

Abstract
The lung is the site of entry for Bacillus anthracis in inhalation anthrax, the deadliest form of the disease. Bacillus anthracis produces virulence toxins required for disease. Alveolar macrophages were considered the primary target of the Bacillus anthracis virulence factor lethal toxin because lethal toxin inhibits mouse macrophages through cleavage of MEK signaling pathway components, but we have reported that human alveolar macrophages are not a target of lethal toxin. Our current results suggest that, unlike human alveolar macrophages, the cells lining the respiratory units of the lung, alveolar epithelial cells, are a target of lethal toxin in humans. Alveolar epithelial cells expressed lethal toxin receptor protein, bound the protective antigen component of lethal toxin, and were subject to lethal-toxin-induced cleavage of multiple MEKs. These findings suggest that human alveolar epithelial cells are a target of Bacillus anthracis lethal toxin. Further, no reduction in alveolar epithelial cell viability was observed, but lethal toxin caused actin rearrangement and impaired desmosome formation, consistent with impaired barrier function as well as reduced surfactant production. Therefore, by compromising epithelial barrier function, lethal toxin may play a role in the pathogenesis of inhalation anthrax by facilitating the dissemination of Bacillus anthracis from the lung in early disease and promoting edema in late stages of the illness.
INTRODUCTION
The lung is the site of entry for Bacillus anthracis in inhalation anthrax, the deadliest form of the disease. Vegetative Bacillus anthracis is not known to cause initial disease in the lung (26). Rather, spores are internalized by resident lung cells, and vegetative bacteria subsequently disseminate to cause systemic disease. Regardless of anthrax disease type, in terminal stages of infection, humans exhibit acute lung injury (3, 62), possibly caused by lethal toxin (LT) produced by vegetative Bacillus anthracis in the blood (31). The pathogenesis and progression of inhalation anthrax depends strongly on the production of LT, a Zn+2-dependent metalloprotease, and edema toxin (ET), a calmodulin-dependent adenylate cyclase (42). LT and ET consist of the anthrax protective antigen (PA) in combination with lethal factor (LF) or edema factor (EF), respectively. PA binds a receptor on the target cell and provides a channel by which LT and ET enter the cytosol (42).
Although LT is an important virulence factor in human anthrax infections, the exact mechanism of action and cellular target of LT during infection are not understood. Macrophages have long been considered a primary target of LT in inhalation anthrax, and much attention has been focused on the relationship between macrophages and LT in anthrax infection and death in mice. LT lyses mouse macrophages and inhibits the innate immune response to Bacillus anthracis, making mice more susceptible to lethal infection. In addition, two recent reports show that mice lacking the capillary morphogenesis protein 2/anthrax toxin receptor 2 (CMG2/ANTXR2), specifically in myeloid cells, are resistant to Bacillus anthracis infection (34, 35), suggesting that myeloid sensitivity to Bacillus anthracis toxins plays some role in anthrax pathogenesis.
However, LT kills mice even if the mouse macrophages are depleted or made insensitive to LT, suggesting that LT-induced macrophage lysis is not directly responsible for death due to LT (23, 41, 42, 46). For example, even when mouse macrophages are rendered insensitive to LT by eliminating functional CMG2/ANTXR2 in their myeloid lineage, the mice are still sensitive to LT. These observations suggest that alveolar macrophages and other myeloid cells are not a required target for LT-induced lethality. Further evidence also supports the conclusion that alveolar macrophages are unlikely to be a primary target of LT. In a previous report, we showed that the main resident phagocyte in the human airway, the human alveolar macrophage (HAM), does not have a functional anthrax toxin receptor, does not bind PA, does not undergo MEK cleavage, and is not killed by Bacillus anthracis (64). Thus, HAM, unlike mouse macrophages, are not a direct cellular target for LT in the lung. As the lung is the portal of entry in inhalational anthrax and is affected in terminal disease, we hypothesize that a different cell in the lung is a target of LT and plays a role in death from inhalation anthrax.
Type I alveolar epithelial cells (AEC I) and type II AEC (AEC II) line the surface of the alveoli, the functional respiratory units of the lung. AEC II cells are precursors of AEC I, produce surfactant, and line ∼5% of the surface area of the alveoli. AEC I are terminally differentiated cells and cover the remaining ∼95% of the alveolar epithelial surface (15). We hypothesized that AEC may be targeted by LT, as alveolar edema is prominent later in disease and the alveolar epithelial barrier must be compromised in the initial stage of anthrax. There is a lack of studies of the effects of LT on human primary AEC. A study on primary human bronchiolar epithelial cells showed that cytoskeletal integrity was reduced after treatment with LT (32). However, primary human AEC are a better model for studies of Bacillus anthracis pathophysiology because edema, when it occurs in terminal disease, involves the alveolar structures and because the initial stages of inhalation anthrax also involve primarily this anatomic unit rather than the more-proximal bronchial airways. Anthrax toxin studies on human primary AEC have likely not been done because of practical considerations rather than a lack of scientific interest, i.e., human lung tissue is a limited resource, the AEC isolation procedures are lengthy (see Materials and Methods), and isolated cells are difficult to store for subsequent experiments (63). However, we decided to pursue this study because we had previous knowledge that primary human alveolar macrophages (HAM) differ significantly in their response to LT compared to cultured immortalized macrophage cell lines (e.g., A549 cells). Accordingly, we reasoned that the response of primary human AEC to LT might yield significant insights into anthrax pathophysiology that cannot be discovered using immortalized cell lines or other types of respiratory cells.
We found that LT exposure of primary human AEC caused MEK cleavage, actin cytoskeleton rearrangement, decreased expression and altered distribution of junction proteins, decreased barrier function, and decreased surfactant production without decreased viability. Our data suggest that the anthrax toxin receptors in the epithelium may play a role in both stages of inhalational anthrax by compromising the epithelial barrier function directly, i.e., through inhibition of barrier integrity (56) and through inhibition of surfactant production (66). These changes would be expected to favor alveolar collapse and edema formation in late-stage disease and, potentially, Bacillus anthracis escape from the alveoli in early stages of the illness.
MATERIALS AND METHODS
Isolation of primary pneumocytes. (i) Digestion.
Human lung was obtained from the National Disease Research Interchange (NDRI; Philadelphia, PA) or the International Institute for the Advancement of Medicine (IIAM; Jessup, PA) under a protocol approved by the University of Oklahoma Health Sciences Institutional Review Board. One lobe was perfused with 2 liters Plasma-Lyte A injection (Baxter Healthcare Corp., Deerfield, IL), lavaged with normal saline containing 100 Kunitz units (KU) of crude DNase from bovine pancreas (Sigma-Aldrich Co., St. Louis, MO) per mg protein. Tissue was digested with 8 U/ml porcine elastase (Worthington Biochemical Corp, Lakewood, NJ) and 150 KU/ml DNase in Hanks balanced salt solution (HBSS) without Ca2+, Mg2+, or phenol red (Mediatech, Inc., Manassas, VA). Following digestion, enzymes were inactivated by addition of HyClone fetal bovine serum (FBS) (Thermo Fisher Scientific, Inc., Waltham, MA) and 0.42% (wt/vol) DNase. The softened tissue was minced, and the slurry was passed sequentially through 1-ply gauze, 2-ply gauze, 150-μm mesh (Small Parts, Inc., Logansport, IN), and 30-μm mesh (17, 20, 24, 63).
(ii) Enrichment.
Following concentration in Dulbecco's modified Eagle medium (DMEM) containing 150 μg/ml DNase, cells were panned for 60 min at 37°C in flasks coated with human IgG. Unattached cells were pelleted and resuspended in DMEM-DNase solution and purified by a Percoll gradient (GE Healthcare Biosciences, Piscataway, NJ) formed by centrifugation of two layers with ρ values of 1.040 and 1.089. The predominant band was negatively selected against CD45+ Dynabeads (Invitrogen Corp., Carlsbad, CA) (12, 49). The average yield was 57 × 106 live, single cells with 91% viability. The identity of freshly isolated AEC II was identified by a modified Papanicolaou stain that colored lamellar bodies blue (12, 30).
(iii) Culture.
AEC were cultured on BioCoat plates (BD Biosciences, Franklin Lakes, NJ) at 37°C, 5% CO2, in a humidified tissue culture incubator in growth medium consisting of DMEM, 10% FBS, 1% solution of penicillin/streptomycin/amphotericin B (Mediatech, Inc., Manassas, VA), 5 μg/liter vancomycin, and 10 μg/liter ceftazidime (both from Sigma-Aldrich Co., St. Louis, MO). The medium was changed every 2 days. Cells were plated from 10 to 75,000 per cm2 on polypropylene dishes coated with 7.5 μg/cm2 rat tail collagen, type I (Sigma-Aldrich Co., St. Louis, MO), and 5 μg/cm2 fibronectin or on BioCoat plates with similar extracellular matrix (ECM). Cell density was optimized to drive differentiation toward a type I or type II phenotype and required additional adjustment with various culture plate sizes. Depending on the application, cells were harvested by scraping treatment with 10 U/cm2 dispase or Cellstripper (Mediatech, Inc., Manassas, VA) or fixed in situ for microscopy with 4% paraformaldehyde.
Freshly cultured cells were used for all experiments because freezing, storage, and thawing led to low recovery of viable cells. Cells were, therefore, limited in quantity, and the number of experimental conditions that were able to be performed for each trial was constrained.
Challenge with anthrax toxin or MEK pathway inhibitors.
At indicated time points, cultured cells were treated with fresh medium containing 2 μg/ml PA or 2 μg/ml LT (2 μg/ml PA plus 2 μg/ml LF), both from List Biological Laboratories, Inc., Campbell, CA. An equal volume of diluent was used as a negative control. Duration of treatment was 24, 72, or 144 h or as indicated. The final concentrations of the chemical inhibitors were 50 μM U0126 (suppresses the extracellular signal-regulated kinase [ERK] pathway), 50 μM SB203580 (suppresses p38), and 50 μM SP600125 (suppresses the Jun N-terminal protein kinase [JNK]), all from Sigma-Aldrich Co., St. Louis, MO. An equal volume of treatment diluent was used as a negative control. Duration of treatment varied from several hours to several days or was as indicated.
Flow cytometry.
Cells were prepared for flow cytometry as previously described (16, 24, 57). Secondary antibodies varied by fluorescent tag and were purchased from the same vendor as the corresponding primary antibodies when possible. AEC stained with annexin conjugates were used for compensation. Data were collected on a Becton Dickinson FACSCalibur cytometer by CellQuest Pro software and reduced by FlowJo analysis software, v. 7.2.5 (Tree Star, Inc., Ashland, OR).
Freshly isolated cells or those treated in culture were fluorescently stained by antibodies against the following surface markers: for characterization, rabbit α-human P2X7-FITC IgG (Sigma-Aldrich Co., St. Louis, MO), rabbit α-human P2X7 IgG (Santa Cruz Biotechnology Inc., Santa Cruz, CA), goat α-human GABAA Rπ IgG, and rabbit α-human GABAA Rπ IgG (both from Abcam, San Francisco, CA) (13); for efficiency of isolation, mouse α-human CD45-FITC IgG1 κ (clone H130) (13) (Becton, Dickinson, and Company, Franklin Lakes, NJ), mouse α-human CD31-PE IgG1 κ (clone L133.1; BD Biosciences), and mouse α-human Vimentin-PE IgG1 (clone RV202; Santa Cruz Biotechnology Inc.); for ANTXR, rabbit α-human TEM8 IgG (Abcam, San Francisco, CA) and goat α-human CMG2 IgG (R&D Systems, Inc., Minneapolis, MN). Protective antigen binding was assessed using fluorescein isothiocyanate (FITC)-labeled PA (List Biological Laboratories, Inc., Campbell, CA) following a 2.5-h incubation at 37°C. Isotype controls were normal rabbit IgG-FITC (Sigma-Aldrich Co., St. Louis, MO), rabbit F(ab′)2 IgG (Abcam, San Francisco, CA), normal goat IgG (R&D Systems, Inc., Minneapolis, MN), normal mouse IgG1-PE (Santa Cruz Biotechnology Inc.), and mouse IgG1 κ conjugated with FITC or phycoerythrin (PE) (both clone MOPC-21; BD Biosciences). The positive controls for AEC markers were Jurkat cells labeled with rabbit α-human P2X7-FITC IgG (Sigma-Aldrich) and pancreatic ductal adenocarcinoma cells (PL-45) labeled with goat α-human GABAA Rπ IgG (Abcam).
Immunofluorescent staining.
Except for early-culture α-human P2X7-FITC staining, all experiments were done on freshly isolated AEC that had been kept at least 10 days (late culture) in plasticware coated with ECM substrate before being fluorescently labeled with conjugated antibodies as previously described (20, 60). Briefly, cells were fixed in situ with 4% paraformaldehyde and then permeabilized with 0.1% Triton X-100. Following 3 5-min washes with phosphate-buffered saline plus 0.1% Triton X-100 (PBST), cells were blocked 60 min at room temperature with 2 μg/μl human IgG or PBST plus 10% FBS and 1% bovine serum albumin (BSA). Primary antibodies or isotype controls were diluted in the wash solution and incubated with the cells for 60 min at room temperature in a closed humidified container. Secondary antibody staining was performed similarly. The final washes were with PBS and 10% FBS. DAPI (4′,6-diamidino-2-phenylindole) was used as a nuclear counterstain. ProLong Gold antifade reagent (Invitrogen Corp., Carlsbad, CA) was added to protect the signal, and the wells were visualized on an Olympus BX51 fluorescence microscope (Olympus America Inc., Center Valley, PA).
(i) Characterization.
On the sixth day after plating, the following antibodies were used to help distinguish early-culture from late-culture phenotypes: primary rabbit α-human P2X7-FITC IgG (Sigma-Aldrich Co.), rabbit α-human P2X7 IgG (Santa Cruz Biotechnology Inc.), secondary donkey α-rabbit IgG (Invitrogen Corp., Carlsbad, CA), and normal rabbit IgG (Santa Cruz Biotechnology Inc.) for the isotype control.
(ii) Actin organization.
Cells were maintained in culture until day 14 in 12-well BioCoat plates. Duplicate wells were fed with fresh medium containing dimethyl sulfoxide (DMSO) diluent (NT), 2 μg/ml LT, a 50 μM mixture of the mitogen-activated protein kinase kinase (MAPKK; MEK) inhibitors UO126, SB203580, and SP600125. Treatment continued for 24, 72, or 144 h without a subsequent medium change. F-actin in the wells was immunofluorescently visualized with phalloidin-Alexa Fluor 555 (Invitrogen Corp.) (48).
(iii) Tight junction and adherens junction stability.
Day 14 AEC cultures were treated for 24, 72, or 144 h with either H2O diluent (NT) or 2 μg/ml LT. Two NT and 2 LT wells at each time point were labeled as described above with the following antibodies, all from Invitrogen Corp.: mouse α-human ZO-1 IgG (clone ZO1-1A12), mouse α-human β-catenin IgG (clone CAT-5H10), mouse α-human E-cadherin IgG (clone HECD-1), and Alexa Fluor 555-conjugated donkey α-mouse IgG. The medium was not changed after addition of the treatment.
Transmission electron microscopy (TEM).
To visualize the lamellar bodies in AEC, fresh cells plated into coated 60-mm polypropylene dishes were grown for 5 days before treatment with PA (2 μg/ml) or LT (2 μg/ml) for 72 h. To observe tight junction formation, cells grown for 14 days in 6-well BioCoat plates were also treated for an additional 72 h. After washing with 0.1 M sodium cacodylate-HCl buffer, pH 7.4, monolayers were fixed in buffer with 2% glutaraldehyde for 40 min at 4°C, followed by 3 wash/fix cycles for 10 min each at 4°C. Cells were prepared for electron microscopy (EM) as described previously (37). Briefly, samples were postfixed for 10 min with 1% OsO4 in cacodylate buffer, washed in the same buffer, treated with 1% tannic acid in 0.05 M cacodylate buffer for 30 min, and rinsed with 0.05 M cacodylate buffer containing 1% Na2SO4. Cells were scraped off the plastic, dehydrated as pellets in a graded series of ethanol solutions, and embedded in epoxy resin. The sections were briefly stained with uranyl acetate and lead citrate and examined with a Hitachi H-7600 electron microscope (Hitachi High Technologies America, Inc. Schaumburg, IL).
Transepithelial electrical resistance (TER).
Barrier integrity of confluent AEC monolayers was determined by the electric cell substrate impedance-sensing (ECIS) system 1600R from Applied BioPhysics Inc., Troy, NY. Cells were plated at 50,000/cm2 into two 8-well 0.8-cm2/well polystyrene culture arrays. Each well contained a 250-μm-diameter microelectrode that enabled measurement of the resistance in ohms created by the monolayer during growth and treatment. Prior to inoculation, the wells were coated with 250 μl/cm2 10 mM l-cysteine and 5 μg/cm2 human fibronectin. On days 4 to 6, culture impedance of each well was measured at 2,000, 4,000, and 8,000 Hz alternating current (AC) (25). The point of confluence for each well was determined to be reached when the resistance remained constant for at least 8 h, usually at days 10 to 13. Treatments consisted of 2 μg/ml PA, 2 μg/ml LT, 10 μM staurosporine (Enzo Biochem, New York, NY) as a positive control, or no treatment and continued for 140 to 144 h beyond confluence. Arrays were recovered and immunofluorescently stained with α-human ZO-1 and DAPI (22).
Quantitative reverse transcription-PCR (qRT-PCR).
Triplicate wells of a day 13 culture were treated with fresh medium containing DMSO diluent (NT), 2 μg/ml LT, or a 50 μM concentration of a mixture of the MEK inhibitors UO126, SB203580, and SP600125 for 3, 10, or 24 h. Wells were washed 2× with PBS and then harvested with the chaotropic salt-containing RLT buffer from the RNeasy Micro RNA isolation and purification kit (Qiagen Inc., Valencia, CA). The final quantity and quality of RNA from the lysates were determined by a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE) measurement. The average yield of total RNA from a single well of a 6-well plate inoculated with 20,000/cm2 was 4.9 μg ± 0.6 μg.
One microgram of RNA was used for reverse transcription by SuperScript III First-Strand Synthesis Supermix (Invitrogen, Inc.). The reaction plates were assembled by a Corbett, Inc. (San Francisco, CA), robot. The qPCR was performed on a Stratagene MX3005p (Agilent Technologies, La Jolla, CA). Efficiencies were determined using the standard curve method with 5-fold dilutions via the Stratagene MXpro software (v. 4.10). The data were imported into GenEx (v. 5.4.0.512; MultiD Analyses AB, Goteborg, Sweden). Preprocessing included normalization against ribosomal protein, large, P0.
Phospholipid measurement.
At day 4 of culture, AEC were left untreated, exposed to stimulant diluent as a negative control, or treated with LT (2 μg/ml) or PA (2 μg/ml). Phorbol 12-myristate 13-acetate (PMA; 50 ng/ml) was used as the positive control and high-dose dexamethasone (1 μM) was used as the negative control for surfactant production. Cell-free culture supernatants and cells were harvested on day 7. Cells were homogenized in a buffer consisting of 5 mM Tris-HCl, pH 7.5, 10 μg/ml pepstatin A (Sigma), 10 μg/ml leupeptin hemisulfate salt (Sigma), and 0.9 mM phenylmethanesulfonyl fluoride (Sigma). Cells were disrupted by a Vibra-Cell sonicator (Sonics and Materials, Inc., Newtown, CT) for 20 s at 25% power and stored at −80°C. Surfactant lipids were extracted from cell-free culture supernatants as described by Bligh and Dyer (5).
Phospholipid content was then determined in lipid extracts using the method of Stewart (55) against dipalmitoylphosphatidylcholine (DPPC) (Avanti Polar Lipids Inc., Alabaster, AL) standard solutions. Briefly, the lipid extracts and DPPC standard solutions were dried under compressed nitrogen gas and then dissolved in chloroform and mixed with 1 ml 2.7% ferric chloride and 3% ammonium thiocyanate in glass tubes. The mixture was vortexed for 1 min and centrifuged at 83 × g for 5 min. The bottom red layer of phospholipids and the ammonium ferrothiocyanate complex were collected, and absorbance was read at 488 nm. The phospholipid level in each culture supernatant was normalized to the protein concentration of its total cell lysate as measured by the bicinchoninic acid (BCA) protein assay microkit (Pierce, Thermo Fisher Scientific, In., Waltham, MA) against the bovine serum albumin (BSA) standard protein.
Isolation of human alveolar macrophages.
Human alveolar macrophages (HAM) were obtained by lavage of nontransplantable human lung with sterile normal saline. Cells were pelleted at 500 × g for 5 min and resuspended in sterile PBS. Viability was determined by trypan blue exclusion, total cell yield was determined by hemacytometer, and cell differential was determined by Diff-Quick staining (Baxter, Miami, FL). The cells obtained were >95% macrophages and were >90% viable. Alternatively, HAM were obtained by bronchoscopy with the signed informed consent of healthy human volunteers according to a protocol approved by the Oklahoma University Health Sciences Center Institutional Review Board and the Institutional Biosafety Committee as previously described (11, 64). HAM were collected in sterile saline solution and washed in sterile PBS. The cells from both collection methods were resuspended in sterile PBS to a final concentration of 2 × 106 macrophages/ml. After isolation of the HAM, cell lysates were prepared immediately for subsequent anthrax toxin receptor quantitation by immunoblotting.
Protein immunoblotting.
The junctional proteins, ZO-1, E-cadherin, and β-catenin, the desmosomal proteins, desmoplakin 1/2 and plakoglobin (γ-catenin), tumor endothelial marker 8 (TEM8)/ANTXR1, CMG2/ANTXR2, and MEK cleavage were assessed by a Western immunoblot. After 10 to 21 days in culture, the AEC were harvested for ANTXR determination or exposed to LT or to PA as described above. For MEK cleavage, AEC were exposed to LT in fresh medium for 24 h; for junction proteins, AEC were incubated with LT in fresh medium for 24, 48, 72 and 144 h; for desmosomal proteins, AEC were incubated with LT or PA in fresh medium for 24 and 72 h. Negative-control cells were exposed to an equivalent volume of sterile LT-free diluent. For all determinations, the AEC were harvested and lysed in 250 μl/well cold RIPA lysis buffer (150 mM NaCl, 50 mM Tris, pH 7.5, 10 mM EDTA, 10 mM EGTA, 1% Triton X-100, 0.5% Na deoxycholate, 0.1% SDS, 0.5 μM microcystin LR, 1.0 mM phenylmethylsulfonyl fluoride [PMSF], 5 μg/ml aprotinin, and 5 μg/ml leupeptin). For ANTXR protein determination, HAM were prepared as described above and lysed in cold RIPA lysis buffer. The total protein concentration of the clarified lysates was measured by a BCA assay (Pierce, ThermoFisher, Rockford, IL), and the lysates were stored at −20°C. AEC and HAM lysates containing 5 to 30 μg lysate protein were mixed with SDS-polyacrylamide gel electrophoresis (PAGE) sample buffer (60 mM Tris, pH 6.8, 10% glycerol, 2.3% SDS) and heated to 95°C for 5 min. For ANTXR blots, immunogen standards for CMG2 protein (R&D Systems, Inc., Minneapolis, MN) and TEM8 protein (Abcam, Cambridge, MA) were used. The samples were separated by SDS-PAGE on 4% to 20% gradient gels (Bio-Rad Laboratories., Hercules, CA), and the separated proteins were electrophoretically transferred onto 0.2-μm-pore-size polyvinylidene fluoride (PVDF) membranes (Westran S; Whatman PLC, Maidstone, Kent, United Kingdom). The membranes were treated with blocking buffer (5% powdered milk in TBS) for at least 1 h and probed overnight at 4°C with primary antibodies diluted in blocking buffer. The following primary antibodies were used: rabbit α-MEK1 (Upstate/Millipore, Billerica, MA), rabbit α-MEK2 (Santa Cruz Biotechnology Inc., Santa Cruz, CA), rabbit α-MEK3 (Santa Cruz Biotechnology Inc.), rabbit α-MEK4 (Santa Cruz Biotechnology Inc.), rabbit α-MEK7 (Abcam), goat α-CMG-2 (R&D Systems), rabbit α-TEM-8 (Abcam), rabbit α-ZO-1 (Invitrogen Corp., Carlsbad, CA), mouse α-β-catenin (CAT-5H10; Invitrogen Corp.), mouse α-E-cadherin (4A-2C7; Invitrogen Corp.), rabbit α-desmoplakin 1 and 2 (Abcam), mouse α-plakoglobin (γ-catenin; 4C12, Abcam), and mouse α-Actin (ACTNO5, Abcam). The membranes were developed with horseradish peroxidase-conjugated, species-specific α-IgG secondary antibodies and chemiluminescent reagents (ECL Plus; Amersham/GE health care, Piscataway, NJ). The following secondary antibodies were used: goat α-rabbit IgG (Cell Signaling Technology, Danvers, MA), horse α-mouse IgG (Cell Signaling Technology), and donkey α-goat IgG (Jackson ImmunoResearch, West Grove, PA). The developed membranes were documented with a charge-coupled device (CCD) digital camera and controlling software, and the blot images were digitally quantified (G:Box Chemi with GeneSnap and GeneTools and ImageQuant software; Syngene, Synoptics Ltd., Cambridge, United Kingdom).
Propidium iodide viability assay.
After various culture periods, as shown in Table 1, LT (2 μg/ml) or medium alone was added to AEC cultured as described above and incubated for various periods from 3 to 66 h. Cells were harvested by collecting supernatants before adding 0.5 ml prewarmed collagenase (2 mg/ml in 1× PBS) to the wells. The plates were incubated in a tissue culture incubator for 25 min to allow cells to detach. Detached cells were collected and added to supernatants. Wells were washed with 2.5 ml PBS, and the wash was added to the collected cells. Cells were centrifuged at 200 × g for 5 min and resuspended in 0.3 ml buffer and placed on ice. Immediately before evaluation by flow cytometry, propidium iodide (PI) was added with vortexing to a final concentration of 1.25 μg/ml. Percent viability was determined as the percentage of cells that excluded PI.
Table 1.
Effect of LT (2 μg/ml) treatment on AEC viability
| Lunga | No. of culture days before LT exposure | LT exposure time (h) | Fold difference in no. of live cells (LT/control)b |
|---|---|---|---|
| A | 2 | 3 | 1.1 |
| B | 2 | 6 | 0.81 |
| A | 2 | 24 | 1 |
| A | 2 | 48 | 0.96 |
| A | 5 | 3 | 0.96 |
| B | 5 | 3 | 0.9 |
| B | 6 | 3 | 1 |
| A | 8 | 3 | 1 |
| A | 8 | 50 | 0.98 |
| A | 9 | 66 | 0.97 |
| B | 14 | 48 | 1 |
Results are from 2 separate lung donor preparations.
A total of 1 to 2 replicates were used for each trial. Ratio of cells excluding PI in the LT-treated condition to control cells.
Mitochondrial dehydrogenase activity.
After 14 days in culture, LT (2 μg/ml), PA (2 μg/ml), staurosporine (10 μM) (Enzo Life Sciences, Plymouth Meeting, PA), or medium only was added to AEC cultured as described above. The plates were incubated in a tissue culture incubator at 37°C for 24, 48, 72, 96, or 144 h. Each well then received 10 μl of Cell Counting Kit-8 solution (Dojindo, Rockville, MD), and the plates were incubated in a tissue culture incubator at 37°C for 2 h. The optical density at 450 nm (OD450) of the wells was determined using a Vmax kinetic microplate reader (Molecular Devices, Sunnyvale, CA) and SoftMax Pro software v. 3 (Molecular Devices). Percent dehydrogenase activity for each treatment was calculated by expressing the OD450 of the toxin-treated wells as a percentage of the OD450 of the untreated wells.
Lactate dehydrogenase viability assay.
After 14 days in culture, LT (2 μg/ml), staurosporine (10 μM) (Enzo Life Sciences, Plymouth Meeting, PA), or medium only was added to AEC cultured as described above. The plates were incubated in a tissue culture incubator at 37°C for 24, 48, 72, 96, or 144 h. Lactate dehydrogenase (LDH) activity was measured using the LDH cytotoxicity assay kit (BioVision) at an absorbance of 490 nm (Vmax kinetic microplate reader; Molecular Devices, Sunnyvale, CA).
Statistical analysis.
Where applicable, the data are expressed as the means and standard errors of the means (SEM). Statistical significance was determined by one-way analysis of variance (ANOVA) with the Bonferroni post hoc correction for multiple comparisons. A P value of <0.05 was considered significant (36, 67).
RESULTS
Highly purified AEC II differentiate into AEC I-like cells during the first 14 days in culture.
Previous reports describe the isolation, culture, and characterization of human AEC (17, 20, 24, 63). We modified these methods to isolate AEC of sufficient purity and to establish cultures that were predominantly AEC II or AEC I, as determined by differentiation markers and phenotypic characteristics under different culture conditions. This allowed us to test the effects of LT on both AEC barrier function, which is predominantly a function of AEC I, and surfactant production, which is predominantly a function of AEC II. We found that cultures of 5 days or less exhibited AEC II-like characteristics, while cells cultured 14 days or longer clearly exhibited AEC I-like features.
Isolation of human AEC is known to result in an initial culture consisting mainly of surfactant-producing AEC II cells, which differentiate to the AEC I phenotype in culture (60). We found this to be the case in our human AEC preparations as well. We purified human AEC using standard methods (17, 20, 24) with modifications as described. Culturing these cells on collagen and fibronectin-coated tissue culture plates at 25,000 cells/cm2 produced a confluent or nearly confluent culture in 10 to 14 days (Fig. 1A).
Fig 1.

Isolation and culture of AEC. (A) Freshly isolated cells were plated at 50,000 live cells/cm2. After 10 to 14 days in culture, the cells were 95 to 100% confluent. Bar = 250 μm. (B) Freshly isolated AEC have lamellar bodies evident as blue inclusions when stained with Harris hematoxylin. Bar = 25 μm. (C) At 10 days in culture, cells showed an increase in diameter, were confluent in areas, exhibited an AEC I-like morphology, and stained positive for the tight junction protein ZO-1. A representative field is shown with ZO-1 stained using α-human ZO-1 (green) and DNA stained with DAPI (blue). Bar = 100 μm. (D) Within the first 24 h after isolation, 4.1% of AEC expressed GABAA Rπ and 49.5% expressed P2X7, as determined by flow cytometry analysis of cells stained with α-human GABAA Rπ, α-human P2X7, or isotype control antibody. Data shown are the results from one donor preparation and are expressed as the means ± SEM. Similar results were obtained from a second donor preparation. (E) Representative flow cytometry histogram of P2X7-positive AEC, day 0. Dotted line, unlabeled cells; dashed line, species-specific negative-control IgG; solid line, marker. (F) At 6 days in culture, immunofluorescent staining with α-human P2X7 determined that 80% of cells were positive for P2X7. A representative field is shown with P2X7 stained by α-human P2X7 (red) and DNA stained with DAPI (blue). Bar = 100 μm. (G) Representative flow cytometry histogram of GABAA Rπ-positive AEC, day 0. Dotted line, unlabeled cells; dashed line, species-specific negative-control IgG; solid line, marker.
To characterize the cultured AEC, we performed serial studies to assess surfactant-containing inclusion bodies and expression of the tight junction component zonula occludens-1 (ZO-1). As expected, lamellar bodies indicative of an AEC II surfactant-producing phenotype were easily identified in almost all AEC immediately after isolation (Fig. 1B) and were markedly decreased by day 14 of culture. This was accompanied by a change in morphology from a cuboidal, AEC II morphology to a flat, AEC I-like appearance (Fig. 1C), consistent with previous reports (60). Also noted during the morphological change to the AEC I-like phenotype was increased expression of the tight junction protein ZO-1 (Fig. 1C).
Expression of surface markers of AEC differentiation was assessed by flow cytometry for the γ-aminobutyric acid receptor, pi subunit (GABAA Rπ), and the purinergic receptor P2X7. These receptors were previously shown to be expressed primarily in rat AEC II (GABAA Rπ) or AEC I (P2X7) (13).
P2X7 was expressed by 49.5% of freshly isolated cells (Fig. 1D and E), though, as expected, 80% of cells expressed the AEC I marker P2X7 by day 6 of culture (Fig. 1F), consistent with AEC I-like differentiation. GABAA Rπ expression was low (mean, 5%) during the entire culture period (Fig. 1D and G). Thus, the GABAA Rπ was uninformative as a marker for differentiation of human AEC. As expected, expression of the nonepithelial markers CD31, CD45, and vimentin was low (≤3% of cells), demonstrating minimal contamination with endothelial, leukocyte, and fibroblast cell types, respectively. For the most part, these results were similar to those reported for isolation of AEC from other species except rat, for which GABAA Rπ is an informative differentiation marker and in which there is no P2X7 expression in the lamellar body-containing AEC II phenotype (13). These findings demonstrate that we successfully isolated and cultured human AEC II and that these cells differentiated to AEC I-like cells in culture.
In subsequent experiments described below, we studied AEC following isolation and culture for different times prior to use. Early-culture AEC were cultured for approximately 5 days in order to study the effects of LT on surfactant production in a primarily type II AEC phenotype. Late-culture AEC were cultured for approximately 14 to 21 days until near confluence to study LT effects on barrier function and tight junction formation in type I-like AEC.
Exposure of AEC to LT causes actin rearrangement, reduced expression of junction protein genes, and decreased epithelial barrier function.
AEC I, which emerge in late cultures, are the predominant cell type forming the alveolar epithelial barrier and junctions in vivo. Late cultures of these cells at or near confluence were exposed to LT (2 μg/ml) for 24 to 144 h. The dose of LT was selected on the basis of previous studies of LT cytotoxicity in vitro and in vivo. We have shown that HAM are relatively resistant to LT up to doses of 1 to 5 μg/ml (64). In contrast, primary human bronchial epithelial cells are sensitive to LT at a dose of 1 μg/ml (32). The in vivo 50% lethal dose (LD50) for mice is 12.5 μg of PA and 2.5 μg of LF administered intravenously (i.v.) (21). When corrected for the estimated blood volume of the animals used, this is equal to 7.8 μg PA and 1.6 μg LF per ml of blood (4). Taking into consideration this information, we performed our experiments with 2 μg/ml LT, defined as 2 μg/ml LF and 2 μg/ml PA.
At 24 h of LT treatment, actin fibers in some of the LT-exposed AEC appeared elongated and thickened compared to those in untreated AEC, as determined by immunohistochemistry using phalloidin-Alexa Fluor 555 (Fig. 2D). In addition, the pattern of fibers changed from being concentrated around the periphery of the cell to being more evenly distributed within the cell. This involved the majority of cells by 144 h of exposure (Fig. 2F). Inhibition of MAPK signaling pathways by cleavage of the MEK component is a known effect of LT. To determine whether inhibition of these pathways by LT may be responsible for the effects seen on actin fibers, we exposed AEC to a 50 μM mixture of chemical inhibitors of the three MAPK pathways for 24 to 144 h. U0126, SB03580, and SP600125 were used to inhibit the ERK, p38, and stress-activated protein kinase (SAPK)/JNK pathways, respectively. By 24 h of exposure, the MAPK pathway inhibitors caused actin rearrangement with stress fiber formation in a majority of the AEC (Fig. 2G). At subsequent times, severe cytotoxicity with disruption of the monolayer occurred (Fig. 2H and I). The findings demonstrate that LT exposure causes actin rearrangement in human AEC and are consistent with LT-induced MAPK pathway inhibition as a potential mechanism.
Fig 2.

AEC respond to LT and the signal pathway inhibitors UO126, SB203580, and SP600125 by rearranging actin. Late-culture AEC were exposed to 2 μg/ml LT (D, E, and F), MAPK pathway inhibitors (MI; 50 μM each) (G, H, and I), or diluent (NT) (A, B, and C) for 24, 72, and 144 h. AEC were fixed and stained with phalloidin-Alexa Fluor 555 (red) and DAPI (blue) and visualized by fluorescence microscopy. Data shown are representative of replicate experiments on two donor preparations. Bar = 250 μm.
Actin supports junction complex structure in epithelial cells (28). We next examined whether the LT-induced actin rearrangement seen in AEC was accompanied by altered expression of junction proteins. Late-culture AEC were exposed to LT (2 μg/ml) for 24 to 144 h, and expression of the junction proteins ZO-1, E-cadherin, and β-catenin was determined by Western immunoblotting. LT exposure of AEC reduced expression of all three junction proteins. ZO-1 was reduced by 1.7-, 2.3-, 2.1-, and 1.8-fold at 24, 48, 72, and 144 h of LT treatment, respectively, and E-cadherin was reduced by 1.5-, 2.6-, 3.6-, and 1.6-fold at these time periods, respectively. Finally, β-catenin expression was reduced by 1.2-, 1.4-, 1.9-, and 1.4-fold at these times, respectively (Fig. 3A).
Fig 3.

AEC respond to LT by reducing the quantity of tight junction proteins and decreasing epithelial barrier function. (A) Western blot analysis was used to determine the relative amounts of ZO-1, β-catenin, E-cadherin, and actin in lysates of cultured AEC treated with LT (2 μg/ml) for 24, 48, 72, and 144 h or untreated. AEC were cultured for 21 days and treated with LT. Data shown are representative of three separate experiments from 2 donor preparations. (B) qRT-PCR was performed to analyze the levels of junction protein mRNA after treatment with LT and MEK inhibitors. Late-culture AEC were treated for 3, 10, and 24 h with LT (2 μg/ml), a combination of the signal pathway inhibitors UO126, SB203580, and SP600125 (50 μM each), or diluent. Relative quantities of RNA for ZO-1, β-catenin (β-Cat), and E-cadherin (E-Cad) were determined for each treatment. Results indicate the mean fold increase or decrease ± SEM of RNA in treated AEC from that in AEC treated with diluent. *, P < 0.05 for the fold difference between treated and untreated cells; **, P < 0.001 for the fold difference between treated and untreated cells. Data represent three replicates. (C) To measure transepithelial electrical resistance (TER), AEC were cultured on gold microelectrodes. Confluence was reached after 6 days, as evidenced by stabilization of TER, and cultures were treated with medium with LT (2 μg/ml), PA (2 μg/ml), staurosporine (Stauro.) (10 μM), or no treatment. After equilibration, TER was measured for 140 h. Traces show the resistance (R) in ohms for the 140 h after treatment, normalized to the same starting R. Small dots, no treatment; large dots, PA; solid line, LT; dots and dashes, staurosporine. (D) LT exposure decreased TER in AEC. The graph depicts the mean R decrease ± SEM versus untreated control AEC at 140 h posttreatment from 3 individuals, with 1 to 4 wells for each treatment. *, P value of <0.05 for treated versus untreated cells; **, P value of <0.001 for treated versus untreated cells. (E) AEC stained for actin (green) and DNA (blue) after 144 h of treatment with medium, LT, or staurosporine and measurement of TER. Bar = 250 μm.
We next examined whether the effect of LT on junction protein gene expression occurs at the level of transcription and whether it may be due to inhibition of MAPK. AEC were exposed to LT or the mixture of the three MAPK pathway inhibitors for 3 to 24 h, and ZO-1, E-cadherin, and β-catenin mRNA levels were measured by qRT-PCR (Fig. 3B). At 3 h, both LT and the MAPK inhibitors appeared to increase endogenous mRNA levels of the junction proteins, though induction of E-cadherin and β-catenin by MAPK inhibitors did not reach statistical significance. However, by 24 h of exposure, MAPK inhibition, but not LT exposure, significantly decreased mRNA levels of the 3 junction protein genes by 2- to 4-fold (P value of <0.001 versus untreated cells), consistent with downregulation at the level of transcription (Fig. 3B). LT induction of ZO-1 and β-catenin mRNA was 1.5- and 1.7-fold over that of control cells (P values of <0.05 and <0.001, respectively), while E-cadherin was unaffected by LT at this time point. The divergent effects of LT and the MAPK inhibitors on junction protein gene mRNA levels suggest that downregulation of junction protein genes cannot be solely ascribed to LT-mediated MAPK inhibition of transcription.
As LT exposure caused actin rearrangement and decreased tight junction protein expression, we next measured transepithelial electrical resistance (TER) to determine whether LT exposure also resulted in physiologic alterations in barrier function in late-culture AEC. Using an electric cell substrate impedance-sensing system (25), we cultured AEC until confluence, as evidenced by stabilization of electrical resistance. We then added LT (2 μg/ml) and measured TER for 6 days (Fig. 3C). Staurosporine (10 μM) was used as a positive control. All treatments were added with fresh medium, which is known to transiently increase TER. After 2 days of treatment, we began to observe a decrease in TER in the LT-treated wells. At 6 days, LT significantly impaired barrier function compared to that of untreated cells cultured for the same period. LT decreased TER by 38% ± 4% from baseline (P value of <0.05 versus untreated cells) (Fig. 3D). As expected, staurosporine also decreased TER significantly (84% ± 3% from baseline; P value of <0.001 versus untreated cells). This decrease in TER is consistent with the loss of junction protein expression described above and with the observed changes in F-actin morphology. Although TER declined, cells in LT-treated cultures remained adherent and confluent throughout the experimental period, as evidenced by the visualization of cultures at the end of the experiment, compared to untreated control cultures and cells in cultures treated with staurosporine to induce apoptosis (Fig. 3E). We concluded that LT exposure reduced AEC barrier function and that this effect was not due to cytotoxicity with cellular detachment.
Exposure of AEC to LT alters immunolocalization of junction proteins.
Since LT impaired junction protein expression and barrier function in AEC, we next examined whether LT affected immunolocalization of these proteins. Late-culture AEC were exposed to LT (2 μg/ml) for 24 to 144 h. Immunolocalization of the junction proteins was performed using antibodies to ZO-1, E-cadherin, and β-catenin and Alexa Fluor 555-conjugated secondary antibody. We found that LT exposure altered the immunolocalization pattern of all three junctional proteins. ZO-1 was evenly distributed in the periphery of unexposed AEC at all time points. In contrast, LT resulted in eccentric peripheral distribution of this protein after 144 h of exposure (Fig. 4A to F). In control cells, E-cadherin was distributed in an even, well-defined, peripheral lace-like structure visible at all time points. It appeared to be present at areas of cell-cell contact. LT exposure resulted in a loss of both the fine detail and the demarcation from the clearer cytoplasm by 72 h of exposure that persisted at 144 h (Fig. 4G to L). The appearance of β-catenin in normal cells was similar to that for E-cadherin in that it was well demarcated, was evenly distributed in the periphery of cells, had a lace-like pattern, and appeared to be present at sites of cell-cell contact. LT exposure also caused a loss of the fine detail, a clear demarcation, and a peripheral distribution of β-catenin after 72 h of exposure that persisted at 144 h (Fig. 4M to R). We concluded that intracellular distribution and fine structure of ZO-1, E-cadherin, and β-catenin are altered by LT in AEC.
Fig 4.

AEC respond to LT by destabilizing tight junctions. Late-culture AEC were exposed to 2 μg/ml LT or diluent (NT) for 24, 72, or 144 h. AEC were fixed and stained with anti-ZO-1 (A to F), anti-E-cadherin (G to L), or anti-β-catenin (M to R) (red) and DAPI (blue) and visualized by fluorescence microscopy. Data shown are representative of two replicates. Bar = 250 μm.
Exposure of AEC to toxins alters intercellular junction morphology.
Our findings of reduced barrier function and junction proteins suggest that LT likely affects formation of the intercellular junctions responsible for barrier function. We therefore examined intercellular junction morphology by transmission electron microscopy (TEM) of LT-treated AEC. Late-culture, nearly confluent AEC were treated with diluent, LT (2 μg/ml), or PA (2 μg/ml) for 72 h, and cells were prepared for TEM. Long junctional complexes with desmosomes and extensive contacts were readily observed in untreated AEC (Fig. 5A). In PA-treated AEC, junctions had extensive contacts but lacked desmosomes (Fig. 5B). In LT-treated AEC, junctions were shorter and were less elaborated than those in the untreated AEC and also lacked desmosomes (Fig. 5C).
Fig 5.

Cell-cell contacts are disrupted by LT. Late-culture AEC were treated with LT (2 μg/ml) or PA (2 μg/ml) for 72 h, fixed, harvested, and prepared for TEM. Representative cell-cell contact images are shown. (A) Untreated AEC had elaborate junctional complexes (arrows), including desmosomes (indicated by the letter D). (B) PA-treated AEC had well-developed junctions (arrows), which appeared normal except for the absence of desmosomes. (C) LT-treated AEC lacked desmosomes and had shorter, less-elaborate junctional complexes (arrows). Bar = 500 nm. (D) Western blot analysis was used to determine the relative amounts of desmoplakin (DP-1/2), plakoglobin (PG), and actin in lysates of cultured AEC treated with LT and PA. AEC were cultured for 21 days and treated with LT (2 μg/ml) or PA (2 μg/ml) or untreated (−) for 24 or 72 h. Data shown are representative of three replicates. (E) qRT-PCR was performed to analyze the levels of desmosomal protein mRNA after treatment with LT and MEK inhibitors. Late-culture AEC were treated for 3, 10, and 24 h with LT (2 μg/ml), a combination of the signal pathway inhibitors UO126, SB203580, and SP600125 (50 μM), or diluent. Relative quantities of RNA for desmoplakin (DP) and plakoglobin (PG) were determined for each treatment. Results indicate the mean fold increase or decrease ± SEM in mRNA in treated AEC compared to that in AEC treated with diluent. *, P value of <0.05 for the fold difference between treated and untreated cells; **, P value of <0.001 for the fold difference between treated and untreated cells. Data represent three replicates.
Exposure of AEC to Bacillus anthracis toxins and MAPK inhibitors downregulates desmosomal protein gene expression.
In order to confirm the TEM findings and to determine whether the loss of desmosomes due to toxins may be due to downregulation of the constituent desmosomal proteins, we measured expression of the desmosomal proteins desmoplakin (DP) and plakoglobin (PG) by Western blotting in AEC exposed to PA (2 μg/ml), LT (2 μg/ml), or diluent as described previously (43, 47). Both PA and LT suppressed expression of DP and PG in AEC. DP-1 was decreased 1.3-fold by LT treatment for 72 h and 2.1-fold by PA treatment for 72 h. DP-2, a splice variant of DP-1 (43), was decreased 1.4-fold by LT treatment for 24 and 72 h and decreased 1.9-fold by PA treatment for 72 h. Plakoglobin was decreased 1.3-fold and 1.4-fold by LT treatment for 24 and 72 h, respectively, and decreased 1.7-fold by PA treatment for 72 h (Fig. 5D). We then examined whether the effect of LT on desmosomal protein genes occurs at the level of transcription and whether it may be due to inhibition of MAPK. AEC were exposed to LT or the mixture of inhibitors of the three MAPK pathway inhibitors U0126, SB03580, and SP600125 for 3 to 24 h, and DP and PG mRNA levels were measured by qRT-PCR. We chose the time of exposure based on the fact that regulation at the mRNA level should precede alterations in translation. After an apparent transient initial stimulation of endogenous mRNA levels of the desmosomal protein genes, by 10 h of exposure, both LT and the MAPK inhibitors appeared to suppress endogenous mRNA levels of DP and PG, though statistical significance was achieved only in cells exposed to the chemical inhibitors (Fig. 5E) (P value of <0.05 versus untreated cells). By 24 h of exposure, suppression of mRNA levels of PG and DP were further enhanced to a 1.5-fold suppression in LT-exposed cells and a 4- to 6-fold suppression in cells exposed to the MAPK inhibitors (Fig. 5E). In this case, suppression of PG by LT was statistically significant (P value of <0.05 versus untreated cells), as was suppression of mRNA levels of both desmosomal protein genes by MAPK inhibitors (P value of <0.001 versus untreated cells). These changes in junction morphology, protein levels, and RNA levels are consistent with, and are likely at least partially responsible for, the loss of barrier function in AEC I exposed to LT. As the patterns of suppression by LT and MAPK inhibitors were similar, LT-mediated MAPK inhibition may play a role in this process.
Exposure to LT decreases surfactant production in AEC II and alters surfactant processing.
Like reduced barrier function of AEC I, decreased surfactant levels are a component of acute lung injury (40, 50). Therefore, to assess whether LT exposure induced signs of acute lung injury in addition to increased barrier permeability, we examined cell-free AEC culture supernatants for secreted surfactant phospholipid using the method of Stewart (55) (Fig. 6). We used early-culture AEC so that a significant number of cells would be AEC II, the major source of surfactant in the lung (16). Cells were untreated or treated with LT (2 μg/ml) or PA (2 μg/ml), and cellular surfactant levels were measured after 72 h. To demonstrate that the cultured AEC were able to respond appropriately, they were also treated with high-dose dexamethasone (Dex) (1 μM) to decrease surfactant or with phorbol myristate (PMA) (50 ng/ml) to increase surfactant. As expected, high-dose Dex suppressed and PMA induced surfactant production in AEC (0.5-fold decrease and 1.4-fold increase, respectively; P value of <0.05 in both comparisons). In the LT- and PA-treated cultures, surfactant levels were decreased by 0.5-fold and 0.6-fold, respectively (P value of <0.05 in both comparisons), suggesting that treatment inhibited either surfactant production or surfactant export by AEC II.
Fig 6.
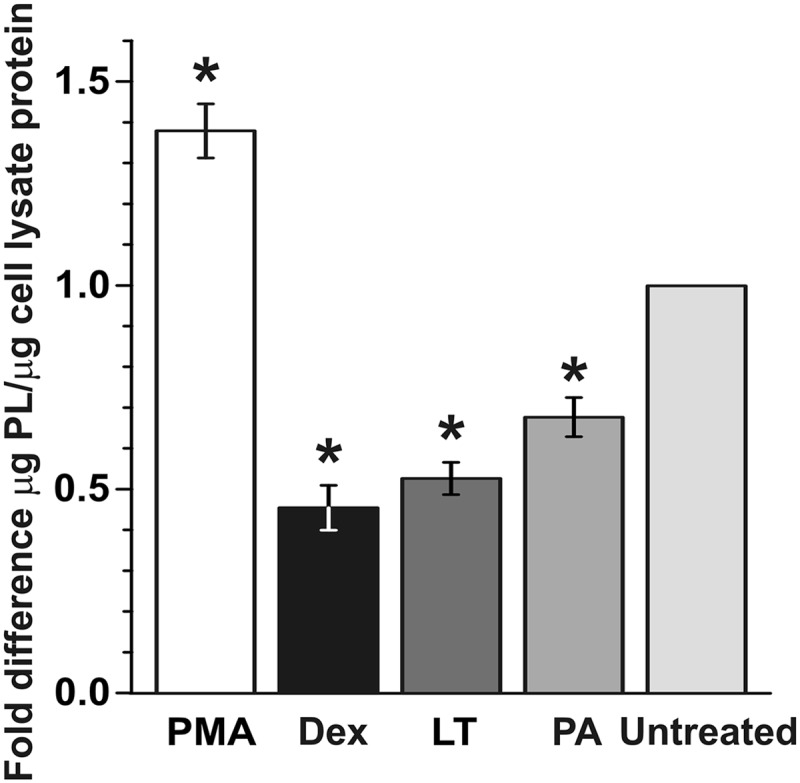
LT decreases surfactant production in AEC. Early-culture AEC were treated with LT (2 μg/ml) for 72 h and harvested. Phorbol 12-myristate (PMA; 50 ng/ml) and dexamethasone (Dex; 1 μM) were used as positive and negative controls, respectively. Surfactant levels in supernatants were measured by a phospholipid assay and normalized to protein content per sample. Data are means ± SEM of the results from 3 experiments on 3 separate individuals, with 2 to 4 trials for each treatment. *, P value of <0.05 versus untreated cells.
To evaluate the possibility that LT treatment altered surfactant processing, we examined cellular morphology by electron microscopy. Early-culture AEC were left untreated or were treated with LT (2 μg/ml) or PA (2 μg/ml) for 72 h. Cells were fixed in the culture plate, harvested by scraping, and prepared for transmission electron microscopy (TEM) in order to evaluate the effect of anthrax toxins on surfactant-containing lamellar bodies. In untreated cells, lamellar bodies consisted of concentrically coiled layers around a central core, a typical arrangement (Fig. 7A and B) (24). In both LT- and PA-treated cells, there appeared to be fewer lamellar bodies per cell, with alterations in lamellar body morphology. The lamellar bodies had increased density in PA-treated cells, and in LT-treated cells, the outer layers were disorganized compared to untreated controls (Fig. 7C to F). In addition, after LT and PA treatment, but not in untreated controls, lamellar bodies were visible inside autophagolysosomes, suggesting dissolution (Fig. 7G to J). These results are consistent with the reduction of extracellular surfactant observed in PA- and LT-treated AEC cultures and may indicate aberrant surfactant processing in toxin-treated cells.
Fig 7.

The structure and location of surfactant-producing lamellar bodies are disrupted by LT. Early-culture AEC were exposed to LT (2 μg/ml) or PA (2 μg/ml) for 72 h, fixed, harvested, and prepared for TEM. Representative fields are shown. White boxes in panels A, C, E, G, and I indicate areas enlarged in panels B, D, F, H, and J, respectively. (A and B) Lamellar bodies in untreated AEC show a distinct layered structure. See the white arrows in panel B. (C and D) Lamellar bodies appear to be denser and less organized in PA-treated AEC, with fewer layers in PA-treated AEC than in untreated AEC; compare the white arrows in panels B and D. (E and F) LT-treated AEC also show disrupted lamellar body structure; compare the white arrows in panels F and B. (G to J) Lamellar bodies were found within autophagosomes in PA-treated (G and H) and LT-treated (I and J) AEC, but equivalent organelles were not found in untreated samples. Bar = 500 nm.
AEC express the anthrax toxin receptors TEM8 and CMG2.
The alterations in actin structure, junction protein expression and function, and surfactant production observed in this study show that primary human AEC are sensitive to Bacillus anthracis LT. In contrast, our previous work showed that primary human alveolar macrophages (HAM) are resistant to LT and do not express functional anthrax toxin receptors (64). To determine if the different LT sensitivities of AEC and HAM may be due to differential expression of functional toxin receptors, we compared anthrax toxin receptor expression in AEC and HAM.
Surface expression of anthrax toxin receptors was determined by flow cytometry using antibodies to the known human anthrax receptors tumor endothelial marker 8 (TEM8/ANTXR1) and capillary morphogenesis protein 2 (CMG2/ANTXR2). AEC were evaluated immediately after isolation (early cultures) and also after culturing for 14 to 21 days (late cultures) to assess receptor expression in both AEC I and AEC II type cells. In early, predominantly AEC II cultures, 8.1% of cells were positive for TEM8/ANTXR1 and 8.7% were positive for CMG2/ANTXR2 (Fig. 8A and B). In late, primarily AEC I cultures, 52.7% of cells were positive for TEM8/ANTXR1 and 65.5% were positive for CMG2/ANTXR2 (Fig. 8A and B).
Fig 8.

AEC express anthrax receptors and bind PA. (A) Freshly isolated and late-culture AEC were stained with α-human TEM8 or α-human CMG2 and analyzed by flow cytometry to measure the percentage of AEC positive for TEM8 or CMG2. Data are the mean percentages ± SEM of cells staining positive for the designated receptor and are from 2 separate preparations, with 2 to 3 trials per receptor. (B) Representative histograms showing staining of AEC with α-human TEM8 or α-human CMG2 antibodies (black) or nonspecific background staining using species-specific IgG negative controls for each antibody (gray). (C) Western blotting was used to determine the relative amounts of TEM8/ANTXR1 and CMG2/ANTXR2 in lysates of late-culture AEC and human alveolar macrophages obtained from bronchoalveolar lavage. The amount of TEM8 and CMG2 per lane was determined by comparison to recombinant protein immunogen standards. Human α-actin was used as a probe to confirm equivalent protein loading per lane and as an additional means of comparison. Data were obtained from 3 individuals for each cell type, and results were calculated as mean fg receptor/cell and as a ratio of receptor to actin. (D) Late-culture AEC were harvested, incubated on ice for 3 h with FITC-labeled PA (2.6 μg/ml), and evaluated for PA binding by flow cytometry. Representative data are shown from one individual. Dotted line, unlabeled AEC; solid line, AEC plus PA-FITC; dashed line, AEC incubated with FITC-labeled rabbit IgG control.
In order to confirm anthrax receptor expression in AEC and to semiquantitatively compare expression in AEC and HAM, anthrax receptor protein levels relative to recombinant protein immunogen standards in late-culture AEC and in HAM were determined by an immunoblot using α-TEM8/ANTRX1 and α-CMG2/ANTXR2 antibodies (Fig. 8C) (64). CMG2/ANTXR2 expression was nearly undetectable in HAM, and a 55-kDa isoform was expressed at low levels (2 fg/cell) in late-culture AEC (Fig. 8C). For TEM8/ANTXR1, overall expression was 2.8-fold higher in AEC than in HAM. Specifically, HAM had 3.7, 4.7, and 0 fg/cell of the 80-kDa, 60-kDa, and 40-kDa isoforms, respectively, while AEC had 2.6, 18.4, and 2.6 fg/cell of these isoforms, respectively. Our finding of multiple isoforms of ANTXR1 in AEC is consistent with their previously reported presence in whole tissue samples (6). When calculated using a ratio of receptor protein to actin, AEC expressed 9-fold more of the dominant 60-kDa isoform than HAM.
We then determined whether anthrax receptors expressed by AEC are capable of binding PA, the component of LT responsible for toxin binding and entry to the cell. To do this, we quantified binding of FITC-labeled PA to AEC using flow cytometry. The mean fluorescence intensity of late-culture AEC incubated with FITC-labeled PA was 20.7, compared to 4.0 for cultures incubated with an FITC-labeled isotype control (Fig. 8D). Therefore, while HAM showed low expression and poor binding capacity (64) of anthrax receptors, these results indicate that AEC express both anthrax toxin receptor 1 and 2 and that the receptors expressed by AEC are functional.
AEC are sensitive to LT-induced MEK cleavage.
Murine macrophage RAW 264.7 cells, which are sensitive to LT, exhibit MEK 1, 2, 6, and 7 cleavage upon exposure to LT. In contrast, HAM are not sensitive to LT and do not show LT-induced MEK cleavage (64). Since AEC express functional anthrax receptors and exhibit altered activity in response to LT, we determined whether AEC are also sensitive to MEK cleavage by LT. Late-culture AEC were exposed to 2 μg/ml LT for 24 h, and the extent of MEK cleavage was determined by an immunoblot. Exposure to LT resulted in >95% cleavage of MEK 1, 2, 3, and 4 and 15% cleavage of MEK 7 (Fig. 9). Early-culture AEC were also sensitive to LT-induced MEK cleavage, with the exception of MEK 7, which was less sensitive to LT-induced cleavage in these cells (data not shown). We were unable to assess cleavage of MEK 6 due to the nonspecificity of the MEK 6 antibody in AEC (data not shown). These findings show that AEC are sensitive to LT-induced MEK cleavage, implicating a possible mechanism by which LT exposure may affect AEC barrier function, junction formation, and surfactant production.
Fig 9.
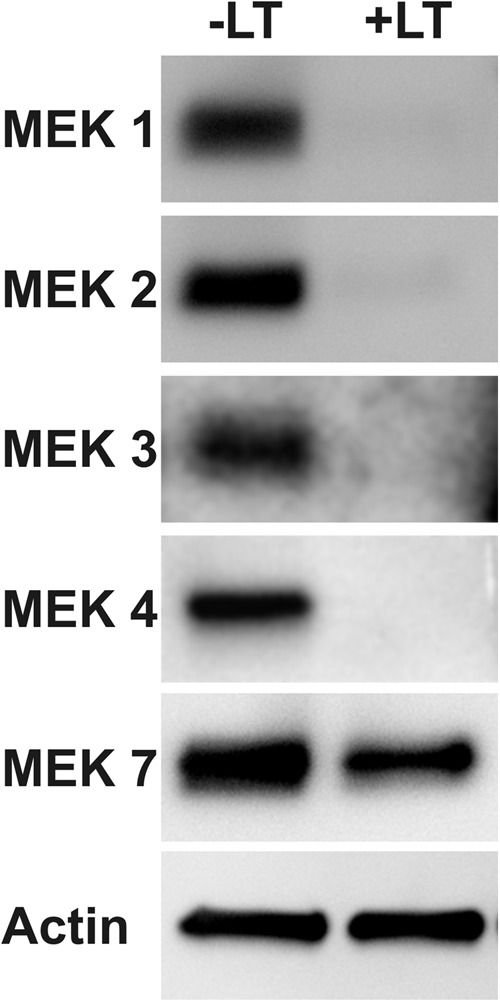
Stimulation of AEC with LT causes MEK cleavage. Late-culture AEC were treated with LT (2 μg/ml) for 24 h, and cell lysates were immunoblotted for the MEK proteins shown. Data shown are representative of experiments using two separate AEC preparations from different donors.
AEC are not killed by LT.
Because LT-induced MEK cleavage has been associated with apoptosis in other cell types (46), we examined whether LT exposure of AEC was associated with cell death. AEC cultured from 2 to 14 days were left untreated or were exposed to LT (2 μg/ml) for various lengths of time (3 to 66 h) to determine if LT caused cell death in either AEC I-like or AEC II cells. This was determined by flow cytometry to assess nuclear exclusion of propidium iodide (PI), by colorimetric assay to assess dehydrogenase activity of living cells, and by lactate dehydrogenase (LDH) release from dead cells (29, 44). Despite the near-complete cleavage of several MEK isoforms, both PI exclusion (Table 1) and LDH release indicated that LT exposure under the tested conditions did not kill the cells by apoptosis or necrosis. Membrane permeability was not increased by LT (Table 1), and LDH release from cells was unchanged by LT through 144 h of exposure, though it was increased by staurosporine, a strong inducer of apoptosis (data not shown).
An assay of mitochondrial dehydrogenase activity (27, 59) of living AEC treated with LT showed a temporary, reversible decrease in metabolic activity at 48, 72, and 96 h of 69% (P < 0.05), 73% (P < 0.001), and 79% (P < 0.05), respectively, from that of untreated controls. By 144 h of LT exposure, dehydrogenase activity increased to 85% (P > 0.05) of untreated controls, an amount which was not significantly different from that for the controls (Fig. 10). A similar pattern occurred with PA treatment, although the decrease in metabolic activity was significant only at 96 h (84% of untreated controls; P < 0.05) and, like with LT, increased to a level not significantly different from that for untreated cells by 144 h (85%; P > 0.05) This transient decrease in mitochondrial activity by LT and PA, coupled with unchanged LDH release, suggests that the AEC are injured by toxin but recover. AEC do not undergo cytolysis and do not die by apoptosis or necrosis when treated with LT.
Fig 10.

LT and PA cause a temporary, reversible decrease in dehydrogenase activity of AEC. Late-culture AEC were left untreated or treated with LT (2 μg/ml PA, 2 μg/ml LF), PA (2 μg/ml), or staurosporine (10 μM). Dehydrogenase activity was assessed at 24, 48, 72, 96, and 144 h using a nontoxic tetrazolium salt assay to detect dehydrogenase activity of living cells. For each time point, mean dehydrogenase activity of untreated cells was considered to represent 100%. Results are the mean percent activity ± SEM of toxin-treated cells versus untreated cells and represent 4 replicate cultures. *, P value of <0.05 versus untreated cells; **, P value of <0.001 versus untreated cells.
These results indicate that the decreases in surfactant production, barrier integrity, and tight junction protein expression in LT-exposed AEC are not due to cell death. We did not specifically investigate markers of apoptosis, such as caspase activation, because secondary necrosis is the outcome of a completed apoptotic program when phagocytic cells are absent (58, 9, 54, 65). Our AEC were not necrotic, and phagocytic cells were absent; therefore, the AEC had not completed an apoptotic program.
DISCUSSION
In this study, we found that AEC are a target of Bacillus anthracis LT. We demonstrated that early- and late-stage AEC have receptors for LT, although the receptor is more highly expressed in late-culture AEC. We found that AEC bind the PA component of LT and undergo MEK cleavage and reductions in barrier function, junction formation, and surfactant production. Because MEKs regulate p38, ERK, and JNK, many transcriptional programs and, hence, cellular functions may be affected by LT-induced MEK cleavage (52). There is evidence that MEK signaling is important in junction integrity. ERK activation has been shown to increase integrity of epithelial tight junctions by inhibiting claudin-2 expression. When ERK was inhibited in this case, junction integrity declined (1, 33). On the other hand, ERK activation can disrupt epithelial junctions in corneal epithelial cells (61). In one study on keratinocytes, a pathway involving MEKs was necessary for a transforming growth factor β (TGF-β)-induced upregulation of the factor Net1A (Rho-A-specific guanine nucleotide exchange factor isoform 2). Net1A was, in turn, necessary to maintain epithelial junctions. Silencing Net1A, coupled with TGF-β stimulation, led to a reduction in E-cadherin and ZO-1 staining in tight junctions. In that study, MEK inhibition was evident in 3 h but junction integrity was reduced at 48 h (45). This is consistent with our findings, in which we observed a time delay between MEK cleavage and reduction of junction integrity, i.e., we measured MEK cleavage at 24 h and observed morphological alterations in junction formation in 72 h, followed by measureable physiologic barrier disruption at 140 h. Our results, taken together with previous studies involving MEK cleavage and junctions, suggest that LT-induced MEK cleavage reduces expression of junction proteins. Protein turnover at junctions is then required before the functional evidence of reduced junction protein expression appears.
Our findings suggest that all of the effects of Bacillus anthracis toxins we demonstrated cannot be attributed solely to LT-induced MEK cleavage. First, we saw effects of PA alone on junction formation and surfactant production. Second, we found that MAPK inhibitors have divergent effects on mRNA levels of junction proteins compared with LT exposure. Thus, some of the effects of LT and PA are likely due to unknown effects of these toxins on AEC or are effects that cannot easily be duplicated by chemical MAPK inhibitors.
The effects of LT on AEC have implications for the lung in the late stages of inhalation anthrax and possibly the early stage. We noted that the addition of PA alone resulted in some effects that were similar to LT but less intense. While we did not specifically study the mechanism of activity of PA, we suggest that a portion of the effects of LT on surfactant production, cell-cell contacts, and lamellar bodies can be attributed to pore formation in the cell membrane by PA. Our finding that PA alone has effects on junction formation and surfactant production is of particular significance as PA levels are higher than LT levels during Bacillus anthracis infection (39).
Lung injury in late-stage anthrax.
Terminal stages of inhalation anthrax are accompanied by significant lung injury during a time when high levels of bacterial toxin components are present (3, 8, 31). We hypothesized that there is a target cell for LT in the lung through which lung injury can be mediated. We previously ruled out HAM as a significant target of LT (64). In this study, we focused on AEC because reduced epithelial barrier function and reduced surfactant levels are characteristic of acute lung injury (40). We demonstrated that AEC have receptors for LT, bind the PA component of LT, and undergo MEK cleavage and that LT causes reduced barrier function, tight junction formation, and reduced surfactant production in these cells. Our results suggest that LT, released by vegetative Bacillus anthracis in the blood, circulates to the lung and is internalized by AEC. MEK cleavage follows internalization. LT-induced MEK cleavage, likely through its effect on MAPK signaling pathways, is associated with alteration of transcription of many different genes (52). In our study, despite extensive cleavage of several MEKs, AEC do not die but exhibit at least two specific responses that potentially represent lung injury in a human host of Bacillus anthracis. The first, cytoskeletal rearrangement with loss of barrier function is likely related to the LT-induced MEK cleavage we observed in these cells. There is evidence that LT reduces actin dynamics in human neutrophils by blocking p38 MAP kinase phosphorylation (19). This suggests that the MEK cleavage we observed in AEC causes the actin rearrangement exhibited by AEC. This is significant, as actin has an important role in the structure of the junction complex structure in epithelial cells (28). Thus, LT effects on actin rearrangement in AEC, which temporally precedes alterations in junction protein immunolocalization and in barrier function, may play an important role in this process. Our findings are consistent with those of Lehman et al. (32), who showed that LT exposure caused cytoskeletal rearrangement and disruption of barrier function in human bronchial epithelial cells. The second response, decrease of surfactant production, has also been associated with MEK pathway inhibition (14), suggesting that the surfactant reduction we observed in AEC also resulted from LT-induced MEK cleavage.
The function of the epithelium and endothelium as a barrier that undergoes an increase in permeability during acute lung injury has been recognized (18, 40, 62). The most common causes of acute lung injury are primary pneumonia and sepsis (40). Interestingly, LT has been proposed as a cause of endothelial barrier dysfunction that leads to pleural effusions (51, 62). However, endothelial barrier injury alone may not be sufficient to induce pulmonary edema. Normally, both the endothelial and epithelial barriers must be disrupted in order to establish pulmonary edema (2, 40). Our results suggest a scenario in which LT is able to disrupt both barriers and cause acute pulmonary edema. Thus, during anthrax infection, LT-induced lung injury may be an independent factor that contributes to lung injury exhibited in human victims of anthrax (7).
In contrast to work done by Park and others in mouse cell lines (46), we found that LT-induced MEK cleavage was not associated with cell death. This is consistent with our previous work in primary mouse alveolar macrophage cells in which we also found that although MEK cleavage occurred after LT exposure, apoptosis and cell death did not occur (64). In terms of barrier function, our findings also are consistent with those seen in endothelial cells in which LT-induced barrier disruption did not require cell death (62).
Early-stage implications.
The role of LT in early-stage anthrax is not understood. It is not known whether there is significant germination of spores and production of LT in the lung before dissemination to the lymph nodes. Studies in the 1950s demonstrated that vegetative bacteria are found in the lymph nodes of the lung within 5 h after intratrachial spore delivery but did not define whether germination occurred in the lung, in transit to the lymph node, or in the lymph node (53). Recent evidence suggests that germination can occur in the lung (38), and studies that modeled the incubation period of anthrax support the possibility of early germination and toxin production (10). If germination occurs in the human lung before dissemination to the lymph nodes, LT may be produced in the lung. In fact, studies that aim to model the incubation period of anthrax take into consideration that early germination and toxin production are a possibility (10). Our findings suggest that if LT is produced early in the lung, disruption of the epithelial barrier may occur before sepsis is established. Early disruption of the epithelial barrier would facilitate entry of spores or bacteria into the pulmonary interstitium. There, pulmonary lymphatic channels provide a route for transit to the mediastinal lymph nodes, as is known to occur in the initial stages of inhalational anthrax. Alternately, direct dissemination through the adjacent endothelium into the pulmonary vasculature may occur, particularly if the endothelium is also disrupted by LT, as has been demonstrated (62). This suggests an additional method of dissemination that does not require transport of the organism to the lymph nodes from the alveolar space.
ACKNOWLEDGMENTS
We thank Karen Ridge of Northwestern University for her helpful advice regarding isolation of primary human AEC.
This work was funded by the NIH for J. P. Metcalf, K. M. Coggeshall, J. D. Ballard (all grant number AI062629), and F. Lupu (grant number GM097747), by the American Heart Association for S. Awasthi (grant-in-aid 11GRNT7220012), and by the Deutsche Forschungsgemeinschaft for A. Braun (grant number SFB587,B4).
Footnotes
Published ahead of print 1 October 2012
REFERENCES
- 1. Aggarwal S, Suzuki T, Taylor WL, Bhargava A, Rao RK. 2011. Contrasting effects of ERK on tight junction integrity in differentiated and under-differentiated Caco-2 cell monolayers. Biochem. J. 433:51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bachofen M, Weibel ER. 1977. Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicemia. Am. Rev. Respir. Dis. 116:589–615 [DOI] [PubMed] [Google Scholar]
- 3. Barakat LA, et al. 2002. Fatal inhalational anthrax in a 94-year-old Connecticut woman. JAMA 287:863–868 [DOI] [PubMed] [Google Scholar]
- 4. Barbee RW, Perry BD, Re RN, Murgo JP. 1992. Microsphere and dilution techniques for the determination of blood flows and volumes in conscious mice. Am. J. Physiol. 263:R728–R733 [DOI] [PubMed] [Google Scholar]
- 5. Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37:911–917 [DOI] [PubMed] [Google Scholar]
- 6. Bonuccelli G, et al. 2005. ATR/TEM8 is highly expressed in epithelial cells lining Bacillus anthracis' three sites of entry: implications for the pathogenesis of anthrax infection. Am. J. Physiol. Cell Physiol. 288:C1402–C1410 [DOI] [PubMed] [Google Scholar]
- 7. Borio L, et al. 2001. Death due to bioterrorism-related inhalational anthrax: report of 2 patients. JAMA 286:2554–2559 [DOI] [PubMed] [Google Scholar]
- 8. Boyer AE, et al. 2009. Kinetics of lethal factor and poly-d-glutamic acid antigenemia during inhalation anthrax in rhesus macaques. Infect. Immun. 77:3432–3441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bresgen N, et al. 2008. Ferritin and FasL (CD95L) mediate density dependent apoptosis in primary rat hepatocytes. J. Cell. Physiol. 217:800–808 [DOI] [PubMed] [Google Scholar]
- 10. Brookmeyer R, Johnson E, Barry S. 2005. Modelling the incubation period of anthrax. Stat. Med. 24:531–542 [DOI] [PubMed] [Google Scholar]
- 11. Chakrabarty K, et al. 2006. Bacillus anthracis spores stimulate cytokine and chemokine innate immune responses in human alveolar macrophages through multiple mitogen-activated protein kinase pathways. Infect. Immun. 74:4430–4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen J, Chen Z, Narasaraju T, Jin N, Liu L. 2004. Isolation of highly pure alveolar epithelial type I and type II cells from rat lungs. Lab. Invest. 84:727–735 [DOI] [PubMed] [Google Scholar]
- 13. Chen Z, et al. 2004. Identification of two novel markers for alveolar epithelial type I and II cells. Biochem. Biophys. Res. Commun. 319:774–780 [DOI] [PubMed] [Google Scholar]
- 14. Chuang CY, et al. 2011. Toll-like receptor 2-mediated sequential activation of MyD88 and MAPKs contributes to lipopolysaccharide-induced sp-a gene expression in human alveolar epithelial cells. Immunobiology 216:707–714 [DOI] [PubMed] [Google Scholar]
- 15. Crapo JD, Barry BE, Gehr P, Bachofen M, Weibel ER. 1982. Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 125:740–745 [DOI] [PubMed] [Google Scholar]
- 16. Cunningham AC, et al. 1994. Constitutive expression of MHC and adhesion molecules by alveolar epithelial cells (type II pneumocytes) isolated from human lung and comparison with immunocytochemical findings. J. Cell Sci. 107(Part 2):443–449 [DOI] [PubMed] [Google Scholar]
- 17. Dobbs LG, Gonzalez RF, Allen L, Froh DK. 1999. HTI56, an integral membrane protein specific to human alveolar type I cells. J. Histochem. Cytochem. 47:129–137 [DOI] [PubMed] [Google Scholar]
- 18. Dudek SM, Garcia JG. 2001. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 91:1487–1500 [DOI] [PubMed] [Google Scholar]
- 19. During RL, et al. 2007. Anthrax lethal toxin paralyzes actin-based motility by blocking Hsp27 phosphorylation. EMBO J. 26:2240–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elbert KJ, et al. 1999. Monolayers of human alveolar epithelial cells in primary culture for pulmonary absorption and transport studies. Pharm. Res. 16:601–608 [DOI] [PubMed] [Google Scholar]
- 21. Ezzell JW, Ivins BE, Leppla SH. 1984. Immunoelectrophoretic analysis, toxicity, and kinetics of in vitro production of the protective antigen and lethal factor components of Bacillus anthracis toxin. Infect. Immun. 45:761–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fang X, et al. 2006. Contribution of CFTR to apical-basolateral fluid transport in cultured human alveolar epithelial type II cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 290:L242–L249 [DOI] [PubMed] [Google Scholar]
- 23. Friedlander AM, Bhatnagar R, Leppla SH, Johnson L, Singh Y. 1993. Characterization of macrophage sensitivity and resistance to anthrax lethal toxin. Infect. Immun. 61:245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fuchs S, et al. 2003. Differentiation of human alveolar epithelial cells in primary culture: morphological characterization and synthesis of caveolin-1 and surfactant protein-C. Cell Tissue Res. 311:31–45 [DOI] [PubMed] [Google Scholar]
- 25. Garcia JG, et al. 2001. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Invest. 108:689–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guidi-Rontani C. 2002. The alveolar macrophage: the Trojan horse of Bacillus anthracis. Trends Microbiol. 10:405. [DOI] [PubMed] [Google Scholar]
- 27. Gutierrez-Canas I, et al. 2003. VIP and PACAP are autocrine factors that protect the androgen-independent prostate cancer cell line PC-3 from apoptosis induced by serum withdrawal. Br. J. Pharmacol. 139:1050–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hartsock A, Nelson WJ. 2008. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 1778:660–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hattersley SM, Greenman J, Haswell SJ. 2011. Study of ethanol induced toxicity in liver explants using microfluidic devices. Biomed. Microdevices 13:1005–1014 [DOI] [PubMed] [Google Scholar]
- 30. Kikkawa Y, Yoneda K. 1974. The type II epithelial cell of the lung. I. Method of isolation. Lab. Invest. 30:76–84 [PubMed] [Google Scholar]
- 31. Kuo SR, et al. 2008. Anthrax toxin-induced shock in rats is associated with pulmonary edema and hemorrhage. Microb. Pathog. 44:467–472 [DOI] [PubMed] [Google Scholar]
- 32. Lehmann M, Noack D, Wood M, Perego M, Knaus UG. 2009. Lung epithelial injury by B. anthracis lethal toxin is caused by MKK-dependent loss of cytoskeletal integrity. PLoS One 4:e4755 doi:10.1371/journal.pone.0004755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lipschutz JH, Li S, Arisco A, Balkovetz DF. 2005. Extracellular signal-regulated kinases 1/2 control claudin-2 expression in Madin-Darby canine kidney strain I and II cells. J. Biol. Chem. 280:3780–3788 [DOI] [PubMed] [Google Scholar]
- 34. Liu S, et al. 2009. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc. Natl. Acad. Sci. U. S. A. 106:12424–12429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu S, et al. 2010. Anthrax toxin targeting of myeloid cells through the CMG2 receptor is essential for establishment of Bacillus anthracis infections in mice. Cell Host Microbe 8:455–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 37. Lupu C, et al. 1999. Cellular effects of heparin on the production and release of tissue factor pathway inhibitor in human endothelial cells in culture. Arterioscler. Thromb. Vasc. Biol. 19:2251–2262 [DOI] [PubMed] [Google Scholar]
- 38. Lyons CR, et al. 2004. Murine model of pulmonary anthrax: kinetics of dissemination, histopathology, and mouse strain susceptibility. Infect. Immun. 72:4801–4809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mabry R, et al. 2006. Detection of anthrax toxin in the serum of animals infected with Bacillus anthracis by using engineered immunoassays. Clin. Vaccine Immunol. 13:671–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matthay MA, Zemans RL. 2011. The acute respiratory distress syndrome: pathogenesis and treatment. Annu. Rev. Pathol. 6:147–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moayeri M, Haines D, Young HA, Leppla SH. 2003. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J. Clin. Invest. 112:670–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moayeri M, Leppla SH. 2009. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol. Aspects Med. 30:439–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mueller H, Franke WW. 1983. Biochemical and immunological characterization of desmoplakins I and II, the major polypeptides of the desmosomal plaque. J. Mol. Biol. 163:647–671 [DOI] [PubMed] [Google Scholar]
- 44. Nizet V, et al. 1996. Group B streptococcal beta-hemolysin expression is associated with injury of lung epithelial cells. Infect. Immun. 64:3818–3826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Papadimitriou E, et al. 2012. Differential regulation of the two RhoA-specific GEF isoforms Net1/Net1A by TGF-beta and miR-24: role in epithelial-to-mesenchymal transition. Oncogene 31:2862–2875 [DOI] [PubMed] [Google Scholar]
- 46. Park JM, Greten FR, Li ZW, Karin M. 2002. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science 297:2048–2051 [DOI] [PubMed] [Google Scholar]
- 47. Peifer M, McCrea PD, Green KJ, Wieschaus E, Gumbiner BM. 1992. The vertebrate adhesive junction proteins beta-catenin and plakoglobin and the Drosophila segment polarity gene armadillo form a multigene family with similar properties. J. Cell Biol. 118:681–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rao JY, et al. 1990. Cellular F-actin levels as a marker for cellular transformation: relationship to cell division and differentiation. Cancer Res. 50:2215–2220 [PubMed] [Google Scholar]
- 49. Richards RJ, Davies N, Atkins J, Oreffo VI. 1987. Isolation, biochemical characterization, and culture of lung type II cells of the rat. Lung 165:143–158 [DOI] [PubMed] [Google Scholar]
- 50. Ridsdale R, Na CL, Xu Y, Greis KD, Weaver T. 2011. Comparative proteomic analysis of lung lamellar bodies and lysosome-related organelles. PLoS One 6:e16482 doi:10.1371/journal.pone.0016482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rolando M, et al. 2009. Injection of Staphylococcus aureus EDIN by the Bacillus anthracis protective antigen machinery induces vascular permeability. Infect. Immun. 77:3596–3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rolando M, et al. 2010. Transcriptome dysregulation by anthrax lethal toxin plays a key role in induction of human endothelial cell cytotoxicity. Cell Microbiol. 12:891–905 [DOI] [PubMed] [Google Scholar]
- 53. Ross JM. 1957. The pathogenesis of anthrax following the administration of spores by the respiratory route. J. Pathol. Bacteriol. 73:485 [Google Scholar]
- 54. Silva MT. 2010. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. 584:4491–4499 [DOI] [PubMed] [Google Scholar]
- 55. Stewart JC. 1980. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 104:10–14 [DOI] [PubMed] [Google Scholar]
- 56. Strengert M, Knaus UG. 2011. Analysis of epithelial barrier integrity in polarized lung epithelial cells, p 195–206 In Turksen K. (ed), Permeability barrier: methods and protocols, vol 763 Springer, New York, NY: [DOI] [PubMed] [Google Scholar]
- 57. Uhal BD, Rannels SR, Rannels DE. 1989. Flow cytometric identification and isolation of hypertrophic type II pneumocytes after partial pneumonectomy. Am. J. Physiol. 257:C528–C536 [DOI] [PubMed] [Google Scholar]
- 58. Vanden Berghe T, et al. 2010. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 17:922–930 [DOI] [PubMed] [Google Scholar]
- 59. Vesterdal LK, et al. 2012. Carbon black nanoparticles and vascular dysfunction in cultured endothelial cells and artery segments. Toxicol. Lett. 214:19–26 [DOI] [PubMed] [Google Scholar]
- 60. Wang J, et al. 2007. Differentiated human alveolar epithelial cells and reversibility of their phenotype in vitro. Am. J. Respir. Cell Mol. Biol. 36:661–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang Y, Zhang J, Yi XJ, Yu FS. 2004. Activation of ERK1/2 MAP kinase pathway induces tight junction disruption in human corneal epithelial cells. Exp. Eye Res. 78:125–136 [DOI] [PubMed] [Google Scholar]
- 62. Warfel JM, Steele AD, D'Agnillo F. 2005. Anthrax lethal toxin induces endothelial barrier dysfunction. Am. J. Pathol. 166:1871–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Witherden IR, Tetley TD. 2001. Isolation and culture of human alveolar type II pneumocytes. Methods Mol. Med. 56:137–146 [DOI] [PubMed] [Google Scholar]
- 64. Wu W, et al. 2009. Resistance of human alveolar macrophages to Bacillus anthracis lethal toxin. J. Immunol. 183:5799–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wu X, Molinaro C, Johnson N, Casiano CA. 2001. Secondary necrosis is a source of proteolytically modified forms of specific intracellular autoantigens: implications for systemic autoimmunity. Arthritis Rheum. 44:2642–2652 [DOI] [PubMed] [Google Scholar]
- 66. Yang L, et al. 2010. Surfactant protein B propeptide contains a saposin-like protein domain with antimicrobial activity at low pH. J. Immunol. 184:975–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zar JH. 1996. Biostatistical analysis. Prentice-Hall, Englewood Cliffs, NJ [Google Scholar]