hooton TM, et al. 2009. Diagnosis, prevention, and treatment of catheter-associated urinary tract infection in adults: 2009 International Clinical Practice Guidelines from the Infectious Diseases Society of America. Clin. Infect. Dis. 50:625–663 [DOI] [PubMed] [Google Scholar]
- 12. Infectious Diseases Society of America 2010. The 10 × '20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin. Infect. Dis. 50:1081–1083 [DOI] [PubMed] [Google Scholar]
- 13. Kim A, Banevicius MA, Nicolau DP. 2008. In vivo pharmacodynamic profiling of doripenem against Pseudomonas aeruginosa by simulating human exposures. Antimicrob. Agents Chemother. 52:2497–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Levasseur P, et al. In vitro antibacterial activity of the ceftazidime-avibactam (NXL104) combination against Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 56:1606–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Livermore DM, et al. 2011. Activities of NXL104 combinations with ceftazidime and aztreonam against carbapenemase-producing Enterobacteriaceae. Antimicrob. Agents Chemother. 55:390–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Louie A, et al. 2012. Pharmacodynamics of beta-lactamase inhibition by NXL104 in combination with ceftaroline: examining organisms with multiple types of beta-lactamases. Antimicrob. Agents Chemother. 56:258–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lucasti C, Popescu I, Ramesh M, Lipka J, Sable C. 2011. Efficacy and safety of ceftazidime/NXL104 plus metronidazole vs. meropenem in the treatment of complicated intra-abdominal infections in hospitalised adults, abstr. P1532. Abstr. 21st European Congress of Clinical Microbiology and Infectious Diseases-27th International Congress of Chemotherapy, Milan, Italy [Google Scholar]
- 18. McSharry JJ, Deziel MR, Zager K, Weng Q, Drusano GL. 2009. Pharmacodynamics of cidofovir for vaccinia virus infection in an in vitro hollow-fiber infection model system. Antimicrob. Agents Chemother. 53:129–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mushtaq S, Warner M, Livermore DM. 2010. In vitro activity of ceftazidime+NXL104 against Pseudomonas aeruginosa and other non-fermenters. J. Antimicrob. Chemother. 65:2376–2381 [DOI] [PubMed] [Google Scholar]
- 20. Nicolau DP, Nightingale CH, Banevicius MA, Fu Q, Quintiliani R. 1996. Serum bactericidal activity of ceftazidime: continuous infusion versus intermittent injections. Antimicrob. Agents Chemother. 40:61–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nikolaou M, Schilling AN, Vo G, Chang KT, Tam VH. 2007. Modeling of microbial population responses to time-periodic concentrations of antimicrobial agents. Ann. Biomed. Eng. 35:1458–1470 [DOI] [PubMed] [Google Scholar]
- 22. Rotstein C, et al. 2008. Clinical practice guidelines for hospital-acquired pneumonia and ventilator-associated pneumonia in adults. Can. J. Infect. Dis. Med. Microbiol. 19:19–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ruzin A, Petersen PJ, Jones CH. Resistance development profiling of piperacillin in combination with the novel β-lactamase inhibitor BLI-489. J. Antimicrob. Chemother. 65:252–257 [DOI] [PubMed] [Google Scholar]
- 24. Sader HS, et al. 2010. Antimicrobial activity of ceftazidime/NXL-104 tested against Gram-negative organisms, including multidrug-resistant subsets, causing infections in USA and European medical centers, abstr E-811. Abstr. 20th Eur. Congr. Clin. Microbiol. Infect. Dis., Vienna, Austria [Google Scholar]
- 25. Stachyra T, et al. 2010. Mechanistic studies of the inactivation of TEM-1 and P99 by NXL104, a novel non-beta-lactam beta-lactamase inhibitor. Antimicrob. Agents Chemother. 54:5132–5138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stevens DL, et al. 2005. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin. Infect. Dis. 41:1373–1406 [DOI] [PubMed] [Google Scholar]
- 27. Turnidge JD. 1998. The pharmacodynamics of beta-lactams. Clin. Infect. Dis. 27:10–22 [DOI] [PubMed] [Google Scholar]
- 28. Vazquez J, et al. 2011. Ceftazidime avibactam (ceftazidime/NXL104) versus imipenem cilastatin for complicated urinary tract infections in hospitalized adults. Abstr 51st Intersci. Conf. Antimicrob. Agents Chemother., Chicago, IL http://www.icaac.org/ [Google Scholar]
- 29. Walkty A, et al. 2011. In vitro activity of ceftazidime combined with NXL104 versus Pseudomonas aeruginosa isolates obtained from patients in Canadian hospitals (CANWARD 2009 study). Antimicrob. Agents Chemother. 55:2992–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wiskirchen DE, Crandon JL, Furtado GH, Williams G, Nicolau DP. In vivo efficacy of a human-simulated regimen of ceftaroline combined with NXL104 against extended-spectrum-beta-lactamase (ESBL)-producing and non-ESBL-producing Enterobacteriaceae. Antimicrob. Agents Chemother. 55:3220–3225 [DOI] [PMC free article] [PubMed] [Google Scholar]