

Abstract
A cDNA and corresponding promoter region for a naturally occurring, feedback-insensitive anthranilate synthase (AS) α-subunit gene, ASA2, has been isolated from an unselected, but 5-methyl-tryptophan-resistant (5MTr), tobacco (Nicotiana tabacum) cell line (AB15–12-1). The ASA2 cDNA contains a putative transit peptide sequence, and Southern hybridization shows that more than one closely related sequence is present in the tobacco genome. The ASA2 cDNA complemented a trpE nonsense mutant Escherichia coli strain, allowing growth on 300 μm 5MT-containing minimal medium without tryptophan, and cell extracts contained feedback-insensitive AS activity. The 5MTr was lost when the E. coli strain was transformed with an ASA2 site-directed mutant (phenylalanine-107-arginine-108 → serine-107-glutamine-108). Identical nucleotide sequences encoding the phenylalanine-107-arginine-108 region have been found in polymerase chain reaction-amplified 326-bp ASA2 genomic fragments of wild-type (5-methyl-tryptophan-sensitive [5MTs]) tobacco and a progenitor species. High-level ASA2 transcriptional expression was detected only in 5MTr-cultured cells, not in 5MTs cells or in plants. Promoter studies indicate that tissue specificity of ASA2 is controlled by the promoter region between −2252 and −607. Since the ASA2 promoter sequences are not substantially different in the 5MTr and 5MTs lines, the increased levels of ASA2 mRNA in the 5MTr lines are most likely due to changes in a regulatory gene affecting ASA2 expression.
Many aromatic compounds are synthesized via the shikimate pathway in higher plants (Haslam, 1993; Herrmann, 1995). Chorismate is located at a branch point of this pathway and is the last common precursor of many of these compounds. AS catalyzes the first committed reaction in the Trp-biosynthesis branch by converting chorismate to anthranilate and is feedback inhibited by the end product, Trp (Haslam, 1993; Romero et al., 1995). That AS is the control point in the Trp branch in plant cells is indicated by (a) pathway-intermediate feeding studies (Widholm, 1974), (b) enzyme activity levels, (c) feedback inhibition of the respective enzyme activities (Singh and Widholm, 1974), and (d) 5MT resistance selection that yielded lines with altered feedback-inhibited AS and higher free Trp (Widholm, 1972a, 1972b).
Plant cell culture systems have been useful for studying the regulatory mechanisms of amino acid biosynthesis (Widholm, 1972a, 1972b). Selecting cells resistant to amino acid analogs such as 5MT can produce lines with increased amounts of the end product, Trp, because of an alteration of the allosteric regulatory enzyme, causing less inhibition of the enzyme by the analog and Trp (Widholm, 1972a, 1972b). Feedback-insensitive AS has been found in 5MT-selected carrot (Daucus carota; Brotherton et al., 1986), Datura innoxia (Ranch et al., 1983), potato (Solanum tuberosum; Carlson and Widholm, 1978), and tobacco (Nicotiana tabacum; Brotherton et al., 1986). The 5MTr tobacco cell line (TX2–4) contained Trp-insensitive AS and higher levels of free Trp than wild-type cells. Similar results were obtained with 5MT-selected cultured potato cells (Carlson and Widholm, 1978). These characteristics were not found in the regenerated tobacco plants but were recovered again in cultured cells induced from leaves of the regenerants (Brotherton et al., 1986). These results suggest that expression of this altered AS is regulated in a tissue-specific manner and that selection for 5MTr produces lines with increased amounts of the feedback-insensitive AS form that is expressed in cultured cells but not in plants.
Recently, AS was purified from cultured plant cells or plant tissues (Poulsen et al., 1993; Bohlmann et al., 1995; Romero and Roberts, 1996) and found to have a subunit composition similar to AS from some microbes, i.e. nonidentical large (α, component I) and small (β, component II) subunits (Zalkin et al., 1984; Yanofsky and Crawford, 1987; Crawford, 1989). AS genes encoding an α-subunit (Niyogi and Fink, 1992; Bohlmann et al., 1995) and β-subunit (Niyogi et al., 1993) have been cloned from Arabidopsis and Ruta graveolens. The two AS genes encoding the α-subunit of the enzyme cloned from Arabidopsis and R. graveolens have been designated ASA1/ASA2 and ASα1/ASα2, respectively (Niyogi and Fink, 1992; Bohlmann et al., 1995). These AS genes have similar gene-expression patterns, since ASA1 and ASα1 expression is induced by wounding and/or elicitor treatment, whereas the ASA2 and ASα2 genes are expressed constitutively at low levels (Niyogi and Fink, 1992; Bohlmann et al., 1995).
Two groups have described a mutant Arabidopsis AS gene encoding a feedback-insensitive α-subunit of the enzyme (Kreps et al., 1996; Li and Last, 1996). Sequencing of these mutant AS genes showed that a single amino acid, Asp-341, was replaced by Asn in both cases. These mutant AS genes were expressed in both cultured cells and leaves, with slightly higher expression in cultured cells (Kreps et al., 1996). In contrast to these Arabidopsis mutants, the ASα1 gene from R. graveolens that encodes a feedback-insensitive α-subunit was isolated from wild-type young shoots (Bohlmann et al., 1996). Based on sequence comparison, it was postulated that a single amino acid substitution in the ASα1 gene, Arg-138 for Gln-138, may cause feedback insensitivity. The residues potentially affecting feedback inhibition in higher plants do not align with those of bacteria (Matsui et al., 1987; Caliguri and Bauerle, 1991) and yeast (Graf et al., 1993), but the altered amino acids are located within or very close to the conserved motifs Leu-Leu-Glu-Ser-X10-Ser and Asn-Pro-Ser-Pro-Tyr-Met, which are important for feedback inhibition (Matsui et al., 1987; Caliguri and Bauerle, 1991; Graf et al., 1993).
The tobacco plants regenerated from the 5MTr suspension cultures described by Brotherton et al. (1986) were not fertile; therefore, the selection was repeated with new cultures to obtain fertile plants (S. Schechter and J.M. Widholm, unpublished data). One of nine plants regenerated from an unselected control culture produced a suspension culture from a leaf that was 5MTr. This resistance was inherited by progeny, and the cell line studied here, AB15–12-1, was initiated from a fifth-generation plant. This cell line has the typical characteristics of 5MTr cells, feedback-insensitive AS and increased free Trp. We report the cloning and characterization of the wild-type tobacco ASA2 gene encoding a feedback-insensitive α-subunit of the enzyme and analysis of the role of promoter regions in tissue-specific expression of this gene. We show that ASA2 mRNA is specifically elevated in 5MTr lines and propose that this increased level of expression is due to enhanced transcription of the ASA2 gene in the 5MTr lines.
MATERIALS AND METHODS
Suspension cultures were maintained by weekly transfers into 50 mL of liquid MX medium (MS basal medium [Murashige and Skoog, 1962] with 1.8 μm 2,4-D). The 5MTr AB15–12-1 cell line originated from a tobacco (Nicotiana tabacum cv Xanthi) plant regenerated from unselected suspension-cultured cells. Originally, nine plants were regenerated by placing the unselected cells on MS agar-solidified medium with 5.7 μm IAA and 2 μm kinetin. Callus and subsequent suspension cultures were initiated from the leaves of these plants. One plant produced a suspension culture that showed resistance to 5MT when 0.5 g fresh weight of cells was incubated for 9 to 12 d in MX liquid medium containing 46 and 137 μm 5MT; other wild-type cells did not grow in the same medium. Because this plant was male-sterile, it was pollinated with wild-type pollen and the resulting progeny were self-pollinated four times to obtain AB15–12-1. Additional tobacco cell-suspension cultures were subsequently initiated from AB15–12-1 seedlings and maintained on MX or 5MT-containing medium as indicated.
Nucleic Acid Analysis
Genomic DNA was isolated from 1-week-old suspension-cultured cells using the procedures of Dellaporta et al. (1983) and purified by CsCl-gradient centrifugation (Ausubel et al., 1989; Sambrook et al., 1989).
Total RNA was prepared using a phenol-extraction method (Wang et al., 1994) from 1-week-old suspension-cultured cells, mature seeds, and leaves, roots, and stems harvested from 3-week-old shoot cultures grown on MS basal medium solidified with 0.8% agar.
DNA and RNA gels were blotted onto a nylon membrane (Hybond N+, Amersham) following a general capillary-transfer method (Sambrook et al., 1989). The Arabidopsis thaliana ASA1 (pKN41) cDNA clone (obtained from K. Niyogi [Niyogi and Fink, 1992]) or other tobacco AS cDNA clones obtained in this work were used as a probe following labeling with a Megaprime DNA-labeling system (Amersham) with [α-32P]dCTP (3000 Ci/mmol). Southern and northern hybridizations were done at 42°C with hybridization solution (50% formamide, 5× SSPE, 5× Denhardt's solution [Sambrook et al., 1989], 0.1% SDS, and 100 μg/mL salmon-sperm DNA). The membranes were washed at high stringency twice at room temperature with 2× SSC and 0.5% SDS for 20 min each time and at 65°C with 0.1× SSC and 0.1% SDS until the background signal disappeared and were then exposed to radiographic film overnight with an intensifying screen.
Cloning of AS cDNA
Tobacco AS cDNAs were isolated using 5′ and 3′ RACE (GIBCO-BRL). Primers 1 and 2 for cloning the 5′ end of the ASA2 cDNA were designed based on the sequence of the Arabidopsis ASA1 genomic clone (GenBank accession no. M92353). The sequences of primers used for cloning are listed in Table I. For 5′ RACE, first-strand cDNA was synthesized with antisense primer 1 at nucleotide position 5403 to 5382 of the Arabidopsis ASA1 genomic clone. The 5′ end of the first-strand cDNA was oligo-dA tailed (200 μm dATP) using terminal deoxynucleotidyl transferase (0.4 unit/μL).
Table I.
Nucleotide sequences of the primers used for cloning
| Primer | Sequence | Locationa |
|---|---|---|
| 1b | 5′-GCGGCTTTGTTCTGGCACTCA-3′ | |
| 2b | 5′-CTGCAAATGTTCGCCGCTCAA-3′ | |
| 3 | 5′-CTAGTTATGGATGAGGACAGG-3′ | +148/+168 |
| 4 | 5′-ACTAGTGGATCCTCTAAAAGCGGGAACTTG | +181/+198 |
| 5 | 5′-TTGCGGGGTACCCTAGTTTCTTTTCTCATGTAC-3′ | +1851/+1831 |
| 6 | 5′-ACTAGTGGATCCTGCCTTCACTCTTCATCTCTAG-3′ | +130/+151 |
| 7 | 5′-ACCTTGAGACCCGGGTTCAACGGATTCAAAGAGAAAGCTTGG-3′ | +327/+286 |
| 8 | 5′-TCCGTTGAACCCGGGTCTCAAGGTTCTAGTGTTGGTCGCTAC-3′ | +304/+345 |
| 9 | 5′-GATCCCATTTTCAAAGTCACC-3′ | −110/−90 |
| 10 | 5′-TGTTCCTTAGCCACAATTTCC-3′ | +392/+372 |
| 11 | 5′-ACGACTGCATTCCTACAAGAG-3′ | +10/−11 |
| 12 | 5′-GGATCCCCCGGGTCCTACAAGAGCACAATA3′ | −1/−18 |
| 13 | 5′-GCATGCCTGCAGCAAATCTATTCGATAGTG-3′ | −2252/−2235 |
| 14 | 5′-GCATGCCTGCAGTAGGCAATACGGCACATA-3′ | −1356/−1339 |
| 15 | 5′-GCATGCCTGCAGTGTATTGCCCATTTCATT-3′ | −606/−589 |
| 16 | 5′-GCATGCCTGCAGTCAGCCAAATGTGTCCAA-3′ | −370/−353 |
Restriction enzyme site for cloning at 5′ end of the primers is underlined. Boldfaced nucleotides in the primers 7 and 8 represent mismatch nucleotides for site-directed mutagenesis.
Locations of primers were numbered based on the nucleotide sequence. Adenine of the translation start codon (ATG) was numbered as a +1.
Degenerated primers 1 and 2 were designed based on the sequence of the Arabidopsis ASA1 genomic clone.
A nested PCR was performed with the adapter primer 5′-GGCCACGCGTCGACTAGTAC(T)17-3′ (GIBCO-BRL) and antisense primer 2 at nucleotides 4430 to 4409 of the Arabidopsis ASA1 genomic clone. The first-strand cDNA was used as a template for the nested PCR. A fragment (approximately 1.1 kb) was amplified and cloned into the pGEM-T vector and sequenced. The procedures used to isolate the 3′ end of the ASA2 cDNA were the same as for 5′ RACE except for the primers and dATP tailing at the 5′ end of the cDNA. Primer 3 was designed based on the sequence of the 5′ ASA2 cDNA clone at nucleotides +148 to +168 (adenine of ATG was counted as +1, since the transcription start site was not determined in this work). An approximately 1.9-kb fragment was amplified and strongly hybridized with the 5′ ASA2 cDNA clone as a probe. This fragment was cloned into the pGEM-T vector and sequenced.
Analysis of the sequence alignment of the two fragments using Clustal (Higgins and Sharp, 1989) shows that the 828-bp overlapping region is identical. There is only one XbaI site in the 828-bp overlapping region, only one NsiI site in the pGEM-T vector, and no NsiI site in either the 5′ or the 3′ fragments. These two restriction enzyme sites were used to construct the full-length tobacco ASA2 cDNA. Sequencing of the cDNA clones was performed by the Genetic Engineering Laboratory of the University of Illinois (Urbana-Champaign) using the dideoxynucleotide-sequencing method.
Complementation Tests and Site-Directed Mutagenesis
The ASA2 cDNA without the presumed transit-peptide sequence (amino acid sequence 1–60) was amplified using Pfu DNA polymerase (Stratagene) with primers 4 and 5 (Table I) containing BamHI and KpnI overhangs, respectively, and ligated into an expression vector (pQE30 containing a 6-His tag-coding sequence [6xHis, Qiagen, Santa Clarita, CA]) followed by restriction-enzyme digestion.
Site-directed mutagenesis was performed by PCR using a primer
containing mismatched nucleotide sequences by changing four nucleotides
(CC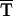 GG
GG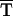 T
T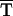 TC
TC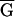 A→CC
A→CC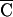 GG
GG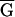 T
T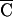 TC
TC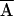 A)
at amino acid residues 105 to 108. The first two mismatched
nucleotides, Pro-105 and Gly-106, do not change the amino acid codon
but create an SmaI site. The last two mismatched nucleotides
change Phe-107 and Arg-108 to Ser-107 and Gln-108. Two PCR products
were obtained using primers 6 and 7 and primers 8 and 5 (Table I).
These two PCR fragments were ligated, digested by SmaI, and
ligated in-frame into the pQE30 vector.
A)
at amino acid residues 105 to 108. The first two mismatched
nucleotides, Pro-105 and Gly-106, do not change the amino acid codon
but create an SmaI site. The last two mismatched nucleotides
change Phe-107 and Arg-108 to Ser-107 and Gln-108. Two PCR products
were obtained using primers 6 and 7 and primers 8 and 5 (Table I).
These two PCR fragments were ligated, digested by SmaI, and
ligated in-frame into the pQE30 vector.
The chimeric constructs were transformed into trpE mutant Escherichia coli (trpE5972, nonsense mutant) using CaCl2 transformation (Sambrook et al., 1989). Complemented strains were plated on M9 minimal medium containing 100 μg/mL ampicillin and 0.1 mm IPTG but no Trp. For the inhibition test, 300 μm 5MT was added to the minimal medium described previously.
Expression of 6xHis-Tagged ASA2 Protein in E. coli
The E. coli trpE mutant (trpE5972) transformed with the 6xHis-tagged ASA2 chimeric construct was grown in 1 L of Luria-Bertani medium supplemented with 100 μg mL−1 ampicillin and 0.1 mm Trp. IPTG (0.1 mm) and PMSF (0.1 mm) were added to a mid-logarithm culture. After 3 h of further incubation at 30°C and 150 rpm, the cells were harvested by centrifugation, resuspended in 20 mL of extraction buffer (Bernasconi et al., 1994) with 0.1 mm PMSF added, disrupted using a French press (two passages at 20,000 p.s.i.), and centrifuged to remove cell debris. The supernatant was held overnight on ice and then combined with 2 volumes of saturated, room-temperature (NH4)2SO4. The resulting protein precipitate was collected by centrifugation at 4°C and resuspended in 10 mL of 50 mm NaH2PO4, 300 mm NaCl, and 2 mm DTT, pH 8.0. The 6xHis-tagged ASA2 protein was partially purified after binding to Ni-nitrilotriacetic acid resin (Qiagen) and elution with 100 mm imidazole in 50 mm NaH2PO4, 300 mm NaCl, 2 mm DTT, and 10% glycerol, pH 6.0.
Arabidopsis ASA1 protein fused with glutathione S-transferase was expressed using the pSCI1674 construct cloned into E. coli MC1061 by Bernasconi et al. (1994). The procedure was the same as described above except that the resuspended (NH4)2SO4 pellet was used without further purification.
AS Enzyme Assay
Cell extracts were prepared as described by Brotherton et al. (1986) from mid-logarithm-phase cell cultures and were used the same day. The resuspended (NH4)2SO4 fraction was desalted using Sephadex G-25.
AS activity was measured using the ethyl-acetate-extraction method used previously for tobacco cell extracts (Widholm, 1971) and incorporating the modifications described by Li and Last (1996), except that the assay buffer was 50 mm Hepes, pH 7.5, 10 mm Gln, 2.0 mm MgCl2, 0.05 mm Na2EDTA, 2.0 mm DTT, and 5% glycerol. The reaction was started by the addition of 50 μL of cell extract. Chorismate and Trp concentrations were as indicated in individual experiments. The production of anthranilate was linear with respect to time for twice the standard incubation time and enzyme concentration (data not shown). The conversion of chorismate to anthranilate never exceeded 1% in any experiment. Chorismate was produced using the fermentation method of Gibson (1970).
ASA2 Genomic Fragments Containing the Phe-107-Arg-108 Region from Wild-Type Nicotiana sp.
ASA2 genomic fragments containing the Phe-108-Arg-108 region were PCR amplified and isolated from three wild-type (5MTs) cultured cell lines, N. tabacum (TXD), Nicotiana tomentosiformis (Nto), and Nicotiana sylvestris (Ns), from leaves harvested from AB15–12-1 shoot cultures, and from two 5MTs (H15–6, NRMX) and one 5MTr (NR5MT) tobacco cell lines. The region from −110 to +392 was first amplified using two sets of primers based on the ASA2 cDNA and promoter sequences 9 and 10 (Table I). Primers 4 and 10 were then used for a nested PCR amplification using the first PCR product as the template to produce the 326-bp genomic fragments (+181 to +392) that were then cloned into pBluescriptSK−, digested with EcoRV, and sequenced.
Cloning of AS Promoter and Construction of Chimeric AS Promoter-GUS Constructs
The ASA2 promoter was isolated using inverse PCR (Ochman et al., 1989). AB15–12-1 genomic DNA was digested with HindIII, circularized with T4 DNA ligase after dilution to 100 ng/mL with distilled, deionized water and used as a template for inverse PCR with primer 3 and antisense primer 11 based on the sequence of the ASA2 cDNA at nucleotide +10 to −11. PCR amplification was performed for 30 cycles (95°C, 1 min; 50°C, 40 s; 72°C, 2 min). A PCR fragment (approximately 2.3 kb) was detected by Southern hybridization with the ASA2 cDNA as a probe and cloned into pGEM-T vector. Sequencing of the ASA2 promoter (approximately 2.3 kb) was performed at the Molecular Analysis and Synthesis Section of the Samuel Roberts Noble Foundation (Ardmore, OK) using the dideoxynucleotide-sequencing method.
Based on database search results, deletions were made by using PCR amplification with seven sets of primers as follows: antisense primer 12 with sense primers 13, 14, 15, or 16 (Table I) that amplified 2252-, 1356-, 606-, or 370-bp ASA2 promoter fragments (including the 5′ UTR), respectively. These PCR products were fused to the GUS reporter gene with NOS terminator. The chimeric constructs were designated 2252, 1356, 606, and 370. Each primer contains a restriction enzyme site overhang for cloning. Primer 12 contains an SmaI site and primers 13, 14, 15, and 16 contain a PstI site. Each restriction enzyme site was underlined in the primer sequences (Table I). These five fragments were cloned into pBI221, replacing the CaMV 35S promoter.
GUS Assay
The constructed plasmid DNAs were isolated using a Plasmid Maxi Kit (Qiagen) and biolistically transferred into tobacco suspension-cultured cells (AB15–12-1) and leaves using a particle-inflow gun (5 μg of DNA and 0.5 mg of 1.0-μm diameter gold particles [Bio-Rad] shot at 80 p.s.i.; Vain et al., 1993). Leaves (180–350 mg fresh weight) harvested from 3-week-old shoot cultures were placed on 5-cm-diameter sterile Whatman no. 1 filter paper discs. Five milliliters of 2-d-old AB15–12-1 suspension-cultured cells (approximately 200 mg fresh weight) was collected on filter paper using a vacuum before bombardment. The samples were transferred with the filter paper onto MX solid medium after bombardment and incubated at 24°C under fluorescent light (60 μE m−2 s−1) for 3 d. The promoter activity was determined with the fluorometric MUG assay (Jefferson, 1987; Jefferson et al., 1987). GUS-specific activity was determined as picomoles of 4-methylumbelliferone per hour per milligram of protein. The protein concentration was determined using a protein dye-binding assay kit (Bio-Rad).
RESULTS
ASA2 cDNA Cloning and Sequence Analysis
We have cloned a full-length ASA2 (2.16 kb including 5′ and 3′ UTR) cDNA using 5′ and 3′ RACE. The nucleotide and deduced amino acid sequences are shown in Figure 1. This AS gene is denoted ASA2, since its amino acid sequence was 72% identical to the sequence of ASα2 of R. graveolens. An apparent 5′ UTR was found 89 nucleotides upstream of a translation start codon (ATG) in the 5′ end of the cDNA fragment. The 3′ RACE results indicate that polyadenylation begins 205 nucleotides downstream of the translation stop codon. No perfect match to the consensus eukaryotic poly(A+) signal sequence AAUAAA was found in the 3′ UTR.
Figure 1.

Nucleotide and the predicted amino acid sequence of the ASA2 cDNA. The coding region corresponds to amino acid sequence 1 (ATG) to 616 (TAG). The putative translation start and stop codons are indicated in boldface. The 89 and 205 bp of the upstream and downstream coding region represent 5′ and 3′ UTRs, respectively.
The ASA2 cDNA clone hybridized under high-stringency conditions to several fragments of genomic DNA digested with nine restriction enzymes (Fig. 2A). Three major bands at approximately 3.3, 4.0, and 24 kb were found in KpnI-digested genomic DNA of the amphidiploid N. tabacum leaves and the two cultured cell lines, whereas the diploid tobacco progenitors, N. sylvestris and N. tomentosiformis, showed different patterns (Fig. 2B). The hybridization pattern of N. tabacum was more like N. tomentosiformis than N. sylvestris, since both had bands at approximately 3.3 and 4.0 kb. Two faint bands hybridized to the ASA2 cDNA clone in N. sylvestris. The 24-kb fragment in N. tabacum shows very weak hybridization and is not found in either N. sylvestris or N. tomentosiformis.
Figure 2.

Genomic DNA-blot analysis. A, Twenty micrograms of AB15–12-1 genomic DNA was digested with nine restriction enzymes. Lanes from left to right correspond to BamHI, EcoRI, EcoRV, HincII, HindIII, KpnI, PstI, ScaI, and XbaI. B, Ten micrograms of each genomic DNA obtained from leaves of N. sylvestris, N. tomentosiformis, N. tabacum, and suspension-cultured cells of the 5MTs and 5MTr tobacco cell line (AB15–12-1) was digested with KpnI. The full-length ASA2 cDNA clone was used as a probe in both A and B. Fragment sizes were determined by a 1-kb ladder (GIBCO-BRL).
Comparison of Amino Acid Sequences of AS Genes
Based on BLAST analysis (Altschul et al., 1990), the five best matches to the predicted amino acid sequence of the tobacco ASA2 gene are aligned in Figure 3. The 1851 nucleotide ASA2-coding region would encode a 616-amino acid polypeptide with a calculated molecular mass of 69,035 D. The tobacco ASA2 gene encodes 60 amino acids beyond the amino terminus of the aligned microbial homologs. The sequence Ile-58-Glu-59-Ala-60-Ser-61 is similar to the consensus cleavage-site motif ([Val/ Ile]X[Ala/Cys]↓Ala) for plant chloroplast transit peptides (von Heijne et al., 1989; Gavel and von Heijne, 1990). The transit peptide sequences of the five plant AS genes show little homology to each other. The tobacco ASA2 gene shows 72 and 70, 67 and 66, and 32% amino acid identity to R. graveolens ASα2 and ASα1, A. thaliana ASA1 and ASA2, and Clostridium thermocellum trpE genes, respectively (Sato et al., 1989; Niyogi and Fink, 1992; Niyogi et al., 1993; Bohlmann et al., 1995).
Figure 3.

Amino acid sequence alignment of AS genes from plants and a prokaryote. TASA2, RASA1, RASA2, AASA1, AASA2, and CTRPE correspond to N. tabacum ASA2, R. graveolens ASα1 and ASα2, A. thaliana ASA1 and ASA2, and C. thermocellum trpE cDNA clones, respectively. Dashes within sequences indicate gaps. Asterisks under the sequence represent identical amino acids among these six different AS sequences. Dots under the sequence indicate similar amino acids. Two consensus motifs, Leu-Leu-Glu-Phe-X10-Ser and Asn-Pro-Ser-Pro-Tyr-Met, affecting feedback inhibition based on microorganisms and yeast, are indicated in boldface. The amino acid(s) substitutions Phe-Arg and Arg, which caused feedback insensitivity in N. tabacum and R. graveolens, are indicated in boldface with a diamond mark at positions 107 and 108, respectively. A single amino acid change in an Arabidopsis mutant (Asp to Asn) is indicated in boldface with a dagger at position 326. A consensus sequence motif for plant chloroplast transit peptides in the tobacco ASA2 is underlined at positions 58 to 61. The amino acid numbers indicated are based on the tobacco ASA2 amino acid sequence.
In bacteria, fungi, and yeast two conserved amino acid motifs, Leu-Leu-Glu-Ser-X10-Ser and Asn-Pro-Ser-Pro-Tyr-Met, are associated with Trp feedback inhibition (Matsui et al., 1987; Caliguri and Bauerle, 1991; Graf et al., 1993). The six AS genes in Figure 3 showed no amino acid changes in these conserved regions except in the Leu-Leu-Glu-Ser-X10-Ser motif, where the Ser and Gln residues found in feedback-sensitive AS of higher plants were changed to Phe-107 and Arg-108 (based on the tobacco ASA2 amino acid sequence) in the tobacco ASA2 and to Arg-108 in the ASα1 gene of R. graveolens. These changes appear to correlate with the feedback insensitivity found in the latter two enzymes.
AS Gene Expression
The tobacco ASA2 cDNA probe detected high levels of a 2.2-kb transcript on northern blots only in 5MTr cultured tobacco cell lines, including 5MTr N. sylvestris but not in 5MTs-cultured cell lines, leaves, roots, stems, and seeds after overnight exposure (Fig. 4).
Figure 4.

Northern analysis. Twenty micrograms of total RNA per lane was used for northern hybridization. A full-length ASA2 cDNA clone (ASA2) and rRNA (rRNA) were used as probes for hybridization. A, Total RNAs were prepared from 1-week-old N. tabacum suspension-cultured cells (SC) of four 5MTs (s) and four 5MTr (r) cell lines, 5MTs N. tabacum leaves (L) harvested from 3-week-old AB15–12-1 shoot cultures, and 1-week-old 5MTr N. sylvestris suspension-cultured cells. B, Different plant organs were used to determine tissue specificity. AB15–12-1 leaves, roots, and stems harvested from 3-week-old shoot cultures and dried mature seeds and suspension-cultured 5MTs cells (TXD) and 5MTr cells (AB15–12-1) were used to prepare total RNA.
Complementation and Inhibition Tests
The E. coli trpE5972 nonsense mutant transformed with the tobacco ASA2 cDNA and with the site-directed mutant ASA2 form (Phe-107-Arg-108 changed to Ser-107-Gln-108) both grew on minimal medium containing ampicillin and IPTG but no Trp (Fig. 5A). The complemented strain transformed with the site-directed mutant, however, did not grow on 300 μm 5MT containing minimal medium without Trp (Fig. 5B), whereas the growth of the strain transformed with the ASA2 cDNA was not inhibited by 300 μm 5MT.
Figure 5.

Complementation of E. coli trpE5972 by the tobacco ASA2 and a site-directed mutant. The pQE30/ASA2 and pQE30/ASA2 mutants were transformed into the trpE nonsense mutant E. coli strain (trpE5972) and plated on M9 minimal medium containing 100 μg/mL ampicillin (A) and 0.1 mm IPTG without and with 300 μm 5MT (B). The picture was taken 2 d after streaking.
AS Kinetic Constants
Trp inhibition of the partially purified product of the E. coli-expressed tobacco ASA2 and Arabidopsis ASA1 genes is shown in Figure 6A. The ASA2 gene product is still 50% active at 100 μm Trp, similar to AS in 5MT-selected tobacco cells and to purified R. graveolens ASα1 protein, which was 80% active at this Trp concentration (Bohlmann et al., 1996).
Figure 6.

Kinetics of AS from wild-type (□) and 5MT-selected (▪) tobacco cells and from E. coli transformed with the Arabidopsis ASA1 gene (⋄) or the tobacco ASA2 gene (♦). A, Relative AS activity in the presence of Trp was measured as described in Methods (with 100 μm chorismate) and is expressed as a percentage of the activity with no Trp added. The specific activity with no Trp for AS from each tobacco line was 73 and 169 nmol anthranilate min−1 mg−1 protein, respectively. B. Lineweaver-Burk plot of AS activity. Velocity is expressed as nanomoles per minute per milligram of protein.
When AS activity was measured in extracts from wild-type and 5MTr tobacco cell-suspension cultures the resistant cultures had a portion of the activity that is more feedback insensitive than that of the wild-type cells (Fig. 6A). The apparent Ki values for Trp are 2 and 300 μm, respectively, for AS from wild-type and 5MT-selected tobacco cells when estimated from the Trp concentration, resulting in 50% inhibition, as shown in Figure 6A. Similar values were reported by Bohlmann et al. (1996) for R. graveolens Trp-sensitive and -insensitive AS (2.8 and >100 μm, respectively). AS from wild-type Arabidopsis had an apparent Ki of 3 μm, whereas AS from a 6-methyl-Trp-selected mutant had a Ki of 8 μm (Li and Last, 1996).
The apparent chorismate Km value was lower for AS from 5MT-selected cells (6 μm) than for the wild-type cells (22 μm) when measured using 10 mm Gln as the second substrate (Fig. 6B). These values are comparable to those obtained for AS from Arabidopsis (10 and 21 μm using Gln [Li and Last, 1996]) and R. graveolens (17 and 29 μm using NH4+ [Bohlmann et al., 1996]). In all three species the chorismate Km value (first value listed) is lower for the feedback-insensitive AS.
Comparison of ASA2 and Wild-Type Gene Sequences
The ASA2 fragment between +181 and +392, which contains the Phe-107-Arg-108 residues, was PCR amplified and cloned from 5MTs and 5MTr genomic DNAs. When the nucleotide sequence of the fragment from AB15–12-1 was aligned with those from the three wild-type Nicotiana sp. cell lines (Fig. 7) and compared with the ASA2 cDNA, the genomic fragments were found to contain a 115-bp intron; therefore, the cloned fragments were 326 bp in length. The nucleotide sequences of this fragment were 100% identical in all cases except for eight nucleotides in N. sylvestris. This eight-nucleotide difference did not change the encoded amino acids. Amino acid sequences in this region showed 66% identity to four other plant AS genes from Arabidopsis and R. graveolens.
Figure 7.

Nucleotide sequence alignment of ASA2 genomic DNA fragments. The 326-bp ASA2 genomic DNA fragments, corresponding to the ASA2 cDNA sequence from +181 to +392, were amplified from the genomic DNAs of tobacco leaves (ASA2) harvested from AB15–12-1 shoot cultures and three wild-type (5MTs) Nicotiana sp. cultured cell lines, N. tabacum (TXD), N. tomentosiformis (Nto), and N. sylvestris (Ns) using primers 4 (sense) and 10 (antisense), which are shown in bold. These genomic DNA fragments contain a 115-bp intron, which is underlined. Eight nucleotides, indicated by (+) above the sequence, are different in the N. sylvestris genomic fragment compared with those in N. tabacum and N. tomentosiformis. The nucleotide sequences corresponding to Phe-107-Arg-108 are indicated in boldface and underlined with the one-letter amino acid codon abbreviations F and R above the sequence.
Promoter-Sequencing Analysis
The 99 nucleotides at the 3′ end of the promoter region and 5′ end of the ASA2 cDNA, including the 5′ UTR, were identical (Fig. 8). The first possible TATA box (TATAAA) is located 121 bp upstream from the translation start site (ATG). BLAST analysis shows that the region from −1187 to −769 exhibits 81 to 83% nucleotide sequence identity to the promoter region of the N. tabacum plant-defense-related Str246C gene (Froissard et al., 1994).
Figure 8.

The nucleotide sequence of the ASA2 promoter region. The first possible TATA box (TATAAA), translation start codon (ATG), is indicated in boldface. The 98 bp at the 3′ end of the ASA2 promoter region, which is identical to the nucleotide sequence at 5′ end of the ASA2 cDNA, is underlined.
Chimeric GUS Gene Expression
When different deleted fragments of the ASA2 promoter region were tested, the transient GUS expression controlled by the promoter region denoted 606 was stronger in both suspension-cultured cells and in leaves than that driven by the CaMV 35S promoter (Fig. 9). The 1356 and 2252 regions still showed high GUS activity in suspension-cultured cells but very low activity in leaves. The 1356 region showed approximately 5 times higher GUS expression than that driven by the CaMV 35S promoter in 5MTr suspension-cultured cells.
Figure 9.

Chimeric ASA2 promoter-GUS constructs and transient GUS assays with tobacco leaves and suspension-cultured cells. Four deleted promoter fragments were fused to the GUS reporter gene with NOS terminator in pUC19 and designated 2252, 1356, 606, and 370. These chimeric constructs and the CaMV 35S-GUS construct as a control were bombarded into 2-d-old AB15–12-1 cultured cells (black bars) and leaves (white bars) harvested from 3-week-old shoot cultures. GUS transient expression was measured using the MUG assay 3 d after bombardment. The average values after subtracting the background from four separate experiments with two replicates are presented. GUS-specific activity was determined as picomoles of 4-methylumbelliferone per hour per milligram of protein.
To determine whether the tissue culture-specific expression controlled by the ASA2 promoter was caused by transcription factor(s) in the 5MTr cell lines, we performed separate experiments using suspension-cultured cells from 5MTs N. tabacum and from 5MTs and 5MTr N. sylvestris. The transient GUS activities in the N. tabacum 5MTr suspension-cultured cells (AB15–12-1) controlled by 35S, 606, 1356, and 2252 promoters were approximately 1.8-, 7.4-, 3.7-, and 3.6-fold that of the 5MTs tobacco suspension-cultured cells, respectively. In the case of N. sylvestris, 3.4-, 10.6-, 6.4-, and 9.1-fold GUS activities were detected in the 5MTr suspension-cultured cells in comparison with the 5MTs suspension-cultured cells, respectively.
That the ASA2 promoter shows tissue-specific expression was also shown by stable transformation experiments with tobacco using Agrobacterium tumefaciens (data not shown).
DISCUSSION
We have cloned the tobacco ASA2 cDNA encoding the α-subunit of a feedback-insensitive AS and the promoter region. Southern hybridization analysis shows that more than one closely related sequence is present in the tobacco genome (Fig. 2). Like all plant AS genes described to date, the tobacco ASA2 has a putative transit peptide sequence; therefore, the enzyme is apparently localized in the plastids.
Our results support the conclusion that ASA2 is a wild-type gene encoding the α-subunit of a feedback-insensitive AS in tobacco. These results include the feedback-insensitive ASA2 gene expression in the trpE mutant E. coli strain (Fig. 5), enzyme kinetic analysis (Fig. 6), the nucleotide sequence of wild-type ASA2 genomic fragments (Fig. 7), and high-level expression in 5MTr suspension-cultured cells (Fig. 4).
Several regions of the AS amino acid sequence have been shown to affect feedback inhibition (Bohlmann et al., 1996; Kreps et al., 1996; Li and Last, 1996). In tobacco ASA2 from both wild-type and 5MTr cell lines, the two amino acids Phe-107 and Arg-108, found in the same region as in ASα1 (Bohlmann et al., 1996), were different from those found in feedback-sensitive AS, Ser-107 and Gln-108. When we changed the ASA2 Phe-107 and Arg-108 residues to Ser-107 and Gln-108 by site-directed mutagenesis, the gene product was still active in E. coli, but this complemented E. coli strain could not grow on 300 μm 5MT containing minimal medium without Trp, whereas the original ASA2 complemented strain could. In addition, the E. coli-expressed ASA2 gene product was feedback insensitive like the AS found in 5MTr tobacco cell extracts. These results indicate that the Phe-107 and Arg-108 residues are important in the control of feedback inhibition. Further mutagenesis and kinetic and binding studies will be necessary to determine the effect of other amino acid changes in this region.
The apparent Trp Ki values for feedback-sensitive AS from tobacco, R. graveolens, and Arabidopsis are similar, as one might expect considering the high degree of identity in the regions known to affect feedback inhibition of bacterial AS, which are also assumed to be important in plant AS (Caliguri and Bauerle, 1991; Bohlmann et al., 1995). The apparent Trp Ki values for the tobacco and R. graveolens feedback-insensitive AS enzymes are similar to each other and are much higher than the Ki value of the mutant Arabidopsis feedback-insensitive AS. This pattern of inhibition constants correlates with the sequence homologies observed and supports the hypothesis that the substitution of Arg-163 in R. graveolens (corresponding to Arg-108 of tobacco) and Phe-107-Arg-108 in tobacco AS are related to higher feedback insensitivity than that found in the Arabidopsis mutant AS when Asp-341 is changed to Asn.
That the apparent Km values for chorismate for tobacco AS are similar to the values obtained for Arabidopsis and R. graveolens AS is also not surprising considering the high degree of similarity, especially in the region known to be involved in catalysis for bacterial AS (Caliguri and Bauerle, 1991; Bohlmann et al., 1995). In all three species the apparent Km for chorismate of the feedback-insensitive AS is lower than the Km for feedback-sensitive AS. This suggests a similar mechanism of inhibition and feedback insensitivity. This would be expected for tobacco and R. graveolens, since a similar sequence difference appears to be involved, but the Arabidopsis mutant contains a different structural change that produces an enzyme that is still appreciably inhibited by Trp.
The results presented here also indicate that the tobacco ASA2 promoter regulates tissue-specific gene expression. This conclusion is supported by measurements of gene expression at the mRNA level (Fig. 4) and by the deletion-analysis studies of the ASA2 promoter region (Fig. 9). The high level of ASA2 gene expression was detected at the mRNA level only in 5MTr suspension-cultured cells, which is similar to the previous results found at the enzyme activity level (Brotherton et al., 1986). In addition, the transient GUS expression was higher in the 5MTr than in the 5MTs suspension-cultured cells and very low-level expression was found in leaves. This tissue-specific expression was controlled by the promoter region between −2252 and −607. These results suggest that there could be changes in the amount or type of regulatory protein(s) in the 5MTr suspension-cultured cells that up-regulate the ASA2 gene expression. Changes in a cis-acting element(s) in the promoter region can probably be excluded, since we have found only a few nucleotide differences in the ASA2 promoter region isolated from three wild-type tobacco cell lines in comparison with ASA2. We also found similar transient expression patterns with these promoters compared with that found with the ASA2 promoter from AB15–12-1 (data not shown).
In Arabidopsis and R. graveolens the ASA1 and ASα1 gene expression is inducible, whereas the ASA2 and ASα2 genes are expressed constitutively. Even though we have denoted our clone ASA2, since the highest amino acid identity was with R. graveolens ASα2 (72%) and since we have isolated a truncated ASA1 cDNA similar to the Arabidopsis ASA1 (data not shown), the tobacco ASA2 is also very similar to R. graveolens ASα1 (70% identity). So far, we have not been able to induce leaf ASA2 mRNA synthesis by treatment with auxin or salicylic acid (within 8 h) or by wounding (within 48 h). To help understand in vivo ASA2 gene expression in plants and the effect of environmental conditions on gene expression, we have recently produced tobacco plants transformed with the GUS reporter gene driven by the 2252-bp ASA2 promoter. The feedback-insensitive R. graveolens ASα1 appears to be involved in the pathway synthesizing anthranilate-derivative alkaloids (Bohlmann et al., 1996). However, we are not aware of any tobacco anthranilate or indole-derived phytoalexins or alkaloids that have been identified; therefore, the function of the ASA2 gene in tobacco has yet to be determined.
In this paper we have reported the cloning of a wildtype tobacco ASA2 gene encoding the α-subunit of a feedback-insensitive AS. Feedback insensitivity in the 5MTr suspension-cultured cells was correlated with the high level of the ASA2 gene expression and with the tissue-specific expression of this gene controlled by defined promoter regions. The role of the feedback-insensitive AS in tobacco is still unknown and further studies are needed to understand the regulation and function of this enzyme in plants.
ACKNOWLEDGMENTS
We thank Krishna Niyogi for cDNA clones, Mani Subramanian for the Arabidopsis ASA1-expressing E. coli line, Charles Yanofsky and the Escherichia coli Genetic Stock Center for the E. coli mutant strains, Rob Alleman for technical assistance with AS kinetic analysis, and Liugen Zhu for technical assistance with the MUG assays.
Abbreviations:
- AS
anthranilate synthase
- CaMV
cauliflower mosaic virus
- IPTG
isopropylthiogalactoside
- MS
Murashige-Skoog
- 5MT
5-methyl-Trp
- 5MTr
5-methyl-Trp-resistant
- 5MTs
5-methyl-Trp-sensitive
- MUG
4-methylumbelliferone glucuronide
- RACE
rapid amplification of cDNA ends
- UTR
untranslated region
Footnotes
This research was supported by funds from the Illinois Council on Food and Agricultural Research and the Illinois Agricultural Experiment Station.
LITERATURE CITED
- Altschul F, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology. Green and. New York: Wiley-Interscience; 1989. [Google Scholar]
- Bernasconi P, Walters EW, Woodworth AR, Siehl DL, Stone TE, Subramanian MV. Functional expression of Arabidopsis thaliana anthranilate synthase subunit I in Escherichia coli. Plant Physiol. 1994;106:353–358. doi: 10.1104/pp.106.1.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlmann J, De Luca V, Eilert U, Martin W. Purification and cDNA cloning of anthranilate synthase from Ruta graveolens: modes of expression and properties of native and recombinant enzymes. Plant J. 1995;7:491–501. doi: 10.1046/j.1365-313x.1995.7030491.x. [DOI] [PubMed] [Google Scholar]
- Bohlmann J, Lin T, Martin W, Eilert U. Anthranilate synthase from Ruta graveolens. Duplicated ASα genes encode tryptophan-sensitive and tryptophan-insensitive isoenzymes specific to amino acid and alkaloid biosynthesis. Plant Physiol. 1996;111:507–514. doi: 10.1104/pp.111.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brotherton JE, Hauptmann RM, Widholm JM. Anthranilate synthase forms in plants and cultured cells of Nicotiana tabacum L. Planta. 1986;168:214–221. doi: 10.1007/BF00402966. [DOI] [PubMed] [Google Scholar]
- Caliguri MG, Bauerle R. Identification of amino acid residues involved in feedback regulation of the anthranilate synthase complex from Salmonella typhimurium. J Biol Chem. 1991;266:8328–8335. [PubMed] [Google Scholar]
- Carlson JE, Widholm JM. Separation of two forms of anthranilate synthase from 5-methyltryptophan-susceptible and -resistant cultured Solanum tuberosum cells. Physiol Plant. 1978;44:251–255. [Google Scholar]
- Crawford IP. Evolution of a biosynthetic pathway: the tryptophan paradigm. Annu Rev Microbiol. 1989;43:567–600. doi: 10.1146/annurev.mi.43.100189.003031. [DOI] [PubMed] [Google Scholar]
- Dellaporta SL, Wood J, Hicks JB. A rapid DNA minipreparation. Version II. Plant Mol Biol Rep. 1983;2:21–42. [Google Scholar]
- Froissard D, Gough C, Czernic P, Schneider M, Toppan A, Roby D, Marco Y. Structural organization of str246C and str246N, plant defense-related genes from Nicotiana tabacum. Plant Mol Biol. 1994;26:515–521. doi: 10.1007/BF00039563. [DOI] [PubMed] [Google Scholar]
- Gavel Y, von Heijne G. A conserved cleavage-site motif in chloroplast transit peptides. FEBS Lett. 1990;261:455–458. doi: 10.1016/0014-5793(90)80614-o. [DOI] [PubMed] [Google Scholar]
- Gibson F. Preparation of chorismic acid. Methods Enzymol. 1970;17:362–364. [Google Scholar]
- Graf R, Mehmann B, Braus HG. Analysis of feedback-resistant anthranilate synthases from Saccharomyces cerevisiae. J Bacteriol. 1993;175:1061–1068. doi: 10.1128/jb.175.4.1061-1068.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslam E (1993) Shikimic Acid: Metabolism and Metabolites. John Wiley & Sons, Chichester, UK, pp 156–254
- Herrmann K. The shikimate pathway as an entry to aromatic secondary metabolism. Plant Physiol. 1995;107:7–12. doi: 10.1104/pp.107.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgin DG, Sharp PM. Fast and sensitive multiple sequence alignments on a microcomputer. Comput Appl Biosci. 1989;5:151–153. doi: 10.1093/bioinformatics/5.2.151. [DOI] [PubMed] [Google Scholar]
- Jefferson RA. Assaying chimeric genes in plants: the GUS gene fusion system. Plant Mol Biol Rep. 1987;5:387–405. [Google Scholar]
- Jefferson RA, Kavanagh TA, Bevan MW. GUS fusions: β- glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 1987;6:3901–3907. doi: 10.1002/j.1460-2075.1987.tb02730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreps JA, Ponappa T, Dong W, Town CD. Molecular basis of α-methyltryptophan resistant in amt-1, a mutant of Arabidopsis thaliana with altered tryptophan metabolism. Plant Physiol. 1996;110:1159–1165. doi: 10.1104/pp.110.4.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Last RJ. The Arabidopsis thaliana trp 5 mutant has a feedback-resistant anthranilate synthase and elevated soluble tryptophan. Plant Physiol. 1996;110:51–59. doi: 10.1104/pp.110.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui K, Miwa K, Sano K. Two single-base-pair substitutions causing desensitization to tryptophan feedback inhibition of anthranilate synthase and enhanced expression of tryptophan genes of Brevibacterium lactofermentum. J Bacteriol. 1987;169:5330–5332. doi: 10.1128/jb.169.11.5330-5332.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murashige T, Skoog F. A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant. 1962;15:473–497. [Google Scholar]
- Niyogi KK, Fink GR. Two anthranilate synthase genes in Arabidopsis: defense-related regulation of the tryptophan pathway. Plant Cell. 1992;4:721–733. doi: 10.1105/tpc.4.6.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niyogi KK, Last RL, Fink GR, Keith B. Suppressors of trp 1 fluorescence identify a new Arabidopsis gene, TRP 4, encoding the anthranilate synthase β subunit. Plant Cell. 1993;5:1011–1027. doi: 10.1105/tpc.5.9.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Ajioka JM, Garza D, Hartl L (1989) Inverse polymerase chain reaction. In HA Erlich, ed, PCR Technology. Stockton Press, New York, pp 105–111
- Poulsen C, Bongaerts RJM, Verpoorte R. Purification and characterization of anthranilate synthase from Catharanthus roseus. Eur J Biochem. 1993;212:431–440. doi: 10.1111/j.1432-1033.1993.tb17679.x. [DOI] [PubMed] [Google Scholar]
- Ranch JP, Rick S, Brotherton JE, Widholm JM. Expression of 5-methyltryptophan resistance in plants regenerated from resistant cell lines of Datura innoxia. Plant Physiol. 1983;71:136–140. doi: 10.1104/pp.71.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero RM, Roberts MF. Anthranilate synthase from Ailanthus altissima cell suspension cultures. Phytochemistry. 1996;41:395–402. [Google Scholar]
- Romero RM, Roberts MF, Phillipson JD. Anthranilate synthase in microorganisms and plants. Phytochemistry. 1995;39:263–276. doi: 10.1016/0031-9422(95)00010-5. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sato S, Nakada Y, Hon-nami K, Yasui K, Shiratsuchi A. Molecular cloning and nucleotide sequence of the Clostridium thermocellum trpE gene. J Biochem. 1989;105:362–366. doi: 10.1093/oxfordjournals.jbchem.a122669. [DOI] [PubMed] [Google Scholar]
- Singh M, Widholm JM. Measurement of the five enzymes which convert chorismate to tryptophan in wheat plants (Triticum aestivum L.) Physiol Plant. 1974;32:240–246. [Google Scholar]
- Vain P, Keen N, Murillo J, Rathus C, Nemes C, Finer JJ. Development of the particle inflow gun. Plant Cell Tissue Organ Cult. 1993;33:237–246. [Google Scholar]
- von Heijne G, Steppuhn J, Herrmann RG. Domain structure of mitochondrial and chloroplast targeting peptides. Eur J Biochem. 1989;180:535–545. doi: 10.1111/j.1432-1033.1989.tb14679.x. [DOI] [PubMed] [Google Scholar]
- Wang CS, Todd JJ, Vodkin LO. Chalcone synthase mRNA and activity are reduced in yellow soybean seed coats with dominant I alleles. Plant Physiol. 1994;105:739–748. doi: 10.1104/pp.105.2.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widholm JM. Control of tryptophan biosynthesis in plant tissue culture: lack of repression of anthranilate and tryptophan synthetases by tryptophan. Physiol Plant. 1971;5:75–79. [Google Scholar]
- Widholm JM. Anthranilate synthetase from 5methyltryptophan-susceptible and -resistant cultured Daucus carota cells. Biochim Biophys Acta. 1972a;279:48–57. doi: 10.1016/0304-4165(72)90240-1. [DOI] [PubMed] [Google Scholar]
- Widholm JM. Cultured Nicotiana tabacum cells with an altered anthranilate synthetase which is less sensitive to feedback inhibition. Biochim Biophys Acta. 1972b;261:52–58. doi: 10.1016/0304-4165(72)90312-1. [DOI] [PubMed] [Google Scholar]
- Widholm JM. Control of aromatic amino acid biosynthesis in cultured plant tissues: effect of intermediates and aromatic amino acids on free levels. Physiol Plant. 1974;30:13–18. [Google Scholar]
- Yanofsky C, Crawford IP (1987) The tryptophan operon. In FC Neidhardt, JL Ingraham, KB Low, B Magasanik, M Schaechter, HE Umbarger, eds, Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. American Society for Microbiology, Washington, DC, pp 1453–1472
- Zalkin H, Paluh JL, van Cleemput M, Moye WS, Yanofsky C. Nucleotide sequence of Saccharomyces cerevisiae genes TRP2 and TRP3 encoding bifunctional anthranilate synthase: indole-3-glycerol phosphate synthase. J Biol Chem. 1984;259:3985–3992. [PubMed] [Google Scholar]