

Abstract
The type IV pilus of Neisseria meningitidis is the major factor for meningococcal adhesion to host cells. In this study, we showed that a mutant of N. meningitidis pilV, a minor pilin protein, internalized less efficiently to human endothelial and epithelial cells than the wild-type strain. Matrix-assisted laser desorption ionization–time of flight mass spectrometry and electrospray ionization tandem mass spectrometry analyses showed that PilE, the major subunit of pili, was less glycosylated at its serine 62 residue (Ser62) in the ΔpilV mutant than in the pilV+ strain, whereas phosphoglycerol at PilE Ser93 and phosphocholine at PilE Ser67 were not changed. Introduction of the pglL mutation, which results in complete loss of O-linked glycosylation from Ser62, slightly reduced N. meningitidis internalization into human brain microvascular endothelial cells, whereas the addition of the ΔpilV mutation greatly reduced N. meningitidis internalization. The accumulation of ezrin, which is part of the cytoskeleton ERM family, was observed with pilV+, ΔpglL, and pilE(S62A) strains but not with the ΔpilV mutant. These results suggested that whereas N. meningitidis pilin originally had an adhesive activity that was less affected by minor pilin proteins, the invasive function evolved with incorporation of the PilV protein into the pili to promote the N. meningitidis internalization into human cells.
INTRODUCTION
Neisseria meningitidis is a Gram-negative diplococcus pathogen that colonizes the human nasopharynx as a unique host. This organism subsequently spreads into the bloodstream, where it causes septicemia and induces meningitis when it passes through the blood-brain barrier (BBB) and reaches the cerebrospinal fluid (CSF). To reach the bloodstream or CSF, N. meningitidis has to adhere to and invade several epithelial and endothelial cells, including the BBB. Since there is no animal model for N. meningitidis infection (21), meningococcal pathogenesis has been mainly investigated using cultured human epithelial and endothelial cells (reviewed in reference 15).
Type IV pili, Opa, and Opc are abundant components of N. meningitidis involved in adherence to host cells (reviewed in references 4, 15, and 34). The N. meningitidis capsule and lipopolysaccharide participate in the resistance to host immune factors and the adhesion, respectively (4, 15, 34). Several minor adhesin or adhesin-like molecules have also been identified by analysis of meningococcal whole-genome sequences (4, 15, 34). In addition, fructose-1,6-bisphosphate aldolase, a cytoplasmic housekeeping enzyme, has been shown to function in N. meningitidis adhesion (14, 33). However, pili protruding from capsulated N. meningitidis are thought to be the most important determinants for interaction with host cells, since the capsule can sterically block the adhesive ability of the other surface-expressed adhesins (34).
Type IV pili are long, hair-like filaments that are helical polymers of one major subunit, pilin (PilE), produced by the N-terminal cleavage of prepilin by prepilin peptidase PilD (10). Although no high-resolution structure for meningococcal pili is available, the structure of gonococcal pili has been reported and shows that the pilus is a helix of pilin monomers with five pilin monomers per turn (9, 22). Several related proteins are associated with the pili of pathogenic neisseriae (reviewed in references 4 and 15). PilQ forms pores for pilus protrusion at the outer membrane, and PilF, PilM, PilN, PilO, and PilP participate in pilus synthesis. PilG, PilH, PilI, PilJ, PilK, and PilW have been suggested to be necessary for functional maturation of the pilus. PilC, which is located at the tip of pili in N. gonorrhoeae (25), is also incorporated into meningococcal pili (20, 36). The meningococcal PilC is encoded in two alleles, pilC1 and pilC2 (19, 20), but only PilC1 is required for adhesion to human cultured cells (20).
Several minor proteins (e.g., PilX, ComP, and PilV) are also incorporated in the neisserial pili and modulate its function (5, 6). In N. gonorrhoeae, ComP and PilV function in natural competence (2, 40) and adhesion to human epithelial cells (39), respectively. In N. meningitidis, PilV also functions in resistance to shear stress (e.g., blood flow) and induction of host actin rearrangement (18). Meningococcal PilX is essential for bacterial agglutination and adhesion to human umbilical vein endothelial cells (HUVEC) (12). In addition, based on X-ray crystal structure results, it has been suggested that surface-exposed motifs in PilX subunits stabilize bacterial aggregants against the disruptive force of pilus retraction (13).
Neisserial pili have been shown to undergo several kinds of posttranslational modification, which has been better characterized in N. gonorrhoeae than in N. meningitidis (reviewed in references 8 and 15). The gonococcal pilin is modified by phosphoethanolamine (PEA), phosphocholine (PC), phosphoglycerol (PG), and O-linked glycosylation. The glycosylation of N. gonorrhoeae PilE involves the serine 63 residue (referred to as Ser63 hereafter) and the disaccharide α-d-galactopyranosyl-(1-3)-2,4-diacetamid-2,4-dideoxy-β-d-glucopyranoside (9) and affects bacterial-cellular interaction (16). Modification of gonococcal PilE with PEA and PC is affected by PilV (1, 11), and a mutation in pilV has been reported to result in decreased pilus bundling in N. gonorrhoeae (39). Several modifications have also been reported in N. meningitidis PilE. Meningococcal PilE was thought to be N-glycosylated at Asp60 (37) but was later shown to be modified at Ser63 with the O-linked trisaccharide Gal(β1-4)-Gal(α1-3)-2,4-diacetamido-2,4,6-trideoxyhexose (DATDH) (17, 27). Glycosylation of PilE results in a small increase in N. meningitidis adhesion to human epithelial cells (17). It was also found that the meningococcal pilin is modified with PG at Ser93 (26) by pilin phosphotransferase B (PptB) (7) and that PG modification stimulated N. meningitidis detachment from host cells and dissemination in the host environment (7). A pilin phosphocholine transferase A (PptA) is found in N. meningitidis (38), but the PilE residue that is modified has not been determined.
We have previously shown that disease-associated N. meningitidis strains are highly infectious for human endothelial and epithelial cells in vitro (30), and these high infectious activities have been analyzed using signature tag mutagenesis (STM) (unpublished data). One STM mutant was defective in the GltT-GltM l-glutamate ABC transporter as previously reported (31). To further investigate the mechanism of N. meningitidis invasion of human cells, we also characterized other STM mutants and identified a mutant with an insertional mutation that inactivated the pilV gene. In the present study, we examined the effect of PilV on N. meningitidis infectious abilities to human cells and on the posttranslational modification of pilin.
MATERIALS AND METHODS
Bacterial growth.
N. meningitidis strains were stocked at −80°C and routinely grown on GC agar plates at 37°C in 5% CO2 (32). Escherichia coli was grown on L plates or in L broth liquid culture at 37 or 30°C. Antibiotics were added as required to the following concentrations: 5 μg of chloramphenicol/ml, 4 μg of erythromycin/ml, and 75 μg of spectinomycin/ml for N. meningitidis and 50 μg of ampicillin/ml, 150 μg of erythromycin/ml, and 75 μg of spectinomycin/ml for E. coli. All of the strains used in the present study are listed in Table 1.
Table 1.
Strains used in this study
| Strain | Genotype | Parent strain | Source or reference |
|---|---|---|---|
| N. meningitidis | |||
| NIID280 | Wild type | 30 | |
| HT1125 | ΔsiaBCD::kan | NIID280 | 30 |
| HT1588 | ΔsiaBCD::kan pilV::Tn-spc | HT1125 | This study |
| HT1688 | ΔsiaBCD::kan ΔpilV::ermC | HT1125 | This study |
| HT1694 | ΔsiaBCD::kan ΔpilV::ermC ggt::pilV+-cat | HT1688 | This study |
| HT1165 | ΔsiaBCD::kan pilE::ermC | HT1125 | 30 |
| HT1741 | ΔsiaBCD::kan pptB::spc | HT1125 | This study |
| HT1747 | ΔsiaBCD::kan ΔpilV::ermC pptB::spc | HT1740 | This study |
| HT1792 | ΔsiaBCD::kan pglL::ermC | HT1125 | This study |
| HT1793 | ΔsiaBCD::kan pglL::spc | HT1125 | This study |
| HT1797 | ΔsiaBCD::kan ΔpilV::ermC pglL::spc | HT1688 | This study |
| HT1744 | ΔsiaBCD::kan pilE+-cat | HT1125 | This study |
| HT1753 | ΔsiaBCD::kan pilE(S33A)-cat | HT1744 | This study |
| HT1754 | ΔsiaBCD::kan pilE(S44A)-cat | HT1744 | This study |
| HT1745 | ΔsiaBCD::kan pilE(S62A)-cat | HT1744 | This study |
| HT1746 | ΔsiaBCD::kan pilE(S67A)-cat | HT1744 | This study |
| HT1755 | ΔsiaBCD::kan pilE(S79A)-cat | HT1744 | This study |
| HT1756 | ΔsiaBCD::kan pilE(S93A)-cat | HT1744 | This study |
| HT1757 | ΔsiaBCD::kan pilE(S94A)-cat | HT1744 | This study |
| HT1758 | ΔsiaBCD::kan pilE(S106A)-cat | HT1744 | This study |
| HT1759 | ΔsiaBCD::kan pilE(S115A)-cat | HT1744 | This study |
| HT1760 | ΔsiaBCD::kan pilE(S149A)-cat | HT1744 | This study |
| HT1749 | ΔsiaBCD::kan pilE(S155A)-cat | HT1744 | This study |
| Escherichia coli | |||
| JM109 | endA1 gyrA96 hsdR17(rK− mK+) mcrB+ recA1 relA1 supE44 thi-1 Δ(lac-proAB) F′[traD36 proAB lacIqZΔM15] | TaKaRa Bio | |
| BL21(DE3) | F− ompT hsdSB (rB− mB−) gal dcm (DE3) | Invitrogen |
Tissue culture.
Human brain microvascular endothelial cells (HBMEC), HUVEC, and the human lung carcinoma epithelial cell line A549 were cultivated as described previously (30).
Determination of host cell-associated and internalized bacteria.
Infection assays using tissue culture cells were performed as described previously (31). Bacterial numbers were statistically compared by using a two-tailed Student t test.
Production of anti-PilV protein rabbit antiserum.
A 300-bp DNA fragment of the pilV gene in HT1125 (DDBJ accession no. AB698916), from which 30 amino acid residues at the N terminus were deleted, was amplified with the primers pilV-11 and pilV-10 (see Table S1 in the supplemental material) by PCR. The DNA fragment was digested with NsiI and XhoI and cloned into the NsiI and XhoI sites in the pET303 CT-His expression vector (Invitrogen), resulting in pHT822. Plasmid pHT822 was transformed into E. coli strain BL21(DE3) (Invitrogen), and the transformant was cultured in 250 ml of MagicMedia (Invitrogen) at 30°C overnight with shaking. The bacteria were harvested and suspended in 5 ml of B-PER reagent (Pierce), followed by incubation at room temperature for 10 min. The bacterial suspension was centrifuged at 13,000 rpm at 4°C for 30 min. The pellet was resuspended in 50 mM phosphate buffer (pH 7.0) containing 300 mM NaCl, 8 M urea, 10% glycerol, and 0.03% n-dodecyl-β-d-maltoside and then applied to a Ni-column. The bound proteins were eluted with 200 mM imidazole and separated by SDS-PAGE. The band corresponding to the PilV recombinant protein was excised from the gel and used to immunize in a rabbit.
Construction of meningococcal mutants.
To construct an N. meningitidis mutant with a pilV deletion, a 2.9-kb DNA fragment from N. meningitidis HT1125 chromosomal DNA containing the pilV gene (0.4 kb) and its upstream (1.7 kb) and downstream (0.8 kb) regions was amplified with the primers pilV-1 and pilV-2 (see Table S1 in the supplemental material) and cloned into the SmaI site of pMW119 (4 kb) (Nippon Gene) to construct pH 805 (6.9 kb). The 6.5-kb DNA region of pHT805, which did not contain the coding region of the pilV gene, was again amplified with the primers pilV-3 and pilV-4 (see Table S1 in the supplemental material) by PrimeSTAR Max DNA polymerase, digested with HpaI, and ligated to an erythromycin-resistant (Ermr) gene (ermC) to construct pH819. pH819 was linearized, and 500 ng was transformed into HT1125. An Ermr clone was selected, resulting in a pilV deletion mutant designated HT1688.
To construct an N. meningitidis pptB insertion mutant, a 1.4-kb DNA fragment from N. meningitidis HT1125 chromosomal DNA containing the pptB gene was amplified with the primers pptB-1 and pptB-2 (see Table S1 in the supplemental material) and cloned in pGEM-T Easy (Promega) to construct pHT864. The plasmid was digested with BssHII (the BssHII site was in the middle of the pglL gene) and blunt ended. The DNA fragment was ligated to a spectinomycin resistance (Spcr) gene (spc) to construct pHT866. pHT866 was linearized, and 500 ng of linearized was transformed into HT1125. A Spcr clone was selected, resulting in a pptB insertion mutant named HT1741.
To construct an N. meningitidis pglL insertion mutant, a 1.8-kb DNA fragment from HT1125 chromosomal DNA containing the pglL gene was amplified with the primers pglL-1 and pglL-2 (see Table S1 in the supplemental material) and cloned in pGEM-T Easy (Promega) to construct pHT904. pHT904 was digested with HpaI (the HpaI site was in the middle of the pglL gene) and ligated to an ermC or spc gene to generate pHT906 or pHT907, respectively. pHT906 and pHT907 were linearized, and 500 ng of each was transformed into HT1125. Ermr or Spcr clone was selected, resulting in a pglL insertion mutant designated HT1792 or HT1793, respectively.
To construct pilE mutants in which each serine residue was replaced with an alanine residue [pilE(S-A)], a 0.7-kb DNA fragment from HT1125 chromosomal DNA containing the wild-type pilE gene coding region, and its downstream region was amplified with both primers pilE-17 and pilE-20 (see Table S1 in the supplemental material) and primers pilE-17 and pilE-18 (see Table S1 in the supplemental material) by using PrimeSTAR Max DNA polymerase (TaKaRa Bio). A 0.7-kb DNA fragment of a chloramphenicol-resistant (Camr) gene (cat) without its own promoter was also amplified with primers cat-13 and cat-12 (see Table S1 in the supplemental material). These three DNA fragments were cloned in pGEM-3z (Promega) using an In-Fusion PCR cloning kit (Clontech) to construct a pilE+-cat transcriptional fusion plasmid designated pHT872. Site-directed mutagenesis to substitute alanine for each serine residue in PilE was performed with pHT872 as a template and the appropriate primers (see Table S1 in the supplemental material) by using a PrimeSTAR mutagenesis basal kit (TaKaRa Bio). The designations of the mutagenized plasmids are listed in Table S2 in the supplemental material. After each mutation was confirmed by nucleotide sequencing, each plasmid was linearized, 500 ng was transformed into N. meningitidis HT1125, and a Camr clone was selected. The replacement of the site-directed mutation in chromosomal pilE gene was confirmed by direct sequencing.
SDS-PAGE and Western blotting.
SDS-PAGE and Western blotting were performed as described previously (29).
TEM.
Negative-stained samples for transmission electron microscopy (TEM) were prepared as described previously (28). Electron microscopy was performed with Hitachi H-7500 transmission electron microscope (28).
Characterization of posttranslationally modified PilE and its proteolytic peptides.
Meningococcal pili were purified as described previously (13). In brief, N. meningitidis grown on three GC agar plates was scraped off the plates and suspended in 10 ml of 150 mM ethanolamine (pH 10.5). Pili were sheared off by vortexing at room temperature for 2 min. The bacteria were centrifuged at 12,000 × g for 10 min at 25°C and further ultracentrifuged at 50,000 × g for 50 min at 25°C. Then, 9 ml of the supernatant was mixed with 1 ml of 150 mM ethanolamine (pH 10.5) saturated with (NH4)2SO4, followed by centrifugation at 21,000 × g for 15 min at 25°C. The pellets were dissolved in 100 μl of 50 mM Tris-HCl (pH 7.5) and stored at −30°C.
The molecular masses of the purified pili were analyzed by liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) with the aid of LTQ linear ion trap MS (Thermo Fisher Scientific) and by matrix-assisted laser desorption ionization–time-of-flight mass spectrometry (MALDI-TOF MS) with a Voyager-DE STR mass spectrometer (Applied Biosystems). The MALDI-TOF spectrometer was operated in positive ion linear and reflector modes and was used with an accelerating voltage of 25 kV, a grid voltage at 92.5%, and a delay time of 350 ns. The pilus solution was initially purified using ZipTip C18 pipette tips (Millipore) and then mixed with an equal volume of α-cyano-4-hydroxycinnamic acid (10 mg/ml in 0.3% trifluoroacetic acid and 50% acetonitrile). A 1-μl sample was allowed to crystallize on the MALDI plate. Proteolytic samples were prepared as follows. For proteolytic samples, peptide samples were analyzed by SDS–15% PAGE and stained with Coomassie brilliant blue. The relevant bands were excised and treated with an in-gel tryptic digestion kit (Thermo Fisher Scientific) according to the manufacturer's protocol. Tryptic peptides were extracted from gel pieces, purified using ZipTip C18 pipette tips, and analyzed by LC-ESI-MS/MS and MALDI-TOF MS.
Determination of ezrin accumulation by immunostaining.
HBMEC were grown on a cover-glass at 37°C in 5% CO2 for 2 days. The confluent HBMEC monolayers were washed twice with phosphate-buffered saline (PBS) and then incubated with serum-free AM [AM(−S)] at 37°C in 5% CO2 for 16 h. The HBMEC monolayers were infected with N. meningitidis in AM(−S) at a multiplicity of infection of 500 for 4 h. The monolayers were washed four times with AM(−S), fixed with 4% paraformaldehyde in PBS for 15 min, and immunostained as described previously (31).
RESULTS
Effect of a mutation in the pilV gene on N. meningitidis infection of HBMEC and other human endothelial and epithelial cell lines.
During studies of a highly infectious N. meningitidis strain (30), an insertional mutant in the pilV gene of N. meningitidis HT1588 (pilV::Tn-spc) was found to reduce the invasion of HBMEC (Fig. 1A). To further investigate the function of PilV in N. meningitidis, the null mutant HT1688 (ΔpilV::ermC) and the ectopically complemented N. meningitidis HT1694 strain with wild-type pilV+ gene (ΔpilV::ermC ggt::pilV+-cat) were constructed (Fig. 1A). In vitro infection of HBMEC by N. meningitidis strains HT1125, HT1688, and HT1694 showed that the HT1688 ΔpilV mutant seemed to adhere about five times less efficiently to HBMEC than did the HT1125 pilV+ strain, but the differences were not statistically significant (Fig. 1B). However, the HT1688 ΔpilV mutant was internalized about 1,000 times less efficiently to HBMEC than was the HT1125 pilV+ strain (Fig. 1C). The deficiencies of the ΔpilV mutant on invasion were recovered to the levels of the pilV+ strain by the complementation of pilV+ gene (HT1694) (Fig. 1C). These results suggested that, while N. meningitidis PilV affected both adhesive and invasive steps in the infection of HBMEC, PilV might play a more significant role in inducing N. meningitidis internalization into host cells.
Fig 1.

Effect of mutation in the pilV gene on N. meningitidis infection of HBMEC. (A) Schematic of N. meningitidis strains with an insertion and deletion mutations in the pilV locus and ectopic complementation of the pilV+ gene at the ggt locus. (B and C) Adherence (B) and internalization (C) of N. meningitidis pilV mutants in HBMEC. The number of bacteria was measured as CFU. Internalized bacteria were determined as the gentamicin-resistant number of bacteria. Each value is the mean ± the standard error of the number of CFU per 104 cells in at least four experiments. The four strains shown in panel A were studied. The symbol under each bar shows the presence (+) or absence (−) of an active pilV gene in the strain studied. A “−/+” indicates the strain had an inactive pilV gene and an active ectopic pilV gene. (D and E) Adherence (D) and internalization (E) of pilV mutant in HBMEC, HUVEC, and A549 cells. Open, filled and gray bars indicate the bacterial number of HT1125, HT1688 and HT1694, respectively. Statistical analyses were performed with the Student t test (n.s., nonsignificant;*, P < 0.05).
The defect in infection of the ΔpilV mutant was also examined in another endothelial cell line, HUVEC, and in the epithelial cell line A549. The numbers of HT1688 bacteria internalized in HUVEC and A549 cells decreased to about 1/100 and 1/1,000 that of HT1125, respectively (Fig. 1E). These results indicated that PilV affected N. meningitidis internalization of human endothelial and epithelial cells.
Expression of PilV, PilE, and PilC and piliation levels in the ΔpilV mutant.
The possible effect of PilV on the levels of PilE (pilin) and PilC proteins was examined by Western blotting of whole-cell extracts (Fig. 2A) and purified pili (Fig. 2B). PilV protein was detected in whole-cell extracts of strains HT1125 (pilV+) and HT1694 (ΔpilV::ermC ggt::pilV+-cat) but not in strain HT1588 or strain HT1688 (Fig. 2A, lanes 1 to 4), indicating that PilV expression was complemented in HT1694. There was not a large difference in the amount of PilE and PilC proteins in the extracts of these four strains (Fig. 2B and C, lanes 1 to 4). In addition, Western blotting of purified pili showed that the amount of PilC relative to that of PilE was not significantly changed by the pilV mutation (Fig. 2E and F, lanes 1 and 2). These results indicated that the levels of PilE and PilC proteins in meningococcal pili were not affected by the ΔpilV mutation.
Fig 2.

(A to C) Western blotting for PilV (A), PilE (B), and PilC (C) proteins in N. meningitidis whole-cell extracts. Bacterial extracts equivalent to an optical density at 600 nm of 0.01 for PilV, 0.0025 for PilE, and 0.05 for PilC were analyzed by Western blotting. Lane 1, HT1125; lane 2, HT1588 (pilV::Tn-spc); lane 3, HT1688 (ΔpilV::ermC); lane 4, HT1694 (ΔpilV::ermC ggt::pilV+-cat); lane 5, HT1792 (pglL::ermC); lane 6, HT1745 [pilE(S62A)]; and lane 7, HT1756 [pilE(S93A)]. The pilE::ermC strain HT1165 (lane 5 in panel B) and strains HT1178 (pilC1::spc), strain HT1182 (ΔpilC2::ermC), and the double mutant HT1186 (pilC1::spc ΔpilC2::ermC) (lanes 8 to 10 in panel C) were used as controls for anti-PilE rabbit serum (32) and anti-PilC rabbit serum (30), respectively. Black arrows indicate PilV, PilE, and PilC proteins, and gray arrows indicate nonspecific bands used as internal controls. (D to F) Western blotting for PilV (D), PilE (E), and PilC (F) proteins in purified N. meningitidis pili. Lane 1, HT1125; lane 2, HT1688 (ΔpilV::ermC); lane 3, HT1792 (pglL::ermC). A 2.5-μl sample of purified pili was analyzed for PilC, and 8-fold- and 20-fold-diluted samples were analyzed for PilV and PilE, respectively. Black arrows indicate PilV, PilE, and PilC proteins. (G to K) Electron micrographs show piliation of N. meningitidis strains (G) HT1125 (wild-type), (H) HT1688 (ΔpilV), (I) HT1792 (pglL::ermC), (J) HT1745 [pilE(S62A)] and (K) HT1756 [pilE(S93A)]. Upper and lower panels show magnifications of 20,000 and 50,000, respectively. Scale bars shown in black (upper panels) and white (lower panels) indicate 100 and 200 nm, respectively.
The level of piliation was also examined by electron microscopy. About 6 to 12 pili and pilus bundles were found in HT1125 (pilV+) (Fig. 2G), with a similar level of piliation observed in HT1688 (ΔpilV) (Fig. 2H). The results indicated that the ΔpilV mutation did not affect the piliation level in N. meningitidis, a finding that was consistent with previous reports on N. meningitidis (3, 18) and N. gonorrhoeae (39).
Posttranslational modification of pilin in the pilV mutant.
Mutation of the N. gonorrhoeae pilV gene has been reported to affect posttranslational modification of PilE at the Ser68 (11). To determine the effect of the pilV mutation on posttranslational modification in N. meningitidis, intact pili were purified from strains HT1125 (pilV+) and HT1688 (ΔpilV::ermC) and analyzed by MALDI-TOF MS (Fig. 3). Pili from strain HT1125 yielded several main peaks with masses from 17,300 to 17,800 (the main peaks are marked with and asterisk in Fig. 3) and a small secondary peak with a mass of 16,828 (Fig. 3A). The same pattern of peaks with masses from 17,300 to 17,800 was also observed in pili from HT1688, but the peak sizes were reduced (Fig. 3B). Intact pili from HT1792 (pglL::spc), which was defective in a trisaccharide transferase for pilin (24), were also analyzed by MALDI-TOF MS. The main peaks with masses from 17,300 to 17,800 seen in pili from strains HT1125 and HT1688 were not observed in pili from strain HT1792 (Fig. 3C). From the PilE primary structure (Fig. 4A) and its reported modification (7, 26), the peak with a mass of 16,828 may be PilE monomers modified with PG (Fig. 3). These results implied that some kinds of posttranslational modification of PilE, including glycosylation, were affected by the ΔpilV mutation in N. meningitidis.
Fig 3.

Whole-protein MS analysis of meningococcal pili. Intact mass analysis of pili purified from strains HT1125 (pilV+) (A), HT1688 (ΔpilV::ermC) (B), and HT1792 (pglL::ermC) (C) by MALDI-TOF MS was performed. Peaks altered by ΔpilV and pglL mutations are marked with an asterisk. The observed mass of 16,828 of the marked peak is in agreement with the calculated mass of the PilE monomer modified with PG, which was suggested by the primary structure and reported modification.
Fig 4.

(A) Putative amino acid sequence of N. meningitidis strain HT1125 PilE (DDBJ accession no. AB698857). The seven N-terminal amino acid residues, shown in gray, are lost in mature pilin, which has been confirmed by analysis of the N-terminal amino acid sequence (data not shown). Serine residues are indicated in boldface, and posttranslationally modified serine residues are shown in color with vertical arrowheads. The tryptic peptides analyzed by LC-ESI-MS/MS and MALDI-TOF MS are shown in red and blue boxes. (B) LC-ESI-MS/MS of a tryptic peptide (calculated m/z = 1,294.6 [M+3H]3+; observed m/z = 1,295.31 [M+3H]3+) of PilE from HT1125. The tandem mass spectrum of the peptide 44SAVTEYYLNHGIWPANNNS*AGVAS**TATDIK73 (S* = glycosylated serine, S** = serine modified with PC) is shown. (C) LC-ESI-MS/MS of a tryptic peptide (calculated m/z = 959.79 [M+3H]3+, observed m/z = 960.36 [M+3H]3+) of PilE from HT1125. The tandem mass spectrum of the peptide 76YVQSVTVANGVVTAQMAXSNVNNKEIK101 (X = serine modified with PG) is shown. The sequence can be read from the annotated b (blue) or y (red) ion series; in either case, some b and y ions were not observed.
Identification of the posttranslational modifications of PilE in N. meningitidis pilV+ strain.
Posttranslational modification of wild-type pili was determined by LC-ESI-MS/MS analysis of tryptic peptides from N. meningitidis HT1125 (pilV+) pili. The data indicated a modified peptide with an observed mass of 1,295.31 [M+3H]3+, which was in good agreement with a calculated mass (1,294.6 [M+3H]3+) for the peptide 44SAVTEYYLNHGIWPANNNSAGVASTATDIK73 modified with a trisaccharide composed of Gal(β1-4)-Gal(α1-3)-2,4-DATDH and PC (Fig. 4B), from ESI-MS analysis. Further sequencing of the peptide by LC-ESI-MS/MS showed that the Ser62 and Ser67 were modified with the trisaccharide and PC, respectively (Fig. 4B). It was interesting that another modified peptide with an observed mass of 1,187.32 [M+3H]3+, which agreed well with a calculated mass (1,186.57 [M+3H]3+) for the peptide 44SAVTEYYLNHGIWPANNNSAGVASTATDIK73 modified with PC at Ser67 and a monosaccharide composed of DATDH at Ser62, was also found (data not shown). Since the peptide with a monosaccharide was separated from the peptide with a trisaccharide by LC analysis (data not shown), some PilE in N. meningitidis HT1125 may be modified at Ser62 with a trisaccharide and some may be modified with a monosaccharide. In addition, since a peptide with an observed mass of 960.36 [M+3H]3+, which agreed with a calculated mass (959.79 [M+3H]3+) for the peptide 76YVQSVTVANGVVTAQMASSNVNNKEIK101 modified with PG was also found, PilE Ser93 was shown to be modified with PG (Fig. 4C).
Therefore, LC-ESI-MS/MS analysis showed that N. meningitidis HT1125 PilE was posttranslationally modified with a trisaccharide or monosaccharide at Ser62, PC at Ser67, and PG at Ser93 (Fig. 4A). These results were almost consistent with those of previous reports (17, 26, 38).
Glycosylation at PilE Ser62 in the ΔpilV mutant.
To further examine the effect of the ΔpilV mutation on O-glycosylation, proteolytic digests of HT1125 and HT1688 PilE were analyzed by MALDI-TOF MS. Five peaks with masses around 3,170, 3,380, 3,560, 3,730, and 3,880 were obtained from both HT1125 and HT1688 PilE peptides (Fig. 5A and B). This result was confirmed by LC-ESI-MS/MS analysis, showing that the peaks with masses of 3,560 and 3,880 corresponded to peptides modified at Ser62 with a monosaccharide and a trisaccharide, respectively (Fig. 5A and B and data not shown). Since peptides corresponding to the other three peaks (marked with asterisks in Fig. 5) were not found by MASCOT analysis (data not shown), these three peaks were probably degradation products generated during MS analysis (Fig. 5). The two peaks with masses of 3,560 and 3,880 were not observed in tryptic digests of PilE from N. meningitidis HT1753 [pilE(S62A)] and HT1792 (pglL::spc) (Fig. 5C and D), showing that PilE was not glycosylated in these two mutants. The intensity of the two peaks with masses of 3,560 and 3,880 in PilE peptides from HT1688 was much lower than PilE peptides from HT1125 (Fig. 5A and B). The peak reductions, including probable degradation products, were almost restored in PilE peptides from HT1694 (ΔpilV::ermC ggt::pilV+-cat) (Fig. 5E). The MALDI-TOF MS results suggested that both monosaccharide and trisaccharide O-glycosylation of PilE Ser62 were reduced similarly in the N. meningitidis ΔpilV mutant.
Fig 5.

MALDI-TOF MS of tryptic peptides of PilE purified from strains HT1125 (wild type) (A), HT1688 (ΔpilV::ermC) (B), HT1753 [pilE(S62A)] (C), HT1792 (pglL::ermC) (D), and HT1694 (ΔpilV::ermC ggt::pilV+-cat) (E). The peaks with an asterisk are considered degradation products generated during MS analysis (see the text).
Effect of ΔpilV mutation on modulation of PilE Ser67 and Ser93.
The effect of the ΔpilV mutation on posttranslational modification of PilE with PC was studied using pili from N. meningitidis strains with a pglL genetic background to eliminate the complicated mass peaks. MALDI-TOF MS analysis of proteolytic digests of pili from pglL and pglL pilV mutants showed the same intensity for the peak with a mass of 3,330 (see Fig. S1 in the supplemental material), suggesting that the ΔpilV mutation did not affect posttranslational modification of PilE at Ser67 with PC.
The effect of the ΔpilV mutation on posttranslational modification with PG was also analyzed by MALDI-TOF MS. A peak with a mass of 2,724 corresponding to a peptide 76YVQSVTVANGVVTAQMASSNVNNKEIK101 with no modification was detected in PilE from HT1125 (see Fig. S2A in the supplemental material). A peak with a mass of 2,880, a mass increase that corresponded to a 76-Da PG, was not found in pilin from pptB and pilE(S93A) mutants (see Fig. S2C and D in the supplemental material). Although the peak with a mass of 2,880 was slightly higher than the peak with a mass of 2,724 in the ΔpilV mutant, there was no striking difference between the pilV+ and ΔpilV strains (see Fig. 2A and B in the supplemental material). These results suggested that the pilV mutation did not affect the posttranslational modification of PilE with PG (see Discussion).
Effect of pilE substitution mutations on meningococcal infectious activities.
Neisserial pilin is posttranslationally modified at serine (11, 22, 26, 27), and there are 11 serine residues in the PilE of HT1125 N. meningitidis strains (Fig. 4A). To analyze the effect of posttranslational modification of PilE on N. meningitidis infection of HBMEC, a series of pilE mutant in which alanine was substituted for each of the serine residues [pilE(S-A) mutants] was constructed. The amount of PilC in whole-cell extracts of pilE(S62A) and pilE(S93A) mutants (Fig. 2C, lanes 6 and 7) was similar to that in the pilE+ wild-type strain (Fig. 2C, lane 1). The amount of PilC protein in both whole-cell extracts and purified pili from the pglL mutant was also largely unchanged (Fig. 2E, lane 7, and Fig. 2F, lane 3). TEM analysis showed that the level of piliation in N. meningitidis pilE(S62A), pilE(S93A), and pglL mutants was similar to that in the wild-type HT1125 strain (Fig. 2G, I, J, and K). These results suggested that posttranslational modification of PilE by O-glycosylation and with PG did not affect the level of pilin-related protein(s) and piliation in N. meningitidis.
The infectious activities of the N. meningitidis pilE(S-A) mutants were examined by in vitro infection of HBMEC (Fig. 6). All of the pilE(S-A) mutants, except the pilE(S93A) mutant, adhered to and internalized in HBMEC as efficiently as the wild-type pilE+ strain. The pilE(S93A) mutant adhered ∼5 times less and internalized ∼50 times less to HBMEC than did the pilE+ strain (Fig. 6). A similar result was found with the pptB mutant (see Fig. S3 in the supplemental material). These results implied that PG modification of PilE affected N. meningitidis adhesion and invasion of human cells (see Discussion).
Fig 6.

(A and B) Adherence (A) and internalization (B) of pilE(S-A) N. meningitidis mutants to HBMEC. The number of bacteria was measured as CFU. Internalized bacteria were determined as the number of gentamicin-resistant bacteria. Each value is the mean ± the standard error of the number of CFU per 104 cells in at least four experiments. The bars show data for strains HT1125 (pilV+ pilE+), HT1688 (ΔpilV::ermC pilE+), and HT1744 (pilV+ pilE+-cat) and the pilE(S-A) substitution mutants. Statistical analyses were performed with the Student t test (n.s., nonsignificant; *, P < 0.05).
Infectious activity of the pglL mutant.
To examine the effect of a reduction in O-glycosylation in PilE on N. meningitidis infection, N. meningitidis pglL mutants were analyzed by in vitro infection of HBMEC (Fig. 7). The N. meningitidis HT1792 pglL mutant was internalized ∼10 times less efficiently in HBMEC than the HT1125 wild-type strain (pglL+) (Fig. 7B). However, the HT1797 pilV pglL double mutant was internalized almost 100 times less efficiently in HBMEC than the HT1792 pglL strain (Fig. 7B). The results suggested that, while the decrease in invasion in the pilV mutant was partly due to the reduction in PilE glycosylation, the decrease was largely due to the absence of PilV protein in meningococcal pili (see Discussion).
Fig 7.

(A and B) Adherence (A) and internalization (B) of N. meningitidis ΔpilV and pglL mutants to HBMEC. The number of bacteria was measured as CFU. Internalized bacteria were determined as the number of gentamicin-resistant bacteria. Each value is the mean ± the standard error of the number of CFU per 104 cells in at least four experiments. The open, filled, gray, and hatched bars indicate the numbers of bacteria for strains HT1125 (pilV+ pglL+), HT1688 (ΔpilV::ermC pglL+), HT1792 (pilV+ pglL::ermC), and HT1797 (ΔpilV::ermC pglL::spc), respectively. Statistical analyses were performed with the Student t test (n.s., nonsignificant; *, P < 0.05).
Ezrin accumulation in HBMEC infected with N. meningitidis mutants.
Host cell cytoskeleton rearrangement in HBMEC after N. meningitidis infection was studied by monitoring the localization of ezrin by indirect immunofluorescence. Ezrin was widely distributed throughout noninfected cells (Fig. 8B). Immunofluorostaining of ezrin and simultaneous observation of bacteria by phase-contrast microscopy showed that ezrin was condensed at the site of bacterial attachment in HBMEC infected with pilV+ strain (Fig. 8C and D). Similar condensation was observed in cells infected with pglL and pilE(S62A) mutants and pilV+-complemented ΔpilV mutant (Fig. 8G, H, I, J, K, and L). In contrast, ezrin condensation was not observed in cells infected with the N. meningitidis ΔpilV mutant (Fig. 8E and F). These results indicated that the presence of PilV in meningococcal pili, rather than O-glycosylation of PilE, was required for bacterium-induced reorganization of the host cell cytoskeleton in N. meningitidis infection (see Discussion).
Fig 8.
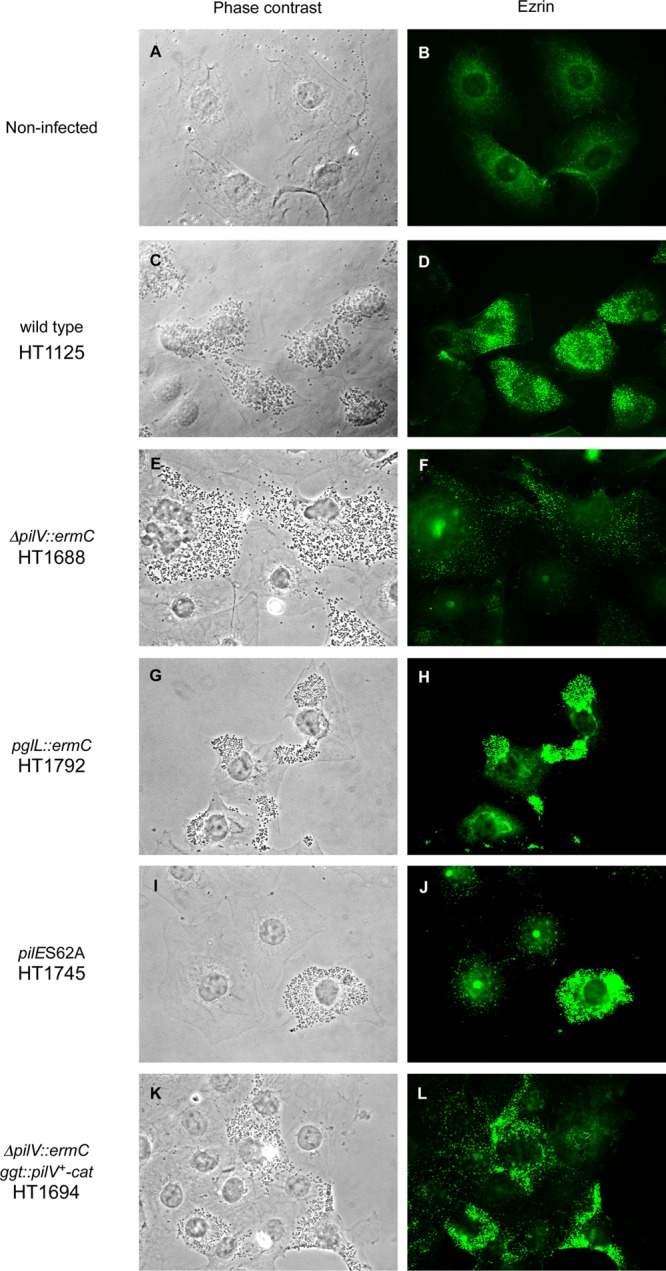
Phase-contrast and immunofluorescence microscopy showing ezrin accumulation in N. meningitidis-infected HBMEC. (A and B) Noninfected HBMEC monolayer. HBMEC monolayers were infected with strains HT1125 (wild type) (C and D), HT1688 (ΔpilV::ermC) (E and F), HT1792 (pglL::ermC) (G and H), HT1745 [pilE(S62A)] (I and J), and HT1694 (ΔpilV::ermC ggt::pilV+-cat) (K and L), respectively. Bacteria and HBMEC were observed by phase-contrast microscopy (left panels), and ezrin was immunostained with anti-ezrin monoclonal antibody and Alexa Fluor 488-conjugated rabbit anti-mouse IgG (right panels).
DISCUSSION
It has been reported that neisserial pilin is modified posttranslationally (11, 22, 26, 27) and that a mutation in the pilV gene changed the modification at PilE Ser68 from PEA to PC in N. gonorrhoeae (11). However, the effect of the pilV mutation on posttranslational modification of PilE and the role of PilV in meningococcal infection of host cells have been unclear. To our knowledge, this is the first report that the pilV mutation in N. meningitidis affected meningococcal invasion of human endothelial and epithelial cells. It is also the first demonstration that O-glycosylation of PilE at Ser62 was reduced by the ΔpilV mutation in N. meningitidis, while modifications of PilE Seri93 with PG and Ser67 with PC were not changed in the ΔpilV mutant.
It has been reported that the glycosylation of pilin could affect the meningococcal adhesion to human endothelial and epithelial cells (17, 37) and gonococcal invasion of primary human cervical epithelial cells (16). These results implied that the reduction of infectious abilities in N. meningitidis pilV mutants was due to a decrease in O-glycosylation at PilE Ser62. However, in HBMEC infected by N. meningitidis strains, pilV mutants internalized about 1/1,000 less efficiently than the wild-type pilV+ strain (Fig. 1C), while the pglL mutant showed a 10-fold reduction in invasive ability compared to the pglL+ strain (Fig. 7). In addition, the N. meningitidis pilE(S62A) mutant infected HBMEC as efficiently the wild-type pilE+ strain (Fig. 6), and ezrin accumulated beneath pilE(S62A) and pglL N. meningitidis mutants very efficiently (Fig. 8C and D). From these results, the reduction of infectious activities of the N. meningitidis ΔpilV mutant may be mostly due to the absence of the PilV protein rather than to the reduction of O-glycosylation in PilE. This hypothesis would be consistent with the result that all of the N. meningitidis pilE(S-A) mutants, except the pilE(S93A) mutant, infected HBMEC as efficiently as the wild-type pilE+ strain (Fig. 6).
How does PilV affect pilin and thereby the infectious abilities of N. meningitidis? It seems likely that the incorporation of PilV in N. meningitidis pili may lead to a conformational change in pilin that opens a currently unidentified site (including O-glycosylated Ser62) to stimulate host cell cytoskeleton rearrangement. Since the defect in adherence in the N. gonorrhoeae missense pilE mutants was suppressed by a mutation in the associated gene pilT (23), a similar effect could have occurred between PilE and an associated protein(s) in neisserial pili. However, we cannot exclude the possibility that PilV in N. meningitidis pili acts as a ligand to stimulate host cell cytoskeleton rearrangement, and the mechanism of enhancement of meningococcal infection by PilV will be further examined.
The N. meningitidis pptB mutant was not internalized into HBMEC (see Fig. S3 in the supplemental material). The same result was observed for the N. meningitidis pilE(S93A) mutant (Fig. 6). However, MS analysis of the tryptic peptide 76YVQSVTVANGVVTAQMASSNVNNKEIK101 from N. meningitidis pilin showed similar PG modification in N. meningitidis pilV+ and ΔpilV strains (see Fig. S3 in the supplemental material). The results suggested that N. meningitidis invasion mediated by PG modification of PilE was independent of PilV. In addition, ezrin accumulated less efficiently with N. meningitidis pptB and pilE(S93A) mutants than with the wild-type strain but more efficiently with the ΔpilV mutant (Fig. 8 and see Fig. S3 in the supplemental material). These defects in N. meningitidis pptB and pilE(S93A) mutants implied that PG modification of PilE in N. meningitidis might modulate meningococcal invasion of human cells by an unknown mechanism independent of PilV.
It was reported that the N. meningitidis pptB and pilE(S93A) mutants internalized in the human intestinal epithelial line Caco-2 about half as efficiently as the wild-type strain and that pptB mutation produced a hyperadherent phenotype (7). However, in the present study, the N. meningitidis pilV mutants were largely defective in invasion and were not hyperadherent to human endothelial and epithelial cells (Fig. 1). These differences might be due to differences in the human cell lines, the experimental conditions, and the N. meningitidis strains. It should be noted that nonencapsulated N. meningitidis mutants were used in all of the experiments described here, whereas an encapsulated N. meningitidis strain was used in the other studies (3, 18). It is known that N. meningitidis adhesion and invasion of host cells is restricted by its capsule (28, 35), which has phase-variable expression in the natural habitat (34). Although it is not clear how N. meningitidis capsule might affect PilV-mediated infection, a conformational change in PilE due to PilV would be partially buried under the capsule layer, so that the effect of PilV on pilin in encapsulated N. meningitidis strains should be less than in nonencapsulated strains.
In our previous study, it was suggested that N. meningitidis internalization was experimentally distinguishable from adhesion to human endothelial and epithelial cells (31). This hypothesis was supported in the present study by the demonstration that invasion was reduced more than adhesion in infections with the N. meningitidis pilV mutants. Although pili have been known as the structures mediating N. meningitidis adhesion to host cells, the adhesive activity might be intrinsic and less affected by accessory proteins like PilV. However, the invasive activity could be extrinsic and modulated by accessory proteins. The adhesive and invasive activities in N. meningitidis pili may be independently regulated, and therefore, may function independently on infection of host cells. Since the GltT-GltM l-glutamate ABC transporter is involved only in N. meningitidis invasion (31), PilV-mediated invasion might function cooperatively with the GltT-GltM l-glutamate ABC transporter in N. meningitidis invasion of host cells.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kaori Otsuki, Masaya Usui, and Aya Abe (RIKEN BSI) for MS analyses and Michiyo Kataoka (National Institute of Infectious Diseases) for TEM analysis.
This study was supported by grants from the Ministry of Education, Science, and Culture of Japan (grants 16790265 and 24590545).
Footnotes
Published ahead of print 17 September 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Aas FE, et al. 2006. Neisseria gonorrhoeae type IV pili undergo multisite, hierarchical modifications with phosphoethanolamine and phosphocholine requiring an enzyme structurally related to lipopolysaccharide phosphoethanolamine transferases. J. Biol. Chem. 281:27712–27723 [DOI] [PubMed] [Google Scholar]
- 2. Aas FE, Lovold C, Koomey M. 2002. An inhibitor of DNA binding and uptake events dictates the proficiency of genetic transformation in Neisseria gonorrhoeae: mechanism of action and links to type IV pilus expression. Mol. Microbiol. 46:1441–1450 [DOI] [PubMed] [Google Scholar]
- 3. Brown DR, Helaine S, Carbonnelle E, Pelicic V. 2010. Systematic functional analysis reveals that a set of seven genes is involved in fine-tuning of the multiple functions mediated by type IV pili in Neisseria meningitidis. Infect. Immun. 78:3053–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carbonnelle E, et al. 2009. Meningococcal interactions with the host. Vaccine 27:B78–B89 [DOI] [PubMed] [Google Scholar]
- 5. Cehovin A, Kroll JS, Pelicic V. 2011. Testing the vaccine potential of PilV, PilX, and ComP, minor subunits of Neisseria meningitidis type IV pili. Vaccine 29:6858–6865 [DOI] [PubMed] [Google Scholar]
- 6. Cehovin A, et al. 2010. Sequence conservation of pilus subunits in Neisseria meningitidis. Vaccine 28:4817–4826 [DOI] [PubMed] [Google Scholar]
- 7. Chamot-Rooke J, et al. 2011. Posttranslational modification of pili upon cell contact triggers Neisseria meningitidis dissemination. Science 331:778–782 [DOI] [PubMed] [Google Scholar]
- 8. Chen A, Seifert HS. 2011. Interactions with host cells causes Neisseria meningitidis pili to become unglued. Front. Microbiol. 2:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Craig L, et al. 2006. Type IV pilus structure by cryo-electron microscopy and crystallography: implications for pilus assembly and functions. Mol. Cell 23:651–662 [DOI] [PubMed] [Google Scholar]
- 10. Freitag NE, Seifert HS, Koomey M. 1995. Characterization of the pilF-pilD pilus-assembly locus of Neisseria gonorrhoeae. Mol. Microbiol. 16:575–586 [DOI] [PubMed] [Google Scholar]
- 11. Hegge FT, et al. 2004. Unique modifications with phosphocholine and phosphoethanolamine define alternate antigenic forms of Neisseria gonorrhoeae type IV pili. Proc. Natl. Acad. Sci. U. S. A. 101:10798–10803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Helaine S, et al. 2005. PilX, a pilus-associated protein essential for bacterial aggregation, is a key to pilus-facilitated attachment of Neisseria meningitidis to human cells. Mol. Microbiol. 55:65–77 [DOI] [PubMed] [Google Scholar]
- 13. Helaine S, Dyer DH, Nassif X, Pelicic V, Forest KT. 2007. 3D structure/function analysis of PilX reveals how minor pilins can modulate the virulence properties of type IV pili. Proc. Natl. Acad. Sci. U. S. A. 104:15888–15893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Henderson B, Martin A. 2011. Bacterial virulence in the moonlight: multitasking bacterial moonlighting proteins are virulence determinants in infectious disease. Infect. Immun. 79:3476–3491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hill DJ, Griffiths NJ, Borodina E, Virji M. 2010. Cellular and molecular biology of Neisseria meningitidis colonization and invasive disease. Clin. Sci. 118:547–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jennings MP, Jen FE, Roddam LF, Apicella MA, Edwards JL. 2011. Neisseria gonorrhoeae pilin glycan contributes to CR3 activation during challenge of primary cervical epithelial cells. Cell Microbiol. 13:885–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marceau M, Forest K, Beretti JL, Tainer J, Nassif X. 1998. Consequences of the loss of O-linked glycosylation of meningococcal type IV pilin on piliation and pilus-mediated adhesion. Mol. Microbiol. 27:705–715 [DOI] [PubMed] [Google Scholar]
- 18. Mikaty G, et al. 2009. Extracellular bacterial pathogen induces host cell surface reorganization to resist shear stress. PLoS Pathog. 5:e1000314 doi:10.1371/journal.ppat.1000314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morand PC, Drab M, Rajalingam K, Nassif X, Meyer TF. 2009. Neisseria meningitidis differentially controls host cell motility through PilC1 and PilC2 components of type IV pili. PLoS One 4:e6834 doi:10.1371/journal.pone.0006834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nassif X, et al. 1994. Roles of pilin and PilC in adhesion of Neisseria meningitidis to human epithelial and endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 91:3769–3773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nassif X, So M. 1995. Interaction of pathogenic neisseriae with nonphagocytic cells. Clin. Microbiol. Rev. 8:376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parge HE, et al. 1995. Structure of the fibre-forming protein pilin at 2.6 Å resolution. Nature 378:32–38 [DOI] [PubMed] [Google Scholar]
- 23. Park HS, et al. 2001. Structural alterations in a type IV pilus subunit protein result in concurrent defects in multicellular behaviour and adherence to host tissue. Mol. Microbiol. 42:293–307 [DOI] [PubMed] [Google Scholar]
- 24. Power PM, Seib KL, Jennings MP. 2006. Pilin glycosylation in Neisseria meningitidis occurs by a similar pathway to wzy-dependent O-antigen biosynthesis in Escherichia coli. Biochem. Biophys. Res. Commun. 347:904–908 [DOI] [PubMed] [Google Scholar]
- 25. Rudel T, Scheurerpflug I, Meyer TF. 1995. Neisseria PilC protein identified as type-4 pilus tip-located adhesin. Nature 373:357–359 [DOI] [PubMed] [Google Scholar]
- 26. Stimson E, et al. 1996. Discovery of a novel protein modification: α-glycerophosphate is a substituent of meningococcal pilin. Biochem. J. 316:29–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stimson E, et al. 1995. Meningococcal pilin: a glycoprotein substituted with digalactosyl 2,4-diacetamido-2,4,6-trideoxyhexose. Mol. Microbiol. 17:1201–1214 [DOI] [PubMed] [Google Scholar]
- 28. Takahashi H, et al. 2008. Modification of lipooligosaccharide with phosphoethanolamine by LptA in Neisseria meningitidis enhances meningococcal adhesion to human endothelial and epithelial cells. Infect. Immun. 76:5777–5789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takahashi H, Hirose K, Watanabe H. 2004. Necessity of meningococcal γ-glutamyl aminopeptidase for the Neisseria meningitidis growth in rat cerebrospinal fluid (CSF) and CSF mimicking medium. J. Bacteriol. 186:244–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Takahashi H, Kim KS, Watanabe H. 2008. Differential in vitro infectious abilities of two common Japan-specific sequence-type (ST) clones of disease-associated ST-2032 and carrier-associated ST-2046 Neisseria meningitidis strains in human endothelial and epithelial cell lines. FEMS Immunol. Med. Microbiol. 52:36–46 [DOI] [PubMed] [Google Scholar]
- 31. Takahashi H, Kim KS, Watanabe H. 2011. Meningococcal internalization into human endothelial and epithelial cells is triggered by the influx of extracellular l-glutamate via GltT l-glutamate ABC transporter in Neisseria meningitidis. Infect. Immun. 79:380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takahashi H, Watanabe H. 2002. A broad-host-range vector of incompatibility group Q can work as a plasmid vector in Neisseria meningitidis: a new genetic tool. Microbiology 148:229–236 [DOI] [PubMed] [Google Scholar]
- 33. Tunio SA, et al. 2010. The moonlighting protein fructose-1,6-bisphosphate aldolase of Neisseria meningitidis: surface localization and role in host cell adhesion. Mol. Microbiol. 76:605–615 [DOI] [PubMed] [Google Scholar]
- 34. Virji M. 2009. Pathogenic neisseriae: surface modulation, pathogenesis and infection control. Nat. Rev. Microbiol. 7:274–286 [DOI] [PubMed] [Google Scholar]
- 35. Virji M, et al. 1992. Expression of the Opc protein correlates with invasion of epithelial and endothelial cells by Neisseria meningitidis. Mol. Microbiol. 6:2785–2795 [DOI] [PubMed] [Google Scholar]
- 36. Virji M, et al. 1995. Functional implications of the expression of PilC proteins in meningococci. Mol. Microbiol. 16:1087–1097 [DOI] [PubMed] [Google Scholar]
- 37. Virji M, et al. 1993. Pilus-facilitated adherence of Neisseria meningitidis to human epithelial and endothelial cells: modulation of adherence phenotype occurs concurrently with changes in primary amino acid sequence and the glycosylation status of pilin. Mol. Microbiol. 10:1013–1028 [DOI] [PubMed] [Google Scholar]
- 38. Warren MJ, Jennings MP. 2003. Identification and characterization of pptA: a gene involved in the phase-variable expression of phosphorylcholine on pili of Neisseria meningitidis. Infect. Immun. 71:6892–6898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Winther-Larsen HC, et al. 2001. Neisseria gonorrhoeae PilV, a type IV pilus-associated protein essential to human epithelial cell adherence. Proc. Natl. Acad. Sci. U. S. A. 98:15276–15281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wolfgang M, van Putten JP, Hayes SF, Koomey M. 1999. The comP locus of Neisseria gonorrhoeae encodes a type IV prepilin that is dispensable for pilus biogenesis but essential for natural transformation. Mol. Microbiol. 31:1345–1357 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.