

Abstract
Adipic acid is a high-value compound used primarily as a precursor for the synthesis of nylon, coatings, and plastics. Today it is produced mainly in chemical processes from petrochemicals like benzene. Because of the strong environmental impact of the production processes and the dependence on fossil resources, biotechnological production processes would provide an interesting alternative. Here we describe the first engineered Saccharomyces cerevisiae strain expressing a heterologous biosynthetic pathway converting the intermediate 3-dehydroshikimate of the aromatic amino acid biosynthesis pathway via protocatechuic acid and catechol into cis,cis-muconic acid, which can be chemically dehydrogenated to adipic acid. The pathway consists of three heterologous microbial enzymes, 3-dehydroshikimate dehydratase, protocatechuic acid decarboxylase composed of three different subunits, and catechol 1,2-dioxygenase. For each heterologous reaction step, we analyzed several potential candidates for their expression and activity in yeast to compose a functional cis,cis-muconic acid synthesis pathway. Carbon flow into the heterologous pathway was optimized by increasing the flux through selected steps of the common aromatic amino acid biosynthesis pathway and by blocking the conversion of 3-dehydroshikimate into shikimate. The recombinant yeast cells finally produced about 1.56 mg/liter cis,cis-muconic acid.
INTRODUCTION
Adipic acid is a chemical precursor used for the production of nylon, lubricants, coatings, plastics, and plasticizers. It belongs to the top 50 bulk chemicals, with an annual production of about 2 × 109 kg. Currently, it is produced mainly by a two-stage process which starts with the oxidation of benzene-derived cyclohexane resulting in a cyclohexanol-cyclohexanone mixture that is further oxidized by nitric acid (47). This chemical process requires a high energy input and further leads to the production of large amounts of the greenhouse gas N2O (46). Moreover, the chemical intermediates are toxic compounds and are partially linked to carcinogenesis (16). Thus, because of the heavy environmental impact and the dependence on finite fossil resources, there is high interest in alternative biotechnological production processes. Currently, no commercial biotechnological process exists. One possible biochemical route is via cis,cis-muconic acid (CCM), which can be hydrogenated to adipic acid (38). Several bacteria are known to convert aromatic compounds into CCM (37, 53). The first synthetic route using renewable resources like glucose has been established in Escherichia coli (10) (Fig. 1). This nonnatural route is based on the expression of three heterologous genes that encode a 3-dehydroshikimate (3-DHS) dehydratase (AroZ), a protocatechuic acid (PCA) decarboxylase (AroY) from Klebsiella pneumoniae, and a catechol 1,2-dioxygenase (CatA) from Acinetobacter calcoaceticus. These enzymes are found in various microorganisms normally enabling the use of aromatic compounds as carbon sources (10, 21). The enzymes convert 3-DHS, an intermediate of the common aromatic amino acid biosynthesis pathway, to CCM via PCA and catechol. This production pathway has been further optimized for improved carbon flow into the aromatic amino acid pathway by increasing the levels of the basal metabolic intermediates phosphoenolpyruvate (PEP) and erythrose-4-phosphate (E4P), which serve as the initial substrates for aromatic amino acid biosynthesis, and by blocking of aromatic amino acid synthesis at the level of 3-DHS by deleting the 3-DHS dehydrogenase gene aroE (11, 16, 49). Moreover, a feedback-resistant mutant form of 3-deoxy-d-arabino-heptulosonate-7-phosphate (DAHP) synthase was overexpressed. The final recombinant E. coli strain produced 36.8 g/liter of CCM, with a yield of 22% (mol/mol) within 48 h of culturing under fed-batch fermentor conditions (38).
Fig 1.

Schematic representation of the de novo CCM biosynthesis pathway. The central intermediates of the pathway shown are PEP (step 1), E4P (step 2), DAHP (step 3), 3-dehydroquinate (3-DHQ; step 4), 3-DHS (step 5), PCA (step 6), catechol (step 7), CCM (step 8), and adipic acid (step 9). Adipic acid can be produced by chemical hydrogenation (dashed arrow). Bold arrows indicate heterologous enzymatic reactions: 3-DHSD, 3-DHS dehydratase; PCA-DC, PCA decarboxylase; CDO, catechol 1,2-dioxygenase. Undesired yeast metabolic reactions are indicated by gray arrows.
Even though the optimized E. coli strain already produces considerable amounts of CCM, it does not allow a cost-competitive industrial production process. This is due to the intrinsic ability of E. coli to grow only under neutral-pH conditions. Purification of CCM from the fermentation broth in its undissociated form is done at low pH values (26). Thus, in the case of E. coli, substantial acidification of the broth after fermentation and a subsequent recycling step with a high salt load would be required. These steps are very cost-intensive and would make the downstream process uneconomical. In contrast, Saccharomyces cerevisiae fermentations can be performed at low pH values. Moreover, S. cerevisiae has further beneficial properties for industrial production processes like high robustness, high resistance to toxic inhibitors and fermentation products, resistance to microbial contamination, and a high level of public acceptance (32, 35, 50).
In this work, we established a heterologous CCM production pathway in S. cerevisiae by the expression of three enzymes. We further optimized this pathway, resulting in the production of 1.56 mg/liter CCM.
MATERIALS AND METHODS
Strains and media.
The yeast and bacterial strains used in this work are listed in Table 1. The bacterial strains were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany. S. cerevisiae strains were grown at 30°C with shaking at 180 rpm in synthetic complete medium (6.7 g/liter Difco yeast nitrogen base without amino acids) supplemented with amino acids as described previously (54) and with 20 g/liter d-glucose as a carbon source (SCD medium), adjusted to pH 6.3. For maintenance of plasmids, selective SCD medium lacked the auxotrophy markers and/or contained 200 mg/liter G418 or 200 mg/liter hygromycin, respectively.
Table 1.
Strains used in this study
| Species or strain | Genotype | Accession no. | Source |
|---|---|---|---|
| Burkholderia xenovorans | DSM-17367 | DSMZa | |
| Acinetobacter sp. | DSM-586 | DSMZ | |
| Bacillus thuringiensis | DSM-6074 | DSMZ | |
| S. cerevisiae strains | |||
| CEN.PK2-1C | MATa leu2-3,112 ura3-52 trp1-289 his3-Δ1 MAL2-8c SUC2 | EUROSCARF, Frankfurt, Germany | |
| CEN.PK2-1CΔaroE | MATa leu2-3,112 ura3-52 trp1-289 his3-Δ1 MAL2-8c SUC2 ARO1Δ1359-1588::loxP | This work |
DSMZ, Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH.
Metabolite analysis.
For metabolite analysis, the yeast cells were removed from samples by centrifugation. Proteins were precipitated by the addition of sulfosalicylic acid to a final concentration of 5% and metabolites were measured by high-performance liquid chromatography (HPLC). The metabolites were separated by HPLC (Dionex) using a Nucleogel Sugar 810 H exchange column (Macherey-Nagel GmbH & Co., Düren, Germany). The column was eluted with 5 mM H2SO4 as the mobile phase at a flow rate of 0.6 ml/min and a temperature of 65°C. The detection of glucose, glycerol, acetic acid, and ethanol was done by means of a Shodex RI-101 refractive-index detector. A UV detector was used for the detection of PCA (220 nm), catechol (220 nm), and CCM (250 nm). For data evaluation, the Chromeleon software (version 6.50) was used.
Plasmid and strain construction.
The plasmids and primers used in this study are listed in Tables 2 and 3. Molecular techniques were performed according to previously published procedures (51). Bacterial genomic DNA was prepared for PCR amplification as described in reference 2. Alternatively, PCRs were performed using broken cells as templates. Codon-optimized gene versions for increased protein expression in S. cerevisiae were obtained from DNA2.0, including the use of a DNA2.0 optimization algorithm. Yeast transformations and reisolation of plasmid DNA from yeast cells were carried out as described previously (1, 17). Genes were cloned by homologous recombination. The coding regions of the respective genes were amplified by PCR from genomic DNA or plasmids by using specific primer pairs with 5′ extensions overlapping vector sequences. PCR fragments were cotransformed into yeast cells together with a linearized vector. All vector-carried genes were under the control of strong promoters (Table 2). Plasmids were amplified in E. coli strain DH5α (Gibco BRL, Gaithersburg, MD). E. coli transformations were performed via electroporation according to the methods of Dower et al. (9). E. coli was grown on Luria-Bertani medium with 40 μg/ml ampicillin for plasmid selection. Strain CEN.PK2-1CΔaroE was constructed by using the cre-loxP-kanMX4-loxP system (19). The loxP-kanMX-loxP deletion cassette was PCR amplified from plasmid pUG6 by using primers with 5′ extensions homologous to the integration site, allowing homologous recombination. The deletion cassette was transformed into CEN.PK2-1C, which was selected for Geneticin (G418) resistance. Positive clones were transformed with plasmid pSH47, which encodes the Cre recombinase. Expression of the enzyme was induced by shifting the transformed cells to galactose-containing medium. The deletion of the aroE domain and the removal of the deletion cassette were verified by PCR analysis.
Table 2.
Plasmids used in this study
| Plasmid | Regulatory elements, markers, genes | GenBank accession no. | Reference |
|---|---|---|---|
| p423-HXT7 | HXT7 promoter, CYC1 terminator, HIS3 | 20 | |
| p425-HXT7 | HXT7 promoter, CYC1 terminator, LEU2 | 20 | |
| p426-HXT7 | HXT7 promoter, CYC1 terminator, URA3 | 20 | |
| pUG6 | loxP-kanMX4-loxP, URA3 | 19 | |
| pRS42K | TEF promoter, TEF terminator, kanMX | 45 | |
| pRS42H | 45 | ||
| TEF promoter, CYC1 terminator, Hygr | |||
| pRS42K-HXT7 | HXT7 promoter, CYC1 terminator, kanMX | This work | |
| pRS42H-HXT7 | HXT7 promoter, FBA1 terminator, Hygr | This work | |
| pRS42H-HXT7-FBA1 | HXT7 promoter, FBA1 terminator | This work | |
| pSH47 | GAL1 promoter, cre recombinase, CYC terminator, URA3 | 19 | |
| p426-HXT7-aroE | HXT7 promoter, aroE from E. coli, CYC1 terminator, URA3 | YP001732122 | This work |
| p425-HXT7-Bx-catA | HXT7 promoter, catA from B. xenovorans, CYC1 terminator, LEU2 | YP558911 | This work |
| p425-HXT7-Ar-catAopt | HXT7 promoter, catA from A. radioresistens codon optimized (DNA2.0), CYC1 terminator, LEU2 | AF380158 | This work |
| p423-HXT7-Bt-aroZ | HXT7 promoter, asbF from B. thuringiensis, CYC1 terminator, HIS3 | NC014171 | This work |
| p423-HXT7-Bt-aroZopt | HXT7 promoter, asbF from B. thuringiensis codon-optimized (DNA2.0), CYC1 terminator, HIS3 | NC014171 | This work |
| p423-HXT7-As-aroZ | HXT7 promoter, aroZ from Acinetobacter sp., CYC1 terminator, HIS3 | Q43922 | This work |
| p423-HXT7-Pa-aroZopt | HXT7 promoter, 3DSD from P. anserina codon-optimized (DNA2.0), CYC1 terminator, HIS3 | CAD60599 | This work |
| pRS42H-HXT7-ARO1ΔaroE | HXT7 promoter, ARO1 from S. cerevisiae lacking the aroE encoding domain (amino acids 1359-1588), FBA1 terminator, Hygr | YDR127W | This work |
| pRS42K-Kp-aroYopt | HXT7 promoter, aroY subunit B, CYC1 terminator; PGK1 promoter, aroY subunit C, PGK1 terminator; TPI1 promoter, aroY subunit D, TAL1 terminator, kanMX; all subunits from K. pneumoniae codon optimized (DNA2.0); the expression cassette can be integrated into the PYK2 locus | AAY57854, AAY57855, AAY57856 | This work |
| pRS42K-Kp-aroYiso.opt | HXT7 promoter, aroY subunit B, CYC1 terminator; PGK1 promoter, aroY subunit C isoform, PGK1 terminator; TPI1 promoter, aroY subunit D, TAL1 terminator, kanMX; all subunits from K. pneumoniae codon optimized (DNA2.0) | AAY57854, AB479384, AAY57856 | This work |
| pRS42K-Sh-aroY | HXT7 promoter, aroY subunit B, CYC1 terminator; PGK1 promoter, aroY subunit C, PGK1 terminator; TPI1 promoter, aroY subunit D, TAL1 terminator, kanMX; all subunits from S. hydroxybenzoicus | AAY67850 AAD50377 AAY67851 | This work |
Table 3.
Primers used in this study
| Primer | 5′–3′ sequence | Targeta |
|---|---|---|
| Pyk2_f | CGCACCTATATCTGCGTGTTG | Plasmid pRS42K-Sh-aroY |
| pHXT7_r | TTTTTGATTAAAATTAAAAAAACTTTTTGT | Plasmid pRS42K-Sh-aroY |
| DecBsh_f | AAACACAAAAACAAAAAGTTTTTTTAATTTTAATCAAAAAATGAGGCTAGTAATTGGAATTTC | Plasmid pRS42K-Sh-aroY |
| DecBsh_r | CGTGAATGTAAGCGTGACATAACTAATTACATGACTCGAGCTAATCATCTTTTGTCCCTTCCCAAGCC | Plasmid pRS42K-Sh-aroY |
| Tcyc_f | CTCGAGTCATGTAATTAGTTATG | Plasmid pRS42K-Sh-aroY |
| pPGK1_r | TGTTTTATATTTGTTGTAAAAAGTAG | Plasmid pRS42K-Sh-aroY |
| DecCsh_f | AAGGAAGTAATTATCTACTTTTTACAACAAATATAAAACAATGGCTAAAGTATACAAAGATTT | Plasmid pRS42K-Sh-aroY |
| DecCsh_r | AAAGAAAAAAATTGATCTATCGATTTCAATTCAATTCAATTTATCTGTTTTGATTTTTTAATAATTC | Plasmid pRS42K-Sh-aroY |
| tPGK_f | ATTGAATTGAATTGAAATCGATAGAT | Plasmid pRS42K-Sh-aroY |
| pTpI1_r | TTTTAGTTTATGTATGTGTTTTTTG | Plasmid pRS42K-Sh-aroY |
| DecDsh_f | TTAAATCTATAACTACAAAAAACACATACATAAACTAAAAATGAAATGTCATAGATGTGGCTC | Plasmid pRS42K-Sh-aroY |
| DecDsh_r | ATAAGGACATGGCCTAAATTAATATTTCCGAGATACTTCCCTATTTTTTCAAGGGTGGTATAGG | Plasmid pRS42K-Sh-aroY |
| tTal_f | GGAAGTATCTCGGAAATATTAAT | Plasmid pRS42K-Sh-aroY |
| tTal_r | ATATTAAGGGTTGTCGACCTGCAGCGTACGAAGTTTAAACGCTAGCCCTAGGGGCGCGCCGACGTTGATTTAAGGTGGTTCC | Plasmid pRS42K-Sh-aroY |
| DecCklp_f | AAGGAAGTAATTATCTACTTTTTACAACAAATATAAAACAATGACCGCTCCAATTCAAGAC | Plasmid pRS42K-Kp-aroYopt |
| DecCklp_r | AAAGAAAAAAATTGATCTATCGATTTCAATTCAATTCAATTACTTAGCAGAACCTTGGTTC | Plasmid pRS42K-Kp-aroYopt |
| DecBklpopt_f | AAACACAAAAACAAAAAGTTTTTTTAATTTTAATCAAAAAATGAAACTGATAATCGGGATGAC | Plasmid pRS42K-Kp-aroYiso.opt |
| DecBklpopt_r | CGTGAATGTAAGCGTGACATAACTAATTACATGACTCGAGTTATTCGATTTCCTGAGCGAATTGTTCAG | Plasmid pRS42K-Kp-aroYiso.opt |
| DecCklpopt_f | AAGGAAGTAATTATCTACTTTTTACAACAAATATAAAACAATGGCCTTTGACGACCTTAGA | Plasmid pRS42K-Kp-aroYiso.opt |
| DecCklpopt_r | AAAGAAAAAAATTGATCTATCGATTTCAATTCAATTCAATTATTTACGATTGGCTAACATAGC | Plasmid pRS42K-Kp-aroYiso.opt |
| DecDklpopt_f | TTAAATCTATAACTACAAAAAACACATACATAAACTAAAAATGATATGCCCAAGATGTGCC | Plasmid pRS42K-Kp-aroYiso.opt |
| DecDklpopt_r | ATAAGGACATGGCCTAAATTAATATTTCCGAGATACTTCCTTATCTCTTATCTTCAGGTAAAAG | Plasmid pRS42K-Kp-aroYiso.opt |
| catA_A.rad.opt_f | AAAAAGTTTTTTTAATTTTAATCAAAAAGTTAACATGCATATGACTGCAGCAAATGTTAAGA | Plasmid p425-HXT7-Ar-catAopt |
| catA_A.rad.opt_r | GAGGGCGTGAATGTAAGCGTGACATAACTAATTACATGACTTAGGCTTGCAATCTTGGTCTATC | Plasmid p425-HXT7-Ar-catAopt |
| AroZ_B.th._opt_f | AAAAACAAAAAGTTTTTTTAATTTTAATCAAAAAGTTAACATGAAGTACTCATTATGCACTATC | Plasmid p423-HXT7-Bt-aroZopt |
| AroZ_B.th._opt_r | TATTAGTCAGTACTTTCATTTCAGCCTTCAAGATATCCTTTTAAGATGTGACAACTTCAAGATTTC | Plasmid p423-HXT7-Bt-aroZopt |
| AroZ_P.ans._opt_f | AAACACAAAAACAAAAAGTTTTTTTAATTTTAATCAAAAAATGCCAAGTAAACTGGCTATTAC | Plasmid p423-HXT7-Pa-aroZopt |
| AroZ_P.ans._opt_r | ATTCTAATAGGTTGTTGAGTTGCCTGTTGTTGGGTAGGAGTTAAAGTGCGGCAGATAGAGATAAC | Plasmid p423-HXT7-Pa-aroZopt |
| S1_ARO11359-1588_F | GCACAATTGGTGAAAGAAAAACTTTTGGACGGAAACAAGTGATTCGTACGCTGCAGGTCGAC | Strain CEN.PK2-1CΔaroE |
| S2_ARO11359-1588_R | AAGCATTGTAAAATATAAAAAAGGATAGATATATTATTGTGCATAGGCCACTAGTGGATCTG | Strain CEN.PK2-1CΔaroE |
| A1_ARO11359-1588_F | CTGTTTGTTGTTGGAAAGCCA | Strain CEN.PK2-1CΔaroE |
| A4_ARO11359-1588_R | AACTTGTAATTATCTAACTGTTGC | Strain CEN.PK2-1CΔaroE |
| CATA_F | AACACAAAAACAAAAAGTTTTTTTAATTTTAATCAAAAAATGAACAGGCAAGCTATCGAC | Plasmid p425-HXT7-Bx-catA |
| CATA_R | GAATGTAAGCGTGACATAACTAATTACATGACTCGAGTCAGGCTTCAGCGCGCAAA | Plasmid p425-HXT7-Bx-catA |
| aro1ΔE_ÜbrexS1 | AACACAAAAACAAAAAGTTTTTTTAATTTTAATCAAAAAATGGTGCAGTTAGCCAAAGTC | Plasmid pRS42H-HXT7-ARO1ΔaroE |
| aro1ΔE_ÜbrexS2 | GAATGTAAGCGTGACATAACTAATTACATGACTCGAGCTACTTGTTTCCGTCCAAAAGTTTT | Plasmid pRS42H-HXT7-ARO1ΔaroE |
| DHSase B.t_S1 | AACACAAAAACAAAAAGTTTTTTTAATTTTAATCAAAAAATGAAATATTCACTATGTACCATT | Plasmid p423-HXT7-Bt-aroZ |
| DHSase B.t._S2 | GAATGTAAGCGTGACATAACTAATTACATGACTCGAGTTAAGAAGTTACTACTTCTAAATTTC | Plasmid p423-HXT7-Bt-aroZ |
Plasmid or strain for the construction of which the primer was used.
Fermentations and feeding experiments.
Cultures of yeast strains (50 ml) were grown in 300-ml shake flasks (Erlenmeyer flasks) at 30°C in a rotary shaker (180 rpm). Precultures were grown into the exponential phase in selective SCD medium. Cells were washed with sterile water and inoculated to an optical density at 600 nm (OD600) of 1.0 in the same medium. Growth and fermentation experiments were performed up to three times by using the same precultures, with the given standard deviations. Samples for OD600 measurement and metabolite analyses were taken at different time points. Anaerobic/oxygen-limited fermentations (100 ml) were done using 100-ml shake flasks (Erlenmeyer flasks) sealed with an airlock and incubation at 30°C. The cell suspension was mixed by using a magnetic stirrer. The oxygen was initially removed by sparging with nitrogen gas. Samples were taken with a syringe using a separate output connection. Since the oxygen level could not be controlled completely, these fermentations are seen as semianaerobic, oxygen-limited conditions. To analyze the rate of turnover of catechol or PCA in yeast cultures, each compound was supplied at a final concentration of 5 or 3.4 mM, respectively.
RESULTS
A heterologous biosynthetic route to CCM in yeast.
In S. cerevisiae, no natural biosynthetic route to CCM exists. Therefore, we aimed to establish a pathway similar to that previously described for E. coli by expression of the heterologous enzymes 3-DHS dehydratase (AroZ), PCA decarboxylase (AroY), and catechol-1,2-dioxygenase (CatA). These enzymes should convert the intermediate 3-DHS of the common aromatic amino acid biosynthesis pathway (4) via PCA and catechol to CCM (Fig. 1). We first set out to identify suitable enzymes for functional expression in yeast. Therefore, for each enzyme activity, we expressed various potential candidate genes in S. cerevisiae and analyzed their activities individually in bioconversion and biotransformation experiments. Finally, we combined the best-performing enzymes into an optimized biosynthetic pathway.
Identification of suitable 3-DHS dehydratases and optimization of the biosynthetic route to 3-DHS.
In the first heterologous reaction, 3-DHS is dehydrated to PCA. Several 3-DHS dehydratases (AroZ) acting in various cellular pathways have been identified in different microbial systems (14, 15, 18, 22, 28, 30, 38, 39, 40). To identify a suitable candidate for the new biosynthetic route in yeast, we chose three different genes, aroZ of the aromatic degradation pathway from Acinetobacter sp. (As-aroZ), aroZ of Podospora anserina (Pa-aroZ) (40), and asbF of the petrobactin biosynthesis pathway from Bacillus thuringiensis (Bt-aroZ) (15, 39). The corresponding protein sequences exhibit a very low overall homology of about 17% (data not shown). The coding sequences of As-aroZ and Bt-aroZ were amplified by PCR from genomic microbial DNA and cloned into high-copy-number yeast vector p423-HXT7. To further optimize gene expression, we additionally cloned a codon-optimized variant of the gene from B. thuringiensis (Bt-aroZopt [opt, optimized]). The gene from P. anserina (Pa-aroZopt) was expressed only in a codon-optimized version. To examine the performance of the different 3-DHS dehydratases, the respective plasmids were transformed into the CEN.PK2-1C yeast strain, the transformants were grown in selective SCD medium, and the production of PCA was assessed by HPLC analysis of the supernatants of batch cultures at different time points. However, only very low PCA levels (<7 mg/liter) could be detected even after cultivation for up to 120 h (Fig. 2 and data not shown). We could not detect higher PCA concentrations when the yeast cells were included in the PCA extraction process, indicating that PCA did not accumulate within the yeast cells.
Fig 2.

Analysis of PCA production with different 3-DHS dehydratases. To analyze the activities of the different 3-DHS dehydratases, the respective expression vectors and the empty control vector (p423-HXT7) were cotransformed with pRS42H-HXT7-ARO1ΔaroE or the empty control vector into yeast wild-type strain CEN.PK2-1C or strain CEN.PK2-1CΔaroE. The cells were grown in selective SCD medium for 120 h, and PCA was measured by HPLC analysis of the supernatants of the cultures. As the levels of PCA varied in different experiments, the results of only a representative experiment are shown.
As low production of PCA might be explained by the competition of the 3-DHS dehydratases with the 3-DHS dehydrogenase activity of the common aromatic amino acid biosynthesis pathway, we blocked the production of phenylalanine, tryptophan, and tyrosine at the level of 3-DHS by the elimination of 3-DHS dehydrogenase activity. In yeast, this reaction is mediated by the pentafunctional enzyme Arom (encoded by ARO1) (12, 13), which mediates the stepwise conversion of 3-DAHP to 5-enolpyruvyl-shikimate-3-phosphate via 3-DHS. Therefore, it was necessary to block only the 3-DHS dehydrogenase activity without affecting the dehydroquinate synthase and dehydroquinate dehydratase activities, which convert 3-DAHP to 3-DHS. The dehydrogenase domain of Arom, which mediates the conversion of 3-DHS to shikimate, has already been identified in a previous work by sequence alignment with the respective aroE-encoded enzyme from E. coli and is the last domain of yeast Arom (amino acids 1359 to 1588) (Fig. 3A) (12). The coding region of the whole dehydrogenase domain was replaced with a stop codon in wild-type strain CEN.PK2-1C by homologous recombination using the cre-loxP-kanMX system (Fig. 3A). After removal of the kanMX cassette, a single loxP site was left behind the truncated gene before the terminator region, resulting in strain CEN.PK2-1CΔaroE. As expected, the strain turned out to be auxotrophic for aromatic amino acids (Fig. 3B).
Fig 3.

Deletion of the 3-DHS dehydratase domain of the Arom enzyme for optimized carbon flow into the CCM pathway. (A) Schematic representation of the ARO1 gene, which encodes the pentafunctional enzyme Arom. The E. coli genes that encode the corresponding enzyme activities are indicated at the bottom. The AroE-encoding region was deleted with a loxP-kanMX-loxP cassette, introducing a stop codon after the AroD-encoding region. A residual loxP site remains in the terminator region after Cre-mediated recombination. 3-DHQ, 3-dehydroquinate; EPSP, 5-enolpyruvyl-shikimate-3-phosphate. (B) Complementation assay to verify that deletion of the AroE domain of Arom did not affect the enzymatic activities of the residual domains. Yeast cells transformed with either p426-HXT7-aroE or the respective empty vector were grown on SCD−Ura medium to an OD600 of 1. Five-microliter volumes of diluted (1:1, 1:100, or 1:1,000) yeast cell suspensions were dropped onto the indicated agar plates on medium with or without tyrosine and phenylalanine. The plates were incubated at 30°C for 3 days.
To confirm that the other enzymatic activities of the truncated Arom enzyme, especially the dehydroquinate synthase and dehydroquinate dehydratase activities, were not affected, the aromatic amino acid auxotrophy of the mutant strain should be complemented by overexpression of the aroE gene from E. coli (Fig. 3B). In E. coli, the individual enzyme activities of Arom are encoded by separate coding regions. After transformation with the overexpression plasmid p426-HXT7-aroE or the respective empty control vector (p426-HXT7), the transformants were tested for the ability to grow on medium lacking tyrosine and phenylalanine. While the mutant transformed with the empty plasmid could not grow, the growth defect could be complemented by aroE from E. coli, indicating that the other domains of AromΔaroE had not lost their activities (Fig. 3B).
To further increase the production of 3-DHS, the truncated ARO1ΔaroE allele was cloned into an overexpression plasmid under the control of the strong HXT7 promoter fragment, resulting in plasmid pRS42H-HXT7-ARO1ΔaroE. The various 3-DHS dehydratase overexpression plasmids were cotransformed with plasmid pRS42H-HXT7-ARO1ΔaroE or the corresponding empty control vector pRS42H-HXT7 into strain CEN.PK2-1CΔaroE. PCA production was measured by HPLC analysis of the supernatants of batch cultures grown in selective SCD medium (Fig. 2). The results show that the block of the aromatic amino acid biosynthesis pathway, together with overexpression of AromΔaroE, had a pronounced effect on PCA production levels. As in E. coli, the five enzymatic reactions of Arom are encoded on separate open reading frames. Alternatively, we also tested the overexpression of aroB (which encodes dehydroquinate synthase) and aroD (which encodes dehydroquinate dehydratase) from E. coli instead of ARO1ΔaroE and found comparable PCA production levels (data not shown). The highest production rates were obtained with the 3-DHS dehydratases genes Bt-aroZopt and Pa-aroZopt. Interestingly, expression of Bt-aroZopt resulted in PCA levels only slightly higher than those obtained with the nonoptimized variant. In contrast, expression of As-aroZ resulted in the lowest PCA levels. On the basis of these results, Bt-aroZopt and Pa-aroZopt represent the most promising candidate genes for the heterologous CCM production pathway.
PCA decarboxylases.
The conversion of PCA to catechol is catalyzed by nonoxidative 3,4-dihydroxybenzoate decarboxylases. These enzymes belong to the family of hydroxyarylic acid decarboxylases/phenol carboxylases and are composed of three different subunits encoded by the B, C, and D genes, which are usually organized in a cluster (33). Hydroxybenzoate-decarboxylating activities have been observed in various microorganisms. These enzymes differ greatly in substrate specificity, and just a few enzymes have been shown to specifically decarboxylate PCA (23, 24, 27, 31, 36, 38, 52). On the basis of the data in the literature, the enzymes from Sedimentibacter hydroxybenzoicus, Enterobacter cloacae, and K. pneumoniae seemed to be the most promising candidates for the CCM pathway, especially with respect to their substrate specificity. For our pathway, we chose the enzymes from S. hydroxybenzoicus and K. pneumoniae. The respective B, C, and D genes were cloned together into the high-copy-number yeast vector pRS42K-HXT7. Each gene was controlled by its own promoter. The promoters were chosen from highly expressed yeast glycolytic genes (Table 2). For S. hydroxybenzoicus (Sh-aroY), we PCR amplified the B, C, and D genes directly from genomic DNA. In contrast, the K. pneumoniae (Kp-aroY) B, C, and D genes were cloned in synthetic, codon-optimized versions. We additionally performed detailed database searches and found another homolog for the C subunit encoded in the genome of K. pneumoniae (AB479384.1). This gene has been previously annotated by H. Ishioka and T. Sonoki in 2009, but experimental data have not been published. Our further analysis revealed that this gene is not organized in a BCD cluster. Sequence alignments showed that the protein sequence shares only up to 27% identity with the previously characterized C subunits of the BCD clusters of various organisms. However, typical core amino acids of the aroY C subunit are conserved (data not shown). Since it might represent a variant leading to an isomeric form of aroY with interesting properties, we included this subunit in our studies. We cloned this gene as a codon-optimized version together with the B and D subunit coding regions of the K. pneumoniae BCD cluster, resulting in the isomeric version named Kp-aroYIso.opt (Iso, isoform).
To analyze the in vivo activities of the different enzymes, we performed fermentations in selective SCD medium with externally added PCA (5 mM) and growing cultures of CEN.PK2-1C transformed with the respective expression plasmids. Previous analysis indicated that several decarboxylases are oxygen sensitive (23, 24, 33, 48). To also test this possibility, we performed the experiments under aerobic and anaerobic conditions. The conversion of PCA to catechol was quantitatively monitored by HPLC analysis of the supernatants of the cultures (Fig. 4). Under anaerobic and aerobic conditions, yeast cells expressing Kp-aroYIso.opt showed the highest catechol production rates and those expressing Sh-aroY showed the lowest ones. Under aerobic conditions, the respective overall activity of Sh-AroY was reduced almost completely whereas the activity of Kp-aroY was reduced 4.5-fold. These observations indicate that these proteins are oxygen sensitive. In contrast, Kp-aroYIso.opt revealed no differences between the activities under the two conditions. Since the final reaction step in the CCM pathway from catechol to CCM requires molecular oxygen (see below) and because it had the highest conversion activity, the Kp-AroYIso.opt variant represented the most promising candidate for the CCM pathway.
Fig 4.

Analysis of PCA decarboxylase activities under anaerobic and aerobic conditions with externally added PCA. To analyze the activities of the different protocatechuate decarboxylases, the respective expression vectors and the empty control vector were transformed into wild-type strain CEN.PK2-1C. Yeast cells were grown in selective SCD medium supplemented with 5 mM PCA under aerobic and anaerobic conditions. Production of catechol was measured by HPLC analysis of the supernatants of the cultures after 120 h. As the levels of catechol varied in different experiments, the results of only a representative experiment are shown.
Catechol 1,2-dioxygenases.
The conversion of catechol to CCM can be mediated by catechol 1,2-dioxygenases. These enzymes catalyze the intradiol cleavage of catechol by incorporation of molecular oxygen. By using sequence database analysis, we identified a catA gene in Burkholderia xenovorans (Bx-catA) (38). The gene was cloned by PCR amplification from genomic DNA of B. xenovorans and subsequent homologous recombination into the vector p425-HXT7. However, numerous 1,2-dioxygenases have been purified and characterized and it was shown that most of these enzymes have a broad substrate specificity, including activity on PCA (6, 44). This side activity would be undesirable for our intended CCM production pathway. Thus, we additionally cloned a codon-optimized version of catA from Acinetobacter radioresistens (Ar-catAopt). The corresponding enzyme has been shown to have no unspecific activity on PCA (5, 7, 8). To verify its expression, as well as its activity with catechol, we performed fermentations with transformed CEN.PK2-1C cells in selective SCD medium with externally added catechol (about 3.4 mM) and measured the levels of catechol and CCM in the supernatants of the cultures. The HPLC analyses showed that both enzymes were actively expressed and able to convert catechol to CCM (Fig. 5). In contrast, no CCM was produced by cells with the empty control vector. As expected, the codon-optimized gene from A. radioresistens encoded the highest CCM production activity. Catechol was not quantitatively converted into CCM, which might indicate the formation of side products. To exclude the possibility that the enzymes have activity on PCA, we performed control experiments using externally added PCA (about 5 mM). For both enzymes, we did not observe any consumption of PCA (data not shown). On the basis of these results, both enzymes should be applicable for use in the CCM production pathway.
Fig 5.
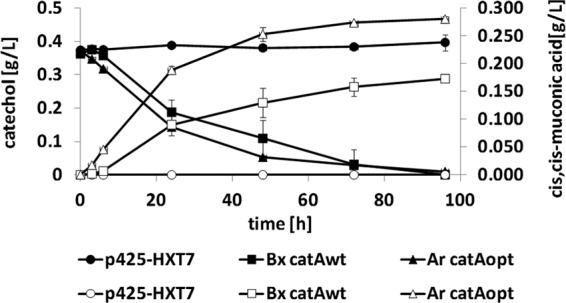
Analysis of catechol 1,2-dioxygenases with externally added catechol. To analyze the activities of the different catechol 1,2-dioxygenases, the respective expression vectors and the empty control vector were transformed into wild-type strain CEN.PK2-1C. The transformants were grown in selective SCD medium supplemented with 3.4 mM catechol. Conversion of catechol into CCM was measured by HPLC analysis of the supernatants of the cultures at up to 96 h. Fermentation experiments were performed three times by using the same precultures, and standard deviations are shown. wt, wild type.
De novo production of CCM from glucose.
Our analysis identified promising enzymes that could be potentially combined in a CCM production pathway. To test this possibility, we transformed the expression vectors for the respective enzymes or their corresponding empty control vectors in different combinations into strain CEN.PK2-1CΔaroE. We further cotransformed the vector pRS42H-HXT7-ARO1ΔaroE to ensure high-level synthesis of the precursor 3-DHS. The different transformants were grown in batch cultures with selective SCD medium under aerobic conditions, and the levels of CCM in the supernatants of the cultures were measured by HPLC analysis (Fig. 6). Our analysis revealed that the highest production rates were obtained by the coexpression of Pa-AroZopt, Kp-AroYIso.opt, and Ar-CatA, which resulted in a final concentration of 1.56 mg/liter CCM after 170 h. In contrast, the combinations that included Sh-AroY revealed no detectable CCM production (data not shown) whereas the combinations with Kp-AroYopt revealed very low CCM levels (up to 0.1 mg/liter). This observation can be explained by the presence of oxygen during the fermentations and is consistent with our analysis of the individual enzymes, which revealed the oxygen sensitivity of both enzymes (Fig. 4). Variation of the 3-DHS dehydratase isoforms had only slight effects on the CCM levels. This is in line with our observations above, which indicated only minor differences between the different enzymes (Fig. 2). In contrast, variation of the catechol 1,2-dioxygenase isoforms led to reduced CCM levels when Bx-CatAwt was used. This observation was expected, as our previous analysis revealed higher activity of the optimized enzyme from A. radioresistens.
Fig 6.

Analysis of de novo synthesis of CCM from glucose. For the expression of different enzyme combinations, the expression plasmids and respective empty control vectors were transformed into the CEN.PK2-1CΔaroE strain. Transformants were grown in selective SCD medium for up to 170 h. (A) The concentrations of CCM in the supernatants of the cultures were determined at different time points by HPLC analysis. (B) PCA and CCM levels after 170 h. As the levels of CCM varied in different experiments, the results of only a representative experiment are shown. wt, wild type.
We further analyzed whether precursors of CCM accumulated in the broth (Fig. 6B). We detected high levels of the precursor PCA in nearly all fermentations with cells expressing 3-DHS dehydratase (AroZ) activity, indicating a bottleneck in AroY or CatA activity. As expected, the highest PCA level was observed in strains expressing only Bt-AroZopt, while additional expression of Kp-AroYIso.opt slightly reduced PCA levels. Although this might indicate further conversion of PCA into catechol, catechol was not detectable. Catechol was not detectable in any of the fermentations, irrespective of the different enzyme combinations. Also surprisingly, expression of Bx-CatAwt together with the other enzymes resulted in not only lower levels of CCM but also lower levels of PCA. These results might be interpreted as meaning that either the different enzymes influence each other when expressed simultaneously or that side products which could not be measured with our HPLC analysis are synthesized. Nevertheless, our results show that it is possible to establish a CCM pathway in S. cerevisiae.
DISCUSSION
In this study, we demonstrate the first de novo production of CCM from glucose in S. cerevisiae. By coexpression of three bacterial enzymes, we assembled a heterologous production pathway that is similar to the previously established pathway in E. coli (38). Our analysis revealed that one key step in the successful engineering of CCM production in S. cerevisiae was the block of the aromatic amino acid synthesis pathway on the level of 3-DHS by deletion of the AroE domain of the pentafunctional Arom enzyme. This allows the conclusion that the heterologous 3-DHS dehydratases are in competition with the 3-DHS dehydrogenase reaction of the aromatic amino acid pathway or that increased levels of 3-DHS favor its conversion into PCA via 3-DHS dehydratases. However, in order to significantly increase the production of PCA, in addition to the deletion of the AroE domain of ARO1 in the genome, the gene for the truncated enzyme with the residual enzyme activities (AromΔaroE) (Fig. 2) or the E. coli aroB and aroD genes had to be overexpressed. These results suggest either that the conversion of 3-DAHP to 3-DHS represents a limiting step in the aromatic amino acid synthesis pathway or that the deletion of the AroE domain at least partially affected the activity of the residual domains.
The different oxygen requirements of AroY and CatA represent a very critical step in the CCM pathway. Whereas molecular oxygen is essential for the final reaction step mediated by CatA, oxygen sensitivity has been reported for several AroY enzymes (23, 24, 33, 48). In agreement with these previous reports, we also observed oxygen sensitivity of Sh-AroY and Kp-AroY (Fig. 4). Whereas Sh-AroY completely lost its activity in the presence of oxygen, Kp-AroY lost at least 80%. Moreover, as we used only oxygen-limited conditions, the activities of both enzymes under strictly anaerobic conditions are probably even higher. By the way, it should be noted that it is not possible to directly compare the activities of Sh-AroY and Kp-AroY, as only in the latter case did we use a codon-optimized gene. AroY is composed of three different subunits which normally are encoded by three open reading frames organized into respective BCD clusters. Interestingly, we found another putative C subunit in the genome of K. pneumoniae which is not part of a BCD cluster. By using this alternative C subunit, we could establish a new enzyme complex (Kp-AroYIso.opt) which did not show oxygen sensitivity. Our results indicate that this alternative C subunit can replace the original C subunit in the aroY BCD cluster of K. pneumoniae. Thus, it is most likely that it is the C subunit which confers oxygen sensitivity on the AroY complex. Moreover, even under oxygen-limited conditions, Kp-AroYIso.opt with the alternative C subunit performed slightly better than Kp-AroY with the original C subunit (Fig. 4). Therefore, in an industrial production process, this new isoform would be highly advantageous because the oxygen level would not need to be controlled so strictly. Interestingly, Niu et al. (38) also used an AroY enzyme from K. pneumoniae for the construction of a CCM pathway in E. coli. In their fermentation experiments, the oxygen level was kept at 10% dissolved oxygen air saturation, which was sufficient for the catechol 1,2-dioxygenase reaction and obviously had no deleterious effects on AroY of K. pneumoniae. Unfortunately, it is not clear which C subunit was used in their study.
Two different isoforms of CatA were tested in our study, one from B. xenovorans and the other a codon-optimized version from A. radioresistens. Whereas the yeast strain expressing no CatA activity did not consume any catechol and did not produce CCM, yeast cells containing either of the two heterologous isoenzymes consumed catechol at an initial concentration of 0.37 g/liter (3.4 mM) completely in about 100 h and in the case of Ar-CatA produced up to 0.28 g/liter (2.0 mM) CCM. Surprisingly, although all of the catechol was also consumed in the case of Bx-CatA, only 0.19 g/liter (1.3 mM) CCM was produced. The only explanation for the incomplete conversion of catechol into CCM would be either that the enzymes can also convert catechol into another product which was not detectable in our HPLC analyses or that their expression induces an activity in yeast that turns catechol into another product. This could also explain the observation that we could never detect catechol in the CCM fermentations, even when the conversion of PCA into catechol should be increased. Even more surprisingly, expression of Bx-CatA, compared to Ar-CatA, together with AroZ and AroY in the complete CCM pathway resulted not only in strongly reduced CCM production levels but also in very low PCA levels (Fig. 6).
When we combined the most promising enzymes into a complete CCM pathway in yeast, the yeast cells could indeed produce CCM. However, it is obvious that there are several bottlenecks in the pathway and that the capacity of carbon flow into and within the pathway must still be optimized substantially. Moreover, as we compared codon-optimized with nonoptimized genes, the choice of the individual enzymes need not necessarily reflect the best combination. The first revealing observation is the accumulation of PCA in the supernatant (Fig. 6) whereas no catechol could be detected. This suggests that either the conversion of PCA to catechol by AroY represents a rate-limiting step or that PCA is secreted out of the cells before it can be converted by AroY. Indeed, throughout our analyses, we observed that all of the intermediates and products of the CCM pathway could be detected in the supernatants of the cultures, and we could not find any indication that they accumulated in the cells. As mere diffusion of the compounds across the plasma membrane is unlikely, this suggests that they can be transported by yeast plasma membrane transporters. The draining of intermediates out of the cells clearly reduces the final CCM production levels. To circumvent this problem, either the transporters responsible must be identified and the genes that encode them deleted or enzymes with higher activities and higher affinities must be found.
Another problem, obviously, is end product repression and feedback inhibition of the committing step in the aromatic amino acid biosynthesis pathway, catalyzed by DAHP synthase (encoded by ARO3 and ARO4) (25, 41). As the AroE domain of the Arom enzyme was deleted from our yeast strain, the resulting auxotrophy had to be complemented by the addition of aromatic amino acids. These probably strongly reduced the expression and activity of DAHP synthase. Interestingly, it was shown previously (34) that genetic modifications leading to feedback-resistant mutant forms of DAHP synthase revealed a >4-fold increase in flux through the aromatic pathway with a >100-fold increase in the extracellular levels of aromatic compounds. Therefore, the overexpression of such mutant forms of DAHP synthase is a very promising strategy by which to further increase CCM yields. In addition, as was also shown in E. coli (10), increasing the supply of the substrates of the aromatic pathway, PEP and E4P, might further increase CCM production.
Nevertheless, compared to the CCM production titers in E. coli of up to 36.8 g/liter (38), the production titers obtained here with S. cerevisiae are much lower. In general, metabolic engineering of S. cerevisiae seems to be much more difficult than that of E. coli (29), as the metabolic network of S. cerevisiae is more rigidly resistant to modifications. This has also been observed, e.g., for the production of isobutanol (3), 1-butanol (43), and biodiesel (42). Much more effort is needed to finally achieve industrially relevant productivity (50). On the other hand, S. cerevisiae has several advantages in industrial production processes, especially in the production of organic acids at low pH values (26, 32, 50). Accordingly, we did not observe any toxic effects of CCM on S. cerevisiae cells up to its solubility level of 200 mg/liter at pH 3 to 6 (data not shown). Altogether, our results might pave the way for the engineering of S. cerevisiae for the industrial production of CCM and its further chemical hydrogenation into adipic acid.
ACKNOWLEDGMENTS
This work was financially supported by Butalco GmbH, Fuerigen, Switzerland. E.B. is a Butalco cofounder and shareholder.
Footnotes
Published ahead of print 21 September 2012
REFERENCES
- 1. Boles E, Zimmermann FK. 1993. Saccharomyces cerevisiae phosphoglucose isomerase and fructose bisphosphate aldolase can be replaced functionally by the corresponding enzymes of Escherichia coli and Drosophila melanogaster. Curr. Genet. 23:187–191 [DOI] [PubMed] [Google Scholar]
- 2. Brat D, Boles E, Wiedemann B. 2009. Functional expression of a bacterial xylose isomerase in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 75:2304–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brat D, Weber C, Lorenzen W, Bode H, Boles E. 2012. Cytosolic re-localization and optimization of valine synthesis and catabolism enables increased isobutanol production with the yeast Saccharomyces cerevisiae. Biotechnol. Biofuels 5:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Braus GH. 1991. Aromatic amino acid biosynthesis in the yeast Saccharomyces cerevisiae: a model system for the regulation of a eukaryotic biosynthetic pathway. Microbiol. Rev. 55:349–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Briganti F, Pessione E, Giunta C, Scozzafava A. 1997. Purification, biochemical properties and substrate specificity of a catechol 1,2-dioxygenase from a phenol degrading Acinetobacter radioresistens. FEBS Lett. 416:61–64 [DOI] [PubMed] [Google Scholar]
- 6. Caglio R, et al. 2009. Fine-tuning of catalytic properties of catechol 1,2-dioxygenase by active site tailoring. Chembiochem 10:1015–1024 [DOI] [PubMed] [Google Scholar]
- 7. Caposio P, et al. 2002. Cloning and characterization of two catechol 1,2-dioxygenase genes from Acinetobacter radioresistens S13. Res. Microbiol. 153(2):69–74 [DOI] [PubMed] [Google Scholar]
- 8. Divari S, et al. 2003. The oxygenase component of phenol hydroxylase from Acinetobacter radioresistens S13. Eur. J. Biochem. 270:2244–2253 [DOI] [PubMed] [Google Scholar]
- 9. Dower WJ, Miller JF, Ragsdale CW. 1988. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16:6127–6145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Draths KM, Frost JW. 1994. Environmentally compatible synthesis of adipic acid from d-glucose. J. Am. Chem. Soc. 116:399–400 [Google Scholar]
- 11. Draths KM, et al. 1992. Biocatalytic synthesis of aromatics from d-glucose: the role of transketolase. J. Am. Chem. Soc. 114:3956–3962 [Google Scholar]
- 12. Duncan K, Edwards RM, Coggins JR. 1987. The pentafunctional arom enzyme of Saccharomyces cerevisiae is a mosaic of monofunctional domains. Biochem. J. 246:375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duncan K, Edwards RM, Coggins JR. 1988. The Saccharomyces cerevisiae ARO1 gene. An example of the co-ordinate regulation of five enzymes on a single biosynthetic pathway. FEBS Lett. 241:83–88 [DOI] [PubMed] [Google Scholar]
- 14. Elsemore DA, Ornston LN. 1995. Unusual ancestry of dehydratases associated with quinate catabolism in Acinetobacter calcoaceticus. J. Bacteriol. 177:5971–5978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fox DT, Hotta K, Kim C-Y, Koppisch AT. 2008. The missing link in petrobactin biosynthesis: asbF encodes a (−)3-dehydroshikimate dehydratase. Biochemistry 47:12251–12253 [DOI] [PubMed] [Google Scholar]
- 16. Frost JW, Draths KM. 1995. Biocatalytic syntheses of aromatics from d-glucose: renewable microbial sources of aromatic compounds. Annu. Rev. Microbiol. 49:557–579 [DOI] [PubMed] [Google Scholar]
- 17. Gietz RD, Woods RA. 2006. Yeast transformation by the LiAc/SS carrier DNA/PEG method. Methods Mol. Biol. 313:107–120 [DOI] [PubMed] [Google Scholar]
- 18. Giles NH, et al. 1985. Gene organization and regulation in the qa (quinic acid) gene cluster of Neurospora crassa. Microbiol. Rev. 49:338–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Güldener U, Heck S, Fiedler T, Beinhauer J, Hegemann JH. 1996. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 24:2519–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hamacher T, et al. 2002. Characterization of the xylose-transporting properties of yeast hexose transporters and their influence on xylose utilization. Microbiology 148:2783–2788 [DOI] [PubMed] [Google Scholar]
- 21. Harwood CS, Parales RE. 1996. The β-ketoadipate pathway and the biology of self-identity. Annu. Rev. Microbiol. 50:553–590 [DOI] [PubMed] [Google Scholar]
- 22. Hawkins AR, Francisco AJ, Roberts CF. 1985. Cloning and characterization of the three enzyme structural genes QUTB, QUTC and QUTE from the quinic acid utilization gene cluster in Aspergillus nidulans. Curr. Genet. 9:305–311 [DOI] [PubMed] [Google Scholar]
- 23. He Z, Wiegel J. 1995. Purification and characterization of an oxygen-sensitive reversible 4-hydroxybenzoate decarboxylase from Clostridium hydroxybenzoicum. Eur. J. Biochem. 229:77–82 [DOI] [PubMed] [Google Scholar]
- 24. He Z, Wiegel J. 1996. Purification and characterization of an oxygen-sensitive, reversible 3,4-dihydroxybenzoate decarboxylase from Clostridium hydroxybenzoicum. J. Bacteriol. 178:3539–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Helmstaedt K, Strittmatter A, Lipscomb WN, Braus GH. 2005. Evolution of 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase-encoding genes in the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 102:9784–9789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hsieh J-H. October 1984. Continuous fermentation process and bioconversion-product recovery. U.S. patent 4,480,034
- 27. Huang J, He H, Wiegel J. 1999. Cloning, characterization, and expression of a novel gene encoding a reversible 4-hydroxybenzoate decarboxylase from Clostridium hydroxybenzoicum. J. Bacteriol. 181:5119–5122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiménez JI, Miñambres B, García JL, Díaz E. 2002. Genomic analysis of the aromatic catabolic pathways from Pseudomonas putida KT2440. Environ. Microbiol. 4:824–841 [DOI] [PubMed] [Google Scholar]
- 29. Kondo A, Ishii J, Hara KY, Hasunuma T, Matsuda F. 19 June. 2012. Development of microbial cell factories for bio-refinery through synthetic bioengineering. J. Biotechnol. (Epub ahead of print.) http://dx.doi.org/10.1016/j.jbiotec.2012.05.021 [DOI] [PubMed]
- 30. Lamb HK, et al. 1990. Spatial and biological characterisation of the complete quinic acid utilisation gene cluster in Aspergillus nidulans. Mol. Gen. Genet. 223:17–23 [DOI] [PubMed] [Google Scholar]
- 31. Liu J, Zhang X, Zhou S, Tao P. 2007. Purification and characterization of a 4-hydroxybenzoate decarboxylase from Chlamydophila pneumoniae AR39. Curr. Microbiol. 54:102–107 [DOI] [PubMed] [Google Scholar]
- 32. Liu ZL. 2011. Molecular mechanisms of yeast tolerance and in situ detoxification of lignocellulose hydrolysates. Appl. Microbiol. Biotechnol. 90:809–825 [DOI] [PubMed] [Google Scholar]
- 33. Lupa B, Lyon D, Gibbs MD, Reeves RA, Wiegel J. 2005. Distribution of genes encoding the microbial non-oxidative reversible hydroxyarylic acid decarboxylases/phenol carboxylases. Genomics 86:342–351 [DOI] [PubMed] [Google Scholar]
- 34. Luttik MA, et al. 2008. Alleviation of feedback inhibition in Saccharomyces cerevisiae aromatic amino acid biosynthesis: quantification of metabolic impact. Metab. Eng. 10:141–153 [DOI] [PubMed] [Google Scholar]
- 35. Margeot A, Hahn-Hagerdal B, Edlund M, Slade R, Monot F. 2009. New improvements for lignocellulosic ethanol. Curr. Opin. Biotechnol. 20:372–380 [DOI] [PubMed] [Google Scholar]
- 36. Matsui T, Yoshida T, Hayashi T, Nagasawa T. 2006. Purification, characterization, and gene cloning of 4-hydroxybenzoate decarboxylase of Enterobacter cloacae P240. Arch. Microbiol. 186:21–29 [DOI] [PubMed] [Google Scholar]
- 37. Mizuno S, Yoshikawa N, Seki M, Mikawa T, Imada Y. 1988. Microbial production of cis,cis-muconic acid from benzoic acid. Appl. Microbiol. Biotechnol. 28:20–25 [Google Scholar]
- 38. Niu W, Draths KM, Frost JW. 2002. Benzene-free synthesis of adipic acid. Biotechnol. Prog. 18:201–211 [DOI] [PubMed] [Google Scholar]
- 39. Pfleger BF, et al. 2008. Structural and functional analysis of AsbF: origin of the stealth 3,4-dihydroxybenzoic acid subunit for petrobactin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 105:17133–17138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rutledge BJ. 1984. Molecular characterization of the Qa-4 gene of Neurospora crassa. Gene 32:275–287 [DOI] [PubMed] [Google Scholar]
- 41. Schnappauf G, Hartmann M, Kunzler M, Braus GH. 1998. The two 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase isoenzymes from Saccharomyces cerevisiae show different kinetic modes of inhibition. Arch. Microbiol. 169:517–524 [DOI] [PubMed] [Google Scholar]
- 42. Shi S, Valle-Rodriguez JO, Khoomrung S, Siewers V, Nielsen J. 2012. Functional expression and characterization of five wax ester synthases in Saccharomyces cerevisiae and their utility for biodiesel production. Biotechnol. Biofuels 5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Steen EJ, et al. 2008. Metabolic engineering of Saccharomyces cerevisiae for the production of n-butanol. Microb. Cell Fact. 7:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Suzuki K, Ichimura A, Ogawa N, Hasebe A, Miyashita K. 2002. Differential expression of two catechol 1,2-dioxygenases in Burkholderia sp. strain TH2. J. Bacteriol. 184:5714–5722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taxis C, Knop M. 2006. System of centromeric, episomal, and integrative vectors based on drug resistance markers for Saccharomyces cerevisiae. Biotechniques 40:73–78 [DOI] [PubMed] [Google Scholar]
- 46. Thiemens MH, Trogler WC. 1991. Nylon production: an unknown source of atmospheric nitrous oxide. Science 251:932–934 [DOI] [PubMed] [Google Scholar]
- 47. Ulrich H. 1988. Raw materials for industrial polymers. Hanser Publishers, Munich, Germany [Google Scholar]
- 48. Valkova N, et al. 2001. Hydrolysis of 4-hydroxybenzoic acid esters (parabens) and their aerobic transformation into phenol by the resistant Enterobacter cloacae strain EM. Appl. Environ. Microbiol. 67:2404–2409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weaver LM, Herrmann KM. 1990. Cloning of an aroF allele encoding a tyrosine-insensitive 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase. J. Bacteriol. 172:6581–6584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weber C, et al. 2010. Trends and challenges in the microbial production of lignocellulosic bioalcohol fuels. Appl. Microbiol. Biotechnol. 87:1303–1315 [DOI] [PubMed] [Google Scholar]
- 51. Wiedemann B, Boles E. 2008. Codon-optimized bacterial genes improve l-arabinose fermentation in recombinant Saccharomyces cerevisiae. Appl. Environ. Microbiol. 74:2043–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yoshida T, Inami Y, Matsui T, Nagasawa T. 2010. Regioselective carboxylation of catechol by 3,4-dihydroxybenzoate decarboxylase of Enterobacter cloacae P241. Biotechnol. Lett. 32:701–705 [DOI] [PubMed] [Google Scholar]
- 53. Yoshikawa N, Mizuno S, Ohta K, Suzuki M. 1990. Microbial production of cis,cis-muconic acid. J. Biotechnol. 14:203–210 [Google Scholar]
- 54. Zimmermann FK. 1975. Procedures used in the induction of mitotic recombination and mutation in the yeast Saccharomyces cerevisiae. Mutat. Res. 31:71–86 [DOI] [PubMed] [Google Scholar]