

Abstract
Clostridium perfringens enterotoxin (CPE) causes the gastrointestinal symptoms of the second most common bacterial food-borne illness. Previous studies suggested that a region named TM1, which has amphipathic characteristics and spans from amino acids 81 to 106 of the native CPE protein, forms a β-hairpin involved in β-barrel pore formation. To further explore the potential role of TM1 in pore formation, the single Cys naturally present in CPE at residue 186 was first altered to alanine by mutagenesis; the resultant rCPE variant, named C186A, was shown to retain cytotoxic properties. Cys-scanning mutagenesis was then performed in which individual Cys mutations were introduced into each TM1 residue of the C186A variant. When those Cys variants were characterized, three variants were identified that exhibit reduced cytotoxicity despite possessing binding and oligomerization abilities similar to those of the C186A variant from which they were derived. Pronase challenge experiments suggested that the reduced cytotoxicity of those two Cys variants, i.e., the F91C and F95C variants, which model to the tip of the β-hairpin, was attributable to a lessened ability of these variants to insert into membranes after oligomerization. In contrast, another Cys variant, i.e., the G103C variant, with impaired cytotoxicity apparently inserted into membranes after oligomerization but could not form a pore with a fully functional channel. Collectively, these results support the TM1 region forming a β-hairpin as an important step in CPE insertion and pore formation. Furthermore, this work identifies the first amino acid residues specifically involved in those two steps in CPE action.
INTRODUCTION
Clostridium perfringens type A food poisoning is currently the second most common bacterial food-borne illness in the United States, where nearly a million cases occur annually (4). This food poisoning typically involves diarrhea and abdominal cramps that self-resolve within 24 h. However, C. perfringens type A food poisoning can be fatal, particularly in the elderly or in medicated patients (1). The symptoms of this food-borne illness are caused by C. perfringens enterotoxin (CPE), a 35-kDa single polypeptide (18, 27). CPE-producing type A strains of C. perfringens also commonly cause human non-food-borne diseases such as antibiotic-associated diarrhea or sporadic diarrhea (3).
The action of CPE on host cells begins with binding of this toxin to receptors. Known CPE receptors include certain members of the claudin family of tight junction proteins (7, 8, 18, 25, 36). Binding of the toxin to claudins initially results in formation of an SDS-sensitive, small (∼90-kDa) complex (38). The CPE small complex then oligomerizes to form a large SDS-resistant CPE complex named CH-1 (26, 29). The CH-1 complex is ∼450 kDa in size but migrates anomalously on SDS-PAGE with an apparent size of ∼155 kDa (26). This complex contains 6 CPE molecules, as well as both receptor and nonreceptor claudins (26). CH-1 initially forms as a prepore on the host cell surface (31, 33); however, at physiologic temperatures, the CH-1 prepore rapidly inserts into plasma membranes to form an active pore (21). Formation of the CPE pore leads to a calcium influx that causes cell death in a toxin dose-dependent manner (5, 6), via either oncosis (high CPE doses) or a classical caspase-3-mediated endocytosis (low CPE doses). In the small intestine, CPE-induced enterocyte death causes epithelial necrosis and desquamation, with this damage producing fluid and electrolyte secretion that results in diarrhea (18).
CPE can also form a second large complex, named CH-2, in sensitive host cells (29). The CH-2 CPE complex is ∼550 kDa, but it migrates anomalously on SDS-PAGE with an apparent size of ∼200 kDa (26). This complex contains 6 CPE molecules, receptor and nonreceptor claudins, along with another tight junction protein named occludin (26). CH-2 formation apparently induces the internalization of occludin and (perhaps in combination with CH-1) claudins into the cytoplasm of CPE-treated cells (28). These effects may contribute to tight junction breakdown and, possibly, cause paracellular permeability changes that contribute to diarrhea (18). However, the primary effect responsible for CPE-induced intestinal fluid and electrolyte secretion is considered to be enterocyte damage resulting from pore formation (32).
Two independent groups recently used X-ray crystallography approaches to solve the structure of CPE (2, 15). Those analyses indicated that the CPE protein consists of two distinct domains, including a C-terminal domain and an N-terminal domain comprised of two halves. Furthermore, based upon its structure, CPE was assigned to the aerolysin-like pore-forming toxin (PFT) family. Coupling this new structural information with results of earlier extensive biochemical studies or CPE mutagenesis experiments (9–12, 35) indicated that the C-terminal domain (residues 196 to 319) of CPE mediates binding of the toxin to claudin receptors.
The N-terminal domain (residues 1 to 195) of the enterotoxin mediates at least two steps in CPE action. First, the region located between the two halves of the cytotoxicity domain is responsible for CPE monomer oligomerization to form the CH-1 prepore (16, 30). Site-directed mutagenesis studies (16, 30) identified CPE residues D48 and I51 of the N-terminal cytotoxicity domain as being particularly critical for CH-1 prepore formation. Second, the CPE region comprising amino acids 81 to 106 in the cytotoxicity domain, named TM1, contains the β4 strand and the only α-helix present in the N-terminal cytotoxicity domain (2, 15). This CPE region possesses two stretches of alternating hydrophobic and hydrophilic residues, which is characteristic of a β-hairpin motif (33). In some other PFTs, similar β-hairpins insert into membranes to form a β-barrel pore (14). Consistent with amino acids 81 to 106 playing a similar role for the CPE protein, an rCPE TM1 variant that specifically lacks amino acids 81 to 106 of native CPE was shown to bind to, and oligomerize on, Caco-2 cells (26, 33). However, this TM1 variant was noncytotoxic (26, 33). Results from pronase challenge and other experiments (26, 33) suggested that the large complexes formed by the TM1 variant remain present on the membrane surface in a prepore; i.e., the CH-1 complex formed by the TM1 variant appeared to be blocked for membrane insertion. This result was consistent with the TM1 region forming a β-hairpin that functions during toxin insertion into membranes.
However, it could be argued that deleting nearly 30 amino acids from the middle of CPE had produced a gross conformational change in the TM1 variant that was responsible for its inability to mediate membrane insertion. Therefore, the current study included Cys-scanning site-directed point mutagenesis of the TM1 region in order (i) to further evaluate whether this CPE region plays a critical role in toxin membrane insertion and pore formation and (ii) to begin identifying individual CPE amino acids important for causing these effects.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth media.
All site-directed mutagenesis and protein expression work was performed with the Escherichia coli strain XL1-Blue. Bacterial cultures for DNA manipulation were grown in Luria-Bertani (LB) medium containing 100 μg/ml of ampicillin. The plasmid pJFKLt-1 was described previously (17) and contains the complete cpe open reading frame (ORF) ligated into the BamHI and EcoRI multiple cloning sites in the pTrcHis A vector (Invitrogen, Carlsbad, CA) to produce rCPE. This plasmid was used as the template for construction of pJSC186A, which encodes a Cys-to-Ala point mutation at CPE residue 186 (see Results). All other mutations in this study were prepared using the template plasmid pJSC186A, which was created in this study as described below. All plasmids used in this study are listed in Table 1.
Table 1.
Plasmids used in this study
| Plasmid | Description | Source or reference |
|---|---|---|
| pTrcHis A | E. coli expression vector | Invitrogen |
| pJKFLt-1 | Contains full-length CPE ORF | 17 |
| pJSD48A | Point mutation for D48A in cpe gene | 30 |
| pJSC186A | Point mutation for C186A in cpe gene | This study |
| Cys substitutions | Point mutation in TM1 regions (80–109 amino acids) by Cys-scanning mutagenesis | This study |
Site-directed mutagenesis.
To create pJSC186A, a C186A mutation was introduced into the cpe ORF present in pJKFLt-1 using primers listed in Table 2 and the QuikChange Lightning site-directed mutagenesis kit (Stratagene, La Jolla, CA). Using pJSC186A and primers listed in Table 2, Cys-scanning mutagenesis was then performed on TM1 and adjacent residues corresponding to amino acids 80 to 109 of native CPE by the same method. Similarly, pJSC186A and primers listed in Table 2 were used to introduce additional mutations, as described in Results, at residues corresponding to F91, F95, and G103 of native CPE. Reaction parameters were in accordance with the manufacturer's instructions. Mutated plasmid DNA resulting from the site-directed mutagenesis reaction was used to transform XL1-Supercompetent Blue E. coli (Agilent). The presence of all mutations in the cpe ORF of pJKFLt-1 or pJSC186A was confirmed by nucleotide sequencing. Plasmids containing each mutated cpe gene were isolated from overnight E. coli cultures with the Qiagen plasmid miniprep kit. Sequencing reactions were performed by the University of Pittsburgh Genomics and Proteomics Core Laboratories. Lasergene (Dnastar) software was used to confirm the presence of the desired mutation.
Table 2.
Primers used in this study
| Variant | Forward primer | Reverse primer |
|---|---|---|
| F87C | 5′ GTATCTATAAATGTAAATTGCTCAGTTGGATTTACTTCTG 3′ | 5′ CAGAAGTAAATCCAACTGAGCAATTTACATTTATAGATAC 3′ |
| S88C | 5′ CTATAAATGTAAATTTTTGCGTTGGATTTACTTCTG 3′ | 5′ CAGAAGTAAATCCAACGCAAAAATTTACATTTATAG 3′ |
| V89C | 5′ CTATAAATGTAAATTTTTCATGTGGATTTACTTCTG 3′ | 5′ CAGAAGTAAATCCACATGAAAAATTTACATTTATAG 3′ |
| G90C | 5′ GTAAATTTTTCAGTTTGCTTTACTTCTGAATTTATAC 3′ | 5′ GTATAAATTCAGAAGTAAAGCAAACTGAAAAATTTAC 3′ |
| F91C | 5′ GTAAATTTTTCAGTTGGATGTACTTCTGAATTTATACAAGC 3′ | 5′ GCTTGTATAAATTCAGAAGTACATCCAACTGAAAAATTTAC 3′ |
| T92C | 5′ GTAAATTTTTCAGTTGGATTTTGTTCTGAATTTATACAAGCATC 3′ | 5′ GATGCTTGTATAAATTCAGAACAAAATCCAACTGAAAAATTTAC 3′ |
| S93C | 5′ CAGTTGGATTTACTTGTGAATTTATACAAGC 3′ | 5′ GCTTGTATAAATTCACAAGTAAATCCAACTG 3′ |
| E94C | 5′ CAGTTGGATTTACTTCTTGCTTTATACAAGCATCTGTAG 3′ | 5′ CTACAGATGCTTGTATAAAGCAAGAAGTAAATCCAACTG 3′ |
| F95C | 5′ GGATTTACTTCTGAATGTATACAAGCATCTGTAG 3′ | 5′ CTACAGATGCTTGTATACATTCAGAAGTAAATCC 3′ |
| I96C | 5′ GTTGGATTTACTTCTGAATTTTGCCAAGCATCTGTAGAATATGG 3′ | 5′ CCATATTCTACAGATGCTTGGCAAAATTCAGAAGTAAATCCAAC 3′ |
| Q97C | 5′ GGATTTACTTCTGAATTTATATGCGCATCTGTAGAATATGG 3′ | 5′ CCATATTCTACAGATGCGCATATAAATTCAGAAGTAAATCC 3′ |
| A98C | 5′ CTTCTGAATTTATACAATGCTCTGTAGAATATGGATTTGG 3′ | 5′ CCAAATCCATATTCTACAGAGCATTGTATAAATTCAGAAG 3′ |
| S99C | 5′ CTGAATTTATACAAGCATGTGTAGAATATGGATTTGG 3′ | 5′ CCAAATCCATATTCTACACATGCTTGTATAAATTCAG 3′ |
| V100C | 5′ CTGAATTTATACAAGCATCTTGCGAATATGGATTTGGAATAAC 3′ | 5′ GTTATTCCAAATCCATATTCGCAAGATGCTTGTATAAATTCAG 3′ |
| E101C | 5′ GAATTTATACAAGCATCTGTATGCTATGGATTTGGAATAAC 3′ | 5′ GTTATTCCAAATCCATAGCATACAGATGCTTGTATAAATTC 3′ |
| Y102C | 5′ CAAGCATCTGTAGAATGCGGATTTGGAATAACTATAG 3′ | 5′ CTATAGTTATTCCAAATCCGCATTCTACAGATGCTTG 3′ |
| G103C | 5′ CAAGCATCTGTAGAATATTGCTTTGGAATAACTATAGGAG 3′ | 5′ CTCCTATAGTTATTCCAAAGCAATATTCTACAGATGCTTG 3′ |
| F104C | 5′ CTGTAGAATATGGATGTGGAATAACTATAGG 3′ | 5′ CCTATAGTTATTCCACATCCATATTCTACAG 3′ |
| G105C | 5′ CTGTAGAATATGGATTTTGCATAACTATAGGAGAAC 3′ | 5′ GTTCTCCTATAGTTATGCAAAATCCATATTCTACAG 3′ |
| I106C | 5′ CTGTAGAATATGGATTTGGATGCACTATAGGAGAACAAAATAC 3′ | 5′ GTATTTTGTTCTCCTATAGTGCATCCAAATCCATATTCTACAG 3′ |
| T107C | 5′ GAATATGGATTTGGAATATGCATAGGAGAACAAAATAC 3′ | 5′ GTATTTTGTTCTCCTATGCATATTCCAAATCCATATTC 3′ |
| I108C | 5′ GAATATGGATTTGGAATAACTTGCGGAGAACAAAATACAATAG 3′ | 5′ CTATTGTATTTTGTTCTCCGCAAGTTATTCCAAATCCATATTC 3′ |
| G109C | 5′ GGATTTGGAATAACTATATGCGAACAAAATAC 3′ | 5′ GTATTTTGTTCGCATATAGTTATTCCAAATCC 3′ |
| F91Y | 5′ GTAAATTTTTCAGTTGGATATACTTCTGAATTTATACAAG 3′ | 5′ CTTGTATAAATTCAGAAGTATATCCAACTGAAAAATTTAC 3′ |
| F91W | 5′ GTAAATTTTTCAGTTGGATGGACTTCTGAATTTATACAAG 3′ | 5′ CTTGTATAAATTCAGAAGTCCATCCAACTGAAAAATTTAC 3′ |
| F95Y | 5′ GGATTTACTTCTGAATATATACAAGCATCTGTAG 3′ | 5′ CTACAGATGCTTGTATATATTCAGAAGTAAATCC 3′ |
| F95W | 5′ GGATTTACTTCTGAATGGATACAAGCATCTGTAG 3′ | 5′ CTACAGATGCTTGTATCCATTCAGAAGTAAATCC 3′ |
| G103A | 5′ CTGTAGAATATGCATTTGGAATAAC 3′ | 5′ GTTATTCCAAATGCATATTCTACAG 3′ |
| G103S | 5′ CAAGCATCTGTAGAATATTCATTTGGAATAACTATAGG 3′ | 5′ CCTATAGTTATTCCAAATGAATATTCTACAGATGCTTG 3′ |
Affinity purification of rCPE variants.
rCPE and derivative rCPE variants each contained a hexahistidine tag fused to its N terminus, which allowed affinity enrichment of these recombinant proteins from lysates of recombinant E. coli transformants. For this purpose, 1 liter of SOB medium was inoculated with 20 ml of an overnight SOB culture of the desired rCPE species-expressing E. coli strains. After shaking of the culture for 3 h at 37°C, expression of the rCPE species was induced by adding isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. After the culture was incubated for 4 h at 37°C with shaking, the bacteria were harvested by centrifugation. Final pellets were resuspended in equilibration/wash buffer (50 mM sodium phosphate, 300 mM NaCl [pH 7.0]). The bacterial suspension was treated with 0.75 mg/ml of lysozyme for 30 min before a Sonicator 3000 (Misonix, Farmingdale, NJ) was used to lyse the bacteria. Cell debris was then pelleted by centrifugation, and the supernatant was collected and incubated with equilibrated TALON resin (Clontech, Mountain View, CA) for 20 min at room temperature (RT). The resin was then washed twice with equilibration/wash buffer, and the rCPE species was eluted with elution buffer (50 mM sodium phosphate, 300 mM NaCl, 50 mM imidazole [pH 7.0]). Protein-containing fractions from the elution were pooled, concentrated, and desalted by a Sephadex G-25 M PD-10 column (GE Healthcare Life Sciences, Uppsala, Sweden). The rCPE species harvested from the each affinity enrichment was quantified by a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, Rockford, IL) and assayed by CPE Western blotting. Purity was assessed by SDS-PAGE and Coomassie blue staining, as described below.
Morphological-damage assay.
Using Eagle minimal essential medium (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (Mediatech, Herndon, VA), 1% minimal essential medium nonessential amino acids (Sigma), 100 μg/ml of ampicillin, 100 μg/ml of streptomycin, and 2 mM l-glutamine, Caco-2 cells were grown at 37°C in 6-well plates in a 5% atmospheric CO2 incubator. Confluent Caco-2 monolayers were washed once with Hanks balanced salt solution (HBSS) prewarmed to 37°C and then treated with 2.5 μg/ml of the specified rCPE species. Cellular morphological damage was determined after a 1-h treatment with toxin that had been preincubated for 15 min at 37°C in the presence or absence of 50 μl of CPE polyclonal antiserum or control rabbit preimmune antiserum. This morphological damage was assessed using a Zeiss Axiovert 25 inverted microscope (magnification, ×200) equipped with a Powershot G5 digital camera (Canon, Inc., Lake Success, NY).
86Rb release experiments.
Formation of a functional CPE membrane pore by each rCPE species was assessed by 86Rb release experiments as described previously (29, 30). In brief, confluent Caco-2 cell monolayers were radiolabeled with 4 μCi/well of 86RbCl (PerkinElmer, Boston, MA) in the culture medium for 4 h at 37°C. After being washed twice with 2 ml of HBSS buffer prewarmed to 37°C, the radiolabeled cells were incubated with up to 32 μg/ml of an rCPE species suspended in 2 ml of HBSS for 15 min at 37°C. The culture supernatant containing released 86Rb was collected, and radioactivity in those samples was quantified with a Cobra Quantum gamma counter (PerkinElmer). Maximal release of 86Rb was determined by treating radiolabeled monolayers with 1 ml each of 0.5% saponin and 1.0 M citric acid. Spontaneous release of 86Rb from cells was measured by incubation with HBSS alone. Data from 86Rb release experiments were converted and plotted as percent maximal release using the following formula: [(experimental release − spontaneous release)/(maximal release − spontaneous release)] × 100.
Limited trypsin proteolysis assay.
A 500-ng aliquot of each purified rCPE species was resuspended in phosphate-buffered saline (PBS) to a final volume of 100 μl. Reaction mixtures were digested at 25°C for 0, 5, 15, 30, or 60 min with 2 μg of trypsin (Sigma). Immediately after this digestion, 100 μl of 2× Laemmli buffer containing 5% β-mercaptoethanol was added to each reaction mixture, and the samples were boiled for 5 min to terminate proteolytic digestion. A 30-μl aliquot of each sample was then loaded onto an SDS-containing 12% polyacrylamide gel, and after electrophoresis, the separated proteins were electrotransferred onto a nitrocellulose membrane (Bio-Rad, Hercules, CA) using Towbins's transfer buffer (15.6 mM Tris-HCl, 120 mM glycine [pH 8.3]). After blocking with 5% milk, the membrane was incubated with anti-CPE polyclonal antibody (26) (used at a 1:5,000 dilution) at 4°C overnight, followed by incubation with anti-rabbit horseradish peroxidase-labeled secondary antibody (used at a 1:10,000 dilution; Sigma). Blots were developed using SuperSignal chemiluminescent substrate (Thermo Scientific, Rockford, IL).
Large-complex formation.
The formation of SDS-resistant large CPE complexes by rCPE species was assessed as described previously (26, 30). Briefly, after two washes with HBSS, confluent Caco-2 cell monolayers were treated with 2.5 μg/ml of rCPE species and incubated at 37°C for 60 min. These cells were then pelleted by centrifugation and washed twice with HBSS (Mediatech, Manassas, VA). All samples were treated for 10 min at RT with Benzonase nuclease to degrade cellular DNA before addition of Laemmli buffer and being loaded on an SDS-containing 4.25% polyacrylamide gel. Electrophoresis of the samples was performed at 15 mA for 3 to 4 h. After protein was transferred to the blot, CPE-containing large complexes present on the blot were detected by CPE Western blotting, as described earlier.
Pronase resistance of CPE large complex.
The pronase susceptibility of large complexes formed by rCPE species was analyzed using a previously published procedure (33, 37). Caco-2 cell monolayers were treated for 1 h at 37°C with 2.5 μg/ml of an rCPE species dissolved in 1 ml of HBSS buffer (Mediatech). Cells were collected and then washed twice with 1 ml of HBSS, followed by treatment at RT for 5 or 60 min with 50 μl of HBSS containing 6, 60, or 600 μg/ml of pronase. Cells were then washed twice with HBSS containing complete inhibitor cocktail (Roche Applied Science, Penzberg, Germany), resuspended in Laemmli buffer, and loaded on an SDS-containing 10% polyacrylamide gel. After electrophoresis and transfer of separated proteins onto a nitrocellulose membrane, the blots were subjected to CPE Western blotting, as detailed above.
RESULTS
Construction and characterization of a C186A rCPE variant.
Because later experiments would use introduced single Cys substitutions to evaluate the cytotoxic importance of the putative β-hairpin identified between residues 81 and 106 of CPE, it was first necessary to change the single natural Cys of native CPE (nCPE) (located at residue 186) to Ala to prevent potential disulfide bond formation in those planned Cys-scanning mutants. After construction of the C186A variant of rCPE (see Materials and Methods), phenotypic experiments were performed to evaluate whether this Cys-to-Ala mutation had affected the characteristics of the C186A variant relative to rCPE or nCPE.
To grossly assess the cytotoxicity of the C186A variant, a cellular morphological damage assay was used (Fig. 1A). As reported previously (26), Caco-2 cells treated with 2.5 μg/ml of nCPE or rCPE developed membrane blebbing and detached from the cell culture well surface. Similar to nCPE or rCPE, the C186A variant caused morphological damage to Caco-2 cells, although both rCPE and the C186A variant were slightly less cytotoxic than nCPE. In contrast, no morphological damage was observed when cells were treated with HBSS buffer alone. When rCPE or the C186A variant was preincubated with polyclonal CPE antiserum (to neutralize CPE activity) before those mixtures were added to Caco-2 cells, no morphological damage developed in the treated Caco-2 cells. In contrast, similar preincubation of rCPE or the 186A variant with rabbit preimmune serum did not prevent the subsequent development of morphological damage to Caco-2 cells (data not shown). These results strongly suggested that the Caco-2 morphological damage observed in cells treated with nCPE, rCPE, or C186A preparations was specifically caused by the added CPE species.
Fig 1.

Comparison of biochemical characteristics of the C186A variant versus rCPE. (A) Cellular morphological damage. To initially assess the cytotoxic properties of the C186A variant, a morphological-damage assay (33) was performed. Confluent Caco-2 cell monolayers were treated at 37°C for 60 min with HBSS or HBSS containing 2.5 μg/ml of nCPE, rCPE, or the C186A variant. After this treatment, Caco-2 cells were photomicrographed at a total magnification of ×400. To test the specificity of cytotoxicity caused by these toxins, the C186A variant or rCPE was preincubated at 37°C with CPE polyclonal antiserum for 15 min; those mixtures were then applied to confluent Caco-2 cells in 6-well plates. Shown are representative results from three repetitions of this experiment. Ab, antibody. (B) Large-complex formation by the C186A variant and rCPE. Confluent Caco-2 cells were treated with 2.5 μg/ml of either rCPE species for 60 min at 37°C. The treated cells were then loaded onto a 4.25% polyacrylamide gel containing SDS. Large-complex formation was evaluated by Western blotting of the gel using rabbit polyclonal anti-CPE serum. “CH1” and “CH2” represent the two CPE large complexes, which migrate anomalously as ∼155- and 200-kDa proteins, respectively, on these gels (26). (C) Limited trypsin digestion assay. To assess gross conformational changes in the C186A variant, both rCPE and the C186A variant (500 ng) were incubated with trypsin (2 μg) at 25°C for the periods indicated above the lanes. After samples were stopped by the addition of Laemmli buffer and boiling for 5 min, proteins in those samples were separated by electrophoresis on an SDS-containing 10% polyacrylamide gel, followed by Western blotting with rabbit anti-CPE polyclonal antibody.
At 37°C, CPE binds and quickly becomes localized in large, SDS-resistant complexes (see the introduction). Since formation of these large complexes, particularly the CH-1 complex, is necessary for CPE-induced cytotoxicity (26, 28, 29), an experiment evaluated the ability of the C186A variant to form the SDS-resistant large CPE-containing complexes. This analysis revealed similar large-complex formation using the C186A variant, nCPE, or rCPE (Fig. 1B).
With Fig. 1A and B results indicating that the C186A variant possesses cytotoxicity and biochemical characteristics similar to those of rCPE, the C186A variant would be predicted to retain a protein structure similar to that of rCPE. To test this expectation, a commonly used limited trypsin digestion analysis (30) was performed to detect whether a gross conformational change had occurred in the C186A variant. The results obtained showed that the C186A variant exhibited the same trypsin digestion pattern as rCPE, strongly suggesting that the C186A variant retains a structure similar to that of rCPE (Fig. 1C).
Cysteine-scanning mutagenesis of the TM1 region in the C186A variant.
Previous studies (33) analyzing the CPE primary sequence had suggested that CPE amino acids 81 to 106, which consist of alternating hydrophobic and hydrophilic residues that are mainly present within an α-helix in the toxin monomer, may unwind to form a β-hairpin during prepore insertion (Fig. 2). Consistent with reports indicating that similar motifs mediate the insertion and pore formation of other β-PFTs (14, 23, 24, 34), we previously reported that deleting this region, named TM1, from rCPE did not interfere with CH-1 formation (33). However, this TM1 variant lost the ability to insert into membranes or form an active pore. While these results support the importance of the region from amino acids 81 to 106 for CPE insertion and pore formation, deleting 30 amino acid residues from rCPE could arguably have produced significant conformational changes that nonspecifically interfered with toxin insertion and pore formation. Therefore, to (i) clarify further the involvement of this region in CPE insertion, (ii) identify the important amino acids in this region, and (iii) determine, in future experiments using fluorescent sulfhydryl-specific probes (23, 34), which specific residues in this region interact with membrane lipids by biophysical methods, Cys-scanning site-directed mutagenesis was performed in the C186A variant. In this work, 30 individual Cys mutations were introduced into the region from amino acids 81 to 109 of the C186A variant by site-directed mutagenesis, and the presence of each desired point Cys mutation was then confirmed by sequencing the mutant cpe genes.
Fig 2.

Location and membrane topology model of putative TM1 transmembrane domain in CPE. (A) Location of the TM1 region (red) in the CPE monomer. (Reprinted from reference 2 with permission from Elsevier.) (B) Analysis of the CPE primary sequence predicted that the TM1 region between amino acids 90 and 106 comprises a β-hairpin with alternating hydrophilic and hydrophobic amino acids (2, 15, 33). A β-hairpin is often used by β-PFTs for membrane insertion and pore formation (23, 34). Circled amino acids were found in this study to be important for CPE cytotoxicity (see Results).
Cytotoxicity screening of the Cys scanning rCPE variants using an 86Rb release assay.
Because the cytotoxicity of some Cys scanning variants might be only minimally attenuated, a sensitive quantitative assay was employed to detect possible toxicity differences between those Cys variants and the C186A variant, from which these Cys variants were derived. The choice for these cytotoxicity analyses was an 86Rb release assay that has been extensively used to measure the plasma membrane permeability alterations responsible for CPE-induced killing of mammalian cells (22, 29, 30).
For this assay, all rCPE species were first enriched by TALON resin (Clontech) and then analyzed by SDS-PAGE. Coomassie blue staining of those gels (data not shown) revealed the presence of a single band of ∼38 kDa, matching the expected molecular mass of an rCPE derivative comprised of the 35-kDa CPE protein plus ∼3 kDa of vector-encoded peptide. Western blotting of those samples (data not shown) confirmed the identity of the Coomassie blue-staining band as an rCPE species. Band intensity was then quantified by Image J software (NIH, Bethesda, MD) to determine the concentration of each rCPE species after normalization to a standard curve prepared using an enriched C186A rCPE variant. After this normalization, 100 ng of protein from each enriched mutant was again analyzed by Western blotting to confirm the accuracy of the normalization process (data not shown).
An aliquot of each enriched rCPE species was used for cytotoxicity screening in an 86Rb release experiment to measure CPE pore formation. The data, shown in Fig. 3A, indicated that the levels of 86Rb release induced by 10 μg/ml of the F91C, F95C, and G103C Cys variants were less than 50%. This result contrasted with the 95% 86Rb release observed using the same amount of the C186A variant.
Fig 3.

86Rb release experiments. (A) Cytotoxicity screening of rCPE mutants by an 86Rb release assay. Confluent Caco-2 cells in 24-well plates were radiolabeled with 4 μCi/well of 86RbCl for 4 h at 37°C and then treated at 37°C with 10 μg/ml of the specified purified 6× His-tagged rCPE species. After 15 min, 86Rb released into the medium was collected and quantified with a Cobra Quantum gamma counter. Data from this experiment were converted into the percentage of maximal 86Rb release (see Materials and Methods). The bars shown in red correspond to significantly attenuated mutants (P < 0.05). Duplicate samples of each variant were tested in three independent experiments. Error bars depict the standard errors. (B) Dose effects of rCPE species on 86Rb release. To further characterize the Cys variants showing reduced 86Rb release in panel A, 86RbCl-labeled Caco-2 cells were treated at 37°C with increasing amounts of the specified rCPE species. Data shown represent the means of three independent experiments, and the error bars show the standard deviations. (C) Cellular morphological damage caused by rCPE species. To confirm the results from the86Rb release experiments, a morphological damage assay was performed as described for Fig. 1. Confluent Caco-2 cell monolayers were treated at 37°C for 60 min with 2.5 μg/ml of each specified rCPE species, including nCPE, rCPE, the C186A variant, the F87C variant, the Q97C variant, and the D48A variant. As a control, some Caco-2 cells were treated with HBSS buffer alone (cHBSS; no added rCPE species).
The three Cys variants exhibiting a substantial reduction in their cytotoxic properties in the initial screening (Fig. 3A) were then chosen for more in-depth characterization at four different toxin doses in the 86Rb release assay, along with rCPE, the C186A variant, three Cys variants (the F87C, Q97C, and G109C variants) exhibiting cytotoxic properties similar to C186A in the initial screening, and a previously prepared negative control, i.e., the inactive rCPE D48A variant (30). The concentration of the C186A variant or rCPE needed to cause 50% of maximal 86Rb release was determined to be 1.5 or 1.2 μg/ml, respectively (Fig. 3B), demonstrating that the C186A variant retains an ability similar to that of rCPE to induce 86Rb release from Caco-2 cells. The 50% 86Rb release values obtained for the F87C, Q97C, and G109C variants were only slightly lower (≤∼2 μg/ml) than the 50% 86Rb release values measured for either the C186A variant or rCPE (Fig. 3B), confirming the initial screening results indicating that these three rCPE Cys variants retained a C186-like ability to cause 86Rb release from the cells. However, compared against the C186A variant from which they were derived, the F91C, F95C, and G103C mutants each exhibited a significantly reduced ability to cause 86Rb release, exhibiting ∼10-, 4-, and 8-fold-lower 50% 86Rb release values, respectively, than the C186A variant (Fig. 3B). As expected from previous studies (30), the negative-control D48A variant failed to appreciably alter plasma membrane permeability of Caco-2 cells, even at the very high concentration (32 μg/ml) of toxin (Fig. 3B). Since these results suggested that residues F91, F95, and G103 in TM1 are important for CPE pore formation, the F91C, F95C, and G103C variants were each characterized further.
Morphological damage caused by the Cys variants.
To confirm the 86Rb release cytotoxicity results shown in Fig. 3B, enriched rCPE and enriched F91C, F95C, G103C, C186A, and F87C variants were each individually applied to confluent Caco-2 cells. Some CPE cytopathology was observed when the Caco-2 cells were treated with any of these rCPE species. However, less damage was caused by the F91C, F95C and C103C variants than by rCPE, the C186A variant, or the fully active F87C variant (Fig. 3C). As expected, the negative control D48A variant, or HBSS alone, caused no damage to Caco-2 cells (Fig. 3C). These morphological damage results are consistent with results of the 86Rb release experiments shown in Fig. 3A and B.
Analysis of rCPE variants for gross conformational changes.
The results described above showed that the cytotoxic properties of the F91C, F95C, and G103C variants are reduced relative to those of the C186A variant or rCPE. Limited trypsin digestion (16, 30) was then performed to help assess whether those three variants might have lost cytotoxic activity due to a gross conformational change resulting from their Cys substitution. In this assay, the attenuated F91C, F95C, and G103C variants each exhibited trypsin digestion patterns (Fig. 4) similar to those of the F87C variant, which retains C186A-like cytotoxic properties, suggesting that none of the three Cys variants with reduced cytotoxic activity had undergone a gross conformational change.
Fig 4.
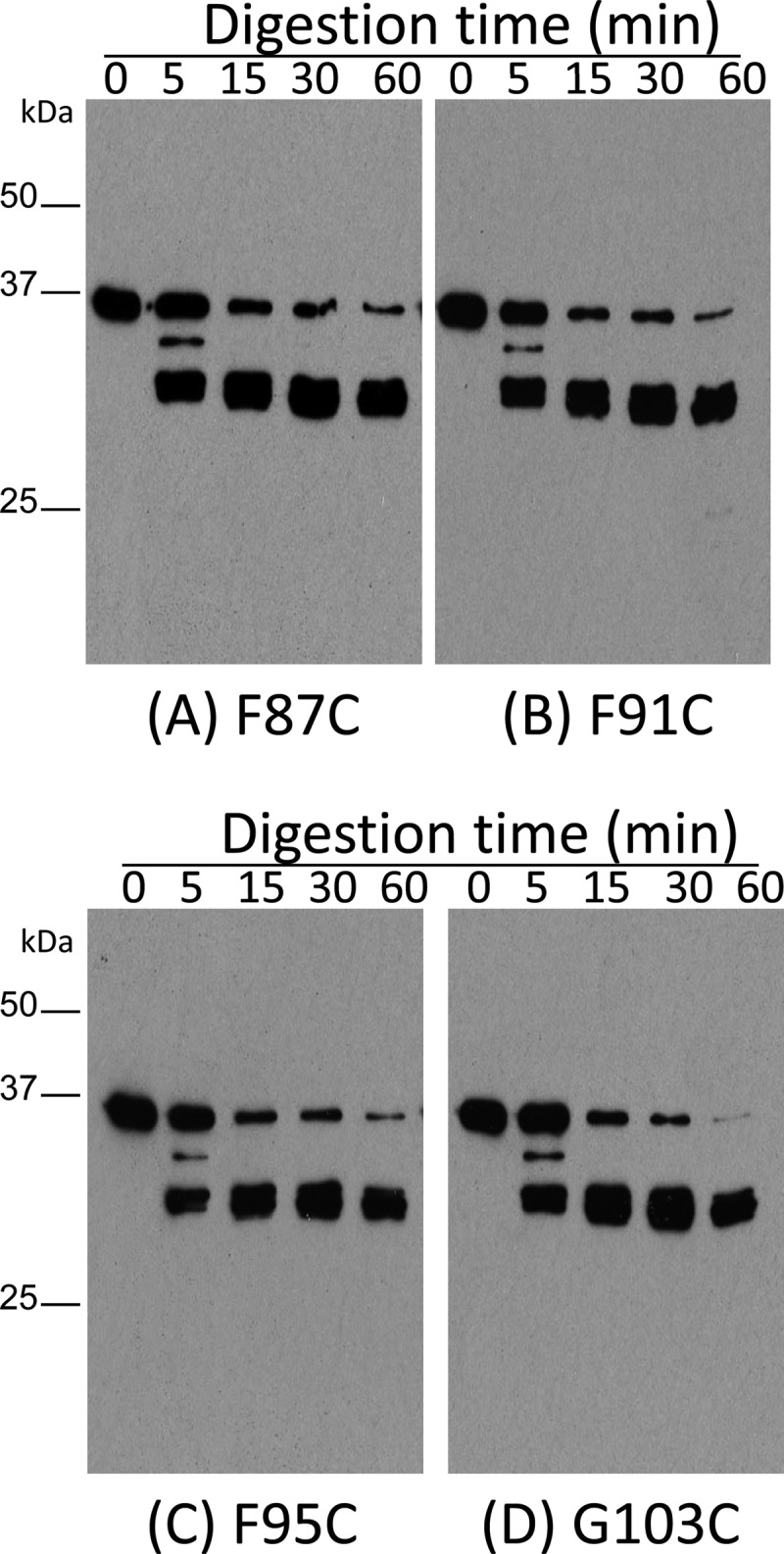
Limited trypsin digestion analysis of rCPE species. To assess gross conformational changes among the Cys variants, 500 ng of the F87C variant (A), the F91C variant (B), the F95C variant (C), or the G103C variant (D) was digested at RT with trypsin (2 μg) for the specified times. The digestion pattern was then detected by Western blotting using rabbit anti-CPE polyclonal antibody.
Large-complex formation by rCPE species.
When applied to sensitive Caco-2 cells, nCPE or rCPE forms SDS-resistant large complex prepores, which then insert into membranes to cause pore formation and eventual cell death (26, 33). Therefore, we next assessed the large complex-forming ability of rCPE, the C186A variant, the three Cys variants with reduced cytotoxic activity, and two fully active Cys variants (the F87C and Q97C variants). The data shown in Fig. 5 indicated that the F91C, F95C, and G103C variants all formed levels of large complexes in Caco-2 cells similar to those formed by either the positive controls or the two Cys rCPE variants with C186A-like cytotoxic properties. Finally, a D48A rCPE variant shown previously (30) not to form large complexes was also included in this experiment as a negative control; as expected, this variant did not form any large CPE complexes (Fig. 5).
Fig 5.
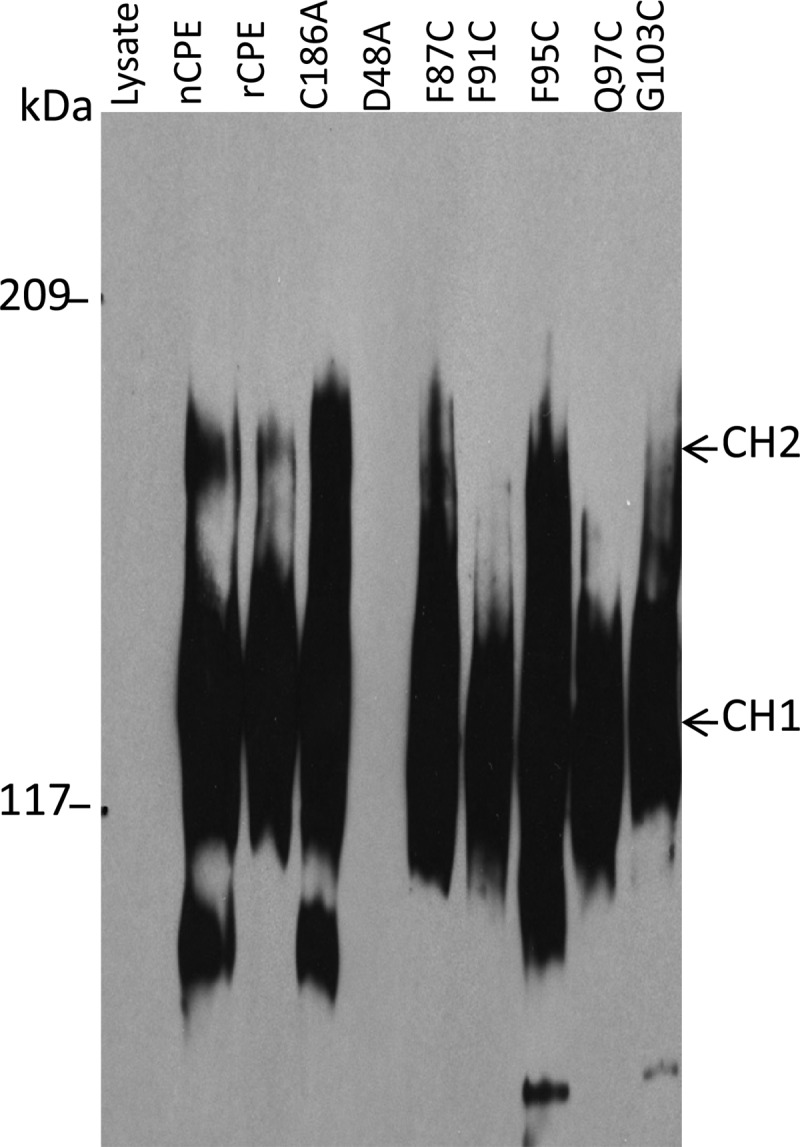
Large-complex formation by rCPE species. After confluent Caco-2 cells were treated for 60 min at 37°C with 2.5 μg/ml of each specified rCPE species, samples were electrophoresed on an SDS-containing 4.25% polyacrylamide gel. Complexes were then electrotransferred onto a nitrocellulose membrane and detected by CPE Western blotting. The “lysate” lane corresponds to Caco-2 cells treated with a mock affinity enrichment preparation with lysates from E. coli cells transformed with the pTrcHis A empty vector.
Pronase resistance of large complexes made by rCPE species.
When the large complexes formed by rCPE or nCPE insert into host cell plasma membranes, those complexes become more resistant to pronase digestion (33, 37). However, the large complexes formed by the TM1 variant of rCPE showed greater susceptibility to pronase digestion, strongly suggesting that those complexes remain exposed as a prepore on the membrane surface due to a membrane insertion defect (33, 37).
Therefore, we assessed the pronase sensitivity of the large complexes present in Caco-2 cells for the three Cys rCPE variants exhibiting reduced cytotoxic properties in the experiments whose results are shown in Fig. 3. These analyses revealed that substantial amounts of the C186A, F87C, and G103C variants present in large complexes in Caco-2 cells remained detectable by Western blotting after those Caco-2 cells had been pronase treated, even using a strong treatment involving 600 μg/ml of pronase for 60 min (Fig. 6A, B, and E). In contrast, the large complexes made by the F91C variant nearly disappeared, while those made by the F95C variant were substantially reduced in abundance, even when those Caco-2 cells were treated with only 6 μg/ml of pronase for 5 min (Fig. 6C and D). If Caco-2 cells containing the large complexes formed by the F91C or F95C variant were incubated at 37°C for 1 h without pronase, no complex degradation was observed (data not shown), indicating that the degradation of the F91C- and F95C-containing large complexes demonstrated in Fig. 6C was a direct result of the addition of pronase to Caco-2 cells. Collectively, these results demonstrated that when present in Caco-2 cells, the large complexes formed by the F91C and F95C variants are much more sensitive to pronase treatment than are the large complexes made by the C186A, F87C, and G103C variants.
Fig 6.

Pronase degradation resistance of large complexes formed by rCPE species. Confluent Caco-2 cells were treated for 1 h at RT with 2.5 μg of each purified rCPE species, including the C186A variant (A), the F87C variant (B), the F91C variant (C), the F95C variant (D), and the G103C variant (E), and then washed to remove unbound toxin. Membrane fractions collected from those cells were then incubated with the concentrations of pronase indicated above each lane at RT for the indicated amounts of time. After digestion, membranes were pelleted and resuspended in Laemmli buffer. The sample proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane, followed by Western blotting with rabbit polyclonal anti-CPE antiserum. Note that CH-2 levels formed by each variant varied from experiment to experiment, as did the formation of bands running below CH-1; similar highly variable bands running below CH-1 have been observed previously in CPE-treated cells (28) and could represent either breakdown of CH-1 or CH-2 or an intermediate in formation of CH-1 or CH-2. Results shown are representative of three repetitions of this experiment.
Phenotypic rescue of variant cytotoxic properties by alternative amino acid substitution.
To assess the nature of the cytotoxicity attenuation observed for the F91C, F95C, and G103C variants, additional rCPE variants were constructed and assayed for cytotoxicity. When the F91 and F95 residues of C186A were changed to tryptophan, the resultant F91W and F95W variants both showed significantly more cytotoxicity than the F91C and F95C variants (Fig. 7). When tyrosine substitutions were introduced into residues F91 or F95 of C186A, the resultant variants were less cytotoxic than the corresponding F91W or F95W variant. Similarly, when G103 in the C186A variant was changed to alanine or serine, the G103S variant (but not the G103A variant) showed significantly more cytotoxicity than the G103C variant.
Fig 7.

Phenotypic rescue of Cys substitution mutants. Residues F91 and F95 of the C186A variant were changed to tryptophan or tyrosine, while residue G103C of the C186A variant was changed to alanine or serine. Confluent Caco-2 cells in 24-well plates were radiolabeled with 4 μCi/well of 86RbCl for 4 h at 37°C and then treated at 37°C with 10 μg/ml of purified 6× His-tagged rCPE species. After 15 min, 86Rb released into the medium was collected and quantified with a Cobra Quantum gamma counter. Data from this experiment were converted into percentage of maximal 86Rb release. The experiment was performed three times, each with duplicate samples. Error bars depict the standard errors. *, significant (P < 0.05) difference between the CPE variant and C186A, as determined using the Student t test; **, significant (P < 0.05) difference between the alternative substitution variant and the equivalent Cys substitution variant.
Analysis of these rCPE variants by limited trypsin digestion and large-complex formation showed that they had not undergone gross conformation changes and that they retained the ability to form large complexes (data not shown). Pronase protection assays indicated (data not shown) that the F91W and F95W variants were more resistant to pronase digestion than the corresponding variants containing Cys substitutions at these residues. The F91Y variant, but not the F95Y variant, was also more resistant to pronase degradation than the corresponding variants containing Cys substitutions at these two residues. Consistent with the Fig. 6 results indicating insertion of the G103C variant, both the G103A and G103S variants were relatively resistant to pronase (data not shown).
DISCUSSION
The largest subfamily of PFTs, which comprise 25 to 30% of all bacterial toxins, forms a β-barrel pore via a process that initiates with the binding of toxin monomers to a receptor (14). Those monomers then oligomerize on the membrane surface in a prepore. Once present in the prepore, each toxin monomer extends one or more β-hairpins into the membrane to form the β-barrel pore. Despite these general similarities, some mechanistic variations exist among β-barrel PFTs (14). For example, cholesterol-dependent cytolysins oligomerize in prepores consisting of ∼36 monomers and then insert two β-hairpins per toxin molecule to form their β-barrel pore (13, 14, 34). Consequently, the pores of cholesterol-dependent cytolysins are very large. In contrast, other pore-forming toxins, e.g., staphylococcal alpha toxin or the aerolysin toxin family, oligomerize in smaller prepores that usually consist only of toxin hexamers or heptamers; the toxin molecules in the prepore then extend a single β-hairpin into membranes to form considerably smaller pores than those made by the cholesterol-dependent cytolysins (14).
CPE has long been recognized as a pore-forming toxin (19, 20). Although it lacks significant primary sequence homology with other PFTs, recent analyses determined that CPE structurally resembles the aerolysin family of β-barrel PFTs (2, 15), a toxin family that includes two other clostridial toxins, namely, C. perfringens epsilon toxin and Clostridium septicum alpha toxin (24). Consistent with CPE belonging to the aerolysin-like toxin family, heteromer gel shift analyses indicated that CPE apparently oligomerizes in a hexameric prepore (26). In addition, CPE forms a relatively small pore similar in size to aerolysin toxin family members (19), rather than the large pore made by cholesterol-dependent cytolysins. Given these similarities between CPE and the aerolysin-like toxin family, it could be expected that CPE action should involve a single β-hairpin that participates in membrane insertion and pore formation.
Results from a previous study (33) had suggested that the TM1 region located between amino acids 81 and 106 comprises the β-hairpin of native CPE; i.e., an alternating pattern of hydrophobic and hydrophilic residues, which is characteristic of a β-hairpin, was identified in the TM1 region. Furthermore, this TM1 region matches the ∼30-amino-acid size of known β-hairpin loops mediating insertion in other β-PFTs, including some other aerolysin-like toxin family members, e.g., C. septicum alpha toxin (23). Further supporting TM1's role as a potential β-hairpin involved in pore formation, deletion of the TM1 region from rCPE produced a mutant protein that could bind and oligomerize into a prepore yet was noncytotoxic because it could not insert into membranes or form pores (33).
It was conceivable that those deletion mutagenesis results may have been due to a gross conformational change introduced into the TM1 mutant protein by deleting 30 amino acids. Although this seemed less likely, since the TM1 protein retained both the binding and oligomerization functions of rCPE, the current study included a Cys-scanning point mutagenesis analysis of the TM1 region since this mild approach was unlikely to introduce a gross conformational change. This mutagenesis work first required deleting the single Cys residue naturally present at residue 186 of CPE and then demonstrating that the resultant variant (C186A) retained rCPE-like toxicity. The full retention of rCPE activities by the C186A variant, as shown in this study, indicates that the Cys186 residue of CPE is not required for biologic activity, which is consistent with the absence, to date, of any biologic functions assigned to the CPE region containing the Cys186 residue.
The retention of biologic activity by the C186A variant allowed us to perform Cys-scanning mutagenesis on each residue located in the TM1 region of the C186A mutant. When tested on Caco-2 cells, three Cys variants with significantly reduced cytotoxicity were identified, i.e., the F91C, F95C, and G103C variants. Results obtained by characterizing, in depth, these three Cys variants with reduced cytotoxicity directly supported the functioning of CPE amino acids 81 to 106, i.e., the TM1 region, as a β-hairpin involved in β-barrel pore formation. Specifically, all three Cys variants with reduced cytotoxicity were able to bind and oligomerize similarly to the fully active C186A variant from which they were derived. However, none of these three Cys variants caused 86Rb release from Caco-2 cells equivalent to that caused by the C186A variant, indicating that these Cys variants were not capable of causing full C186A-like pore formation in host cells. Results from the trypsin sensitivity experiments suggested that the reduced cytotoxicity of these three Cys variants was not due to gross conformational disruptions.
Although the F91C, F95C, and G103C Cys variants each caused reduced 86Rb release from Caco-2 cells, there appears to be mechanistic diversity behind this effect. Once oligomerized into large complexes in Caco-2 cells, the F91C and F95C variants exhibited substantially more sensitivity to pronase challenge than did the C186A variant (or rCPE), strongly suggesting that these two Cys variants remained frozen in a prepore on the membrane surface due to their impaired membrane insertion abilities. However, after oligomerizing in Caco-2 cells, the G103C variant exhibited more limited pronase sensitivity, similar to the C186A variant or rCPE. This result suggests that the G103C variant is capable of inserting into membranes but apparently forms a pore with a defective channel. This represents the first evidence supporting the importance of the CPE region from amino acids 81 to 106 not only for membrane insertion but also as a participant in pore function, as would be expected if this CPE region is a β-hairpin involved in β-barrel pore formation and function.
This study reports the first analysis of contributions by specific TM1 region amino acids to CPE action. The most noteworthy finding concerned the G103 residue, which is predicted (Fig. 2) to reside within the CPE channel. Consistent with this modeling, the G103C variant apparently showed membrane insertion similar to that shown by the C186A variant yet was sharply attenuated for causing 86Rb release from Caco-2 cells. This result is consistent with the involvement of the G103 residue in CPE pore function. In contrast, the F91 and F95 residues of CPE should lie at or near the tip of the β-hairpin, where they might be involved in penetrating the membrane bilayer. Their location near the tip of the β-hairpin could explain the impaired ability of the F91C and F95C variants to penetrate into membranes, as suggested by the pronase sensitivity results obtained in the current study. Presumably, this reduced insertion ability accounts for the impaired pore formation observed for these two variants, as evident from their lower ability to cause 86Rb release from Caco-2 cells. This issue will be the subject of planned further studies.
To further understand the cytotoxic deficiencies of the F91C, F95C, and G103C variants shown in Fig. 3, attempts were made to restore cytotoxicity by introducing alternative amino acid substitutions into the C186A variant. Substituting tryptophan at residue 91 or 95 of C186A yielded variants with more cytotoxicity than the F91C or F95C variants, indicating that the presence of an aromatic ring at these positions is important for cytotoxicity. However, the F91Y variant and, particularly, the F95Y variant showed less cytotoxic ability than the corresponding F91W or F95W variant, suggesting that hydrophilicity at these residues is also important for cytotoxicity; this finding is consistent with the proposed structure of TM1, shown in Fig. 2, in which F91 and F95 are proposed to interact with membrane lipids. When residue 103 of C186A was changed to serine or alanine, the resultant G103S variant showed substantially more cytotoxicity than the G103C variant, while the cytotoxic properties of the G103C and G103A variants were similar. This result suggests that hydrophilicity at residue 103 can increase pore function; that conclusion is consistent with the Fig. 2 model and the Fig. 6 results obtained for the G103C variant that suggest that residue G103 lines the CPE pore, where it is in contact with the aqueous environment.
In summary, the current findings directly support the TM1 region functioning as a β-hairpin during CPE pore formation and also identify the first specific amino acid residues that appear to be important for CPE membrane insertion or pore function. Furthermore, these results also support the assignment of CPE to the aerolysin toxin family, which includes toxins known to utilize a β-hairpin for membrane insertion and pore formation. While the TM1 region shares no primary sequence homology with proven β-hairpins mediating insertion of other β-barrel pore-forming toxins, including those of the aerolysin toxin family, this is not particularly surprising given the considerable sequence diversity that exists among the proven β-hairpins mediating insertion of β-PFTs (14). However, it is notable that the β-hairpins of CPE and other aerolysin toxin family members reside in equivalent structural domains (2, 14, 15). Ongoing studies are now under way to use these Cys mutants with fluorescent sulfhydryl probes to further study CPE membrane insertion and pore formation.
ACKNOWLEDGMENT
This research was supported by grant R37-AI19844-29 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Published ahead of print 10 September 2012
REFERENCES
- 1. Bos J, et al. 2005. Fatal necrotizing enteritis following a foodborne outbreak of enterotoxigenic Clostridium perfringens type A infection. Clin. Infect. Dis. 15:78–83 [DOI] [PubMed] [Google Scholar]
- 2. Briggs DC, et al. 2011. Structure of the food-poisoning Clostridium perfringens enterotoxin reveals similarity to the aerolysin-like pore-forming toxins. J. Mol. Biol. 413:138–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carman RJ. 1997. Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea of man and other animals. Rev. Med. Microbiol. 8(Suppl 1):S43–S45 [Google Scholar]
- 4. CDC 2011, posting date Clostridium perfringens. CDC, Atlanta, GA: http://www.cdc.gov/foodborneburden/clostridium-perfringens.html [Google Scholar]
- 5. Chakrabarti G, McClane BA. 2005. The importance of calcium influx, calpain, and calmodulin for the activation of CaCo-2 cell death pathways by Clostridium perfringens enterotoxin. Cell. Microbiol. 7:129–146 [DOI] [PubMed] [Google Scholar]
- 6. Chakrabarti G, Zhou X, McClane BA. 2003. Death pathways activated in CaCo-2 cells by Clostridium perfringens enterotoxin. Infect. Immun. 71:4260–4270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fujita K, et al. 2000. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction membrane protein. FEBS Lett. 476:258–261 [DOI] [PubMed] [Google Scholar]
- 8. Gao Z, McClane BA. 2012. Use of Clostridium perfringens enterotoxin and the enterotoxin receptor-binding domain (C-CPE) for cancer treatment: opportunities and challenges. J. Toxicol. 2012:981626 doi:10.1155/2012/981626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hanna PC, Mietzner TA, Schoolnik GK, McClane BA. 1991. Localization of the receptor-binding region of Clostridium perfringens enterotoxin utilizing cloned toxin fragments and synthetic peptides. The 30 C-terminal amino acids define a functional binding region. J. Biol. Chem. 266:11037–11043 [PubMed] [Google Scholar]
- 10. Hanna PC, Wnek AP, McClane BA. 1989. Molecular cloning of the 3′ half of the Clostridium perfringens enterotoxin gene and demonstration that this region encodes receptor-binding activity. J. Bacteriol. 171:6815–6820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harada M, et al. 2007. Role of tyrosine residues in modulation of claudin-4 by the C-terminal fragment of Clostridium perfringens enterotoxin. Biochem. Pharmacol. 73:206–214 [DOI] [PubMed] [Google Scholar]
- 12. Horiguchi Y, Akai T, Sakaguchi G. 1987. Isolation and function of a Clostridium perfringens enterotoxin fragment. Infect. Immun. 55:2912–2915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hotze EM, Tweten RK. 2012. Membrane assembly of the cholesterol-dependent cytolysin pore complex. Biochim. Biophys. Acta 1818:1028–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iacovache I, Bischofberger M, van der Goot FG. 2010. Structure and assembly of pore-forming proteins. Curr. Opin. Struct. Biol. 20:241–246 [DOI] [PubMed] [Google Scholar]
- 15. Kitadokoro K, et al. 2011. Crystal structure of Clostridium perfringens enterotoxin displays features of beta-pore-forming toxins. J. Biol. Chem. 286:19549–19555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kokai-Kun JF, Benton K, Wieckowski EU, McClane BA. 1999. Identification of a Clostridium perfringens enterotoxin region required for large complex formation and cytotoxicity by random mutagenesis. Infect. Immun. 67:6534–6541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kokai-Kun JF, McClane BA. 1997. Deletion analysis of the Clostridium perfringens enterotoxin. Infect. Immun. 65:1014–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McClane BA, Robertson SL, Li J. Clostridium perfringens, p 465–489 In Doyle MP, Buchanan RL. (ed), Food microbiology: fundamentals and frontiers, 4th ed., in press ASM Press, Washington, DC [Google Scholar]
- 19. McClane BA. 1994. Clostridium perfringens enterotoxin acts by producing small molecule permeability alterations in plasma membranes. Toxicology 87:43–67 [DOI] [PubMed] [Google Scholar]
- 20. McClane BA. 1984. Osmotic stabilizers differentially inhibit permeability alterations induced in Vero cells by Clostridium perfringens enterotoxin. Biochim. Biophys. Acta 777:99–106 [DOI] [PubMed] [Google Scholar]
- 21. McClane BA, Wnek AP. 1990. Studies of Clostridium perfringens enterotoxin action at different temperatures demonstrate a correlation between complex formation and cytotoxicity. Infect. Immun. 58:3109–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McClane BA, Wnek AP, Hulkower KI, Hanna PC. 1988. Divalent cation involvement in the action of Clostridium perfringens type A enterotoxin. J. Biol. Chem. 263:2423–2435 [PubMed] [Google Scholar]
- 23. Melton JA, Parker MW, Rossjohn J, Buckley JT, Tweten RK. 2004. The identification and structure of the membrane-spanning domain of the Clostridium septicum alpha toxin. J. Biol. Chem. 279:14315–14322 [DOI] [PubMed] [Google Scholar]
- 24. Popoff MR. 2011. Epsilon toxin: a fascinating pore-forming toxin. FEBS J. 278:4602–4615 [DOI] [PubMed] [Google Scholar]
- 25. Robertson SL, Smedley JG, III, McClane BA. 2010. Identification of a claudin-4 residue important for mediating the host cell binding and action of Clostridium perfringens enterotoxin. Infect. Immun. 78:505–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robertson SL, et al. 2007. Compositional and stoichiometric analysis of Clostridium perfringens enterotoxin complexes in Caco-2 cells and claudin 4 fibroblast transfectants. Cell. Microbiol. 9:2734–2755 [DOI] [PubMed] [Google Scholar]
- 27. Sarker MR, Carman RJ, McClane BA. 1999. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol. Microbiol. 33:946–958 [DOI] [PubMed] [Google Scholar]
- 28. Singh U, Mitic LL, Wieckowski E, Anderson JM, McClane BA. 2001. Comparative biochemical and immunochemical studies reveal differences in the effects of Clostridium perfringens enterotoxin on polarized CaCo-2 cells versus Vero cells. J. Biol. Chem. 276:33402–33412 [DOI] [PubMed] [Google Scholar]
- 29. Singh U, Van Itallie CM, Mitic LL, Anderson JM, McClane BA. 2000. CaCo-2 cells treated with Clostridium perfringens enterotoxin form multiple large complex species, one of which contains the tight junction protein occludin. J. Biol. Chem. 275:18407–18417 [DOI] [PubMed] [Google Scholar]
- 30. Smedley JG, III, McClane BA. 2004. Fine-mapping of the N-terminal cytotoxicity region of Clostridium perfringens enterotoxin by site-directed mutagenesis. Infect. Immun. 72:6914–6923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smedley JG, III, Fisher DJ, Sayeed S, Chakrabarti G, McClane BA. 2004. The enteric toxins of Clostridium perfringens. Rev. Physiol. Biochem. Pharmacol. 152:183–204 [DOI] [PubMed] [Google Scholar]
- 32. Smedley JG, III, et al. 2008. Noncytotoxic Clostridium perfringens enterotoxin (CPE) variants localize CPE intestinal binding and demonstrate a relationship between CPE-induced cytotoxicity and enterotoxicity. Infect. Immun. 76:3793–3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smedley JG, III, Uzal FA, McClane BA. 2007. Identification of a prepore large-complex stage in the mechanism of action of Clostridium perfringens enterotoxin. Infect. Immun. 75:2381–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tweten RK. 2005. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immun. 73:6199–6209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Van Itallie CM, Betts L, Smedley JG, III, McClane BA, Anderson JM. 2008. Structure of the claudin-binding domain of Clostridium perfringens enterotoxin. J. Biol. Chem. 283:268–274 [DOI] [PubMed] [Google Scholar]
- 36. Veshnyakova A, et al. 2012. Mechanism of Clostridium perfringens enterotoxin interaction with claudin-3/-4 protein suggests structural modifications of the toxin to target specific claudins. J. Biol. Chem. 287:1698–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wieckowski E, Kokai-Kun JF, McClane BA. 1998. Characterization of membrane-associated Clostridium perfringens enterotoxin following pronase treatment. Infect. Immun. 66:5897–5905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wieckowski EU, Wnek AP, McClane BA. 1994. Evidence that an ∼50kDa mammalian plasma membrane protein with receptor-like properties mediates the amphiphilicity of specifically bound Clostridium perfringens enterotoxin. J. Biol. Chem. 269:10838–10848 [PubMed] [Google Scholar]