

Abstract
The Toll-like receptor 2 (TLR2)/TLR1 receptor complex responds to amyloid fibrils, a common component of biofilm material produced by members of the phyla Firmicutes, Bacteroidetes, and Proteobacteria. To determine whether this TLR2/TLR1 ligand stimulates inflammatory responses when bacteria enter intestinal tissue, we investigated whether expression of curli amyloid fibrils by the invasive enteric pathogen Salmonella enterica serotype Typhimurium contributes to T helper 1 and T helper 17 responses by measuring cytokine production in the mouse colitis model. A csgBA mutant, deficient in curli production, elicited decreased expression of interleukin 17A (IL-17A) and IL-22 in the cecal mucosa compared to the S. Typhimurium wild type. In TLR2-deficient mice, IL-17A and IL-22 expression was blunted during S. Typhimurium infection, suggesting that activation of the TLR2 signaling pathway contributes to the expression of these cytokines. T cells incubated with supernatants from bone marrow-derived dendritic cells (BMDCs) treated with curli fibrils released IL-17A in a TLR2-dependent manner in vitro. Lower levels of IL-6 and IL-23 production were detected in the supernatants of the TLR2-deficient BMDCs treated with curli fibrils. Consistent with this, three distinct T-cell populations—CD4+ T helper cells, cytotoxic CD8+ T cells, and γδ T cells—produced IL-17A in response to curli fibrils in the intestinal mucosa during S. Typhimurium infection. Notably, decreased IL-6 expression by the dendritic cells and decreased IL-23 expression by the dendritic cells and macrophages were observed in the cecal mucosa of mice infected with the curli mutant. We conclude that TLR2 recognition of bacterial amyloid fibrils in the intestinal mucosa represents a novel mechanism of immunoregulation, which contributes to the generation of inflammatory responses, including production of IL-17A and IL-22, in response to bacterial entry into the intestinal mucosa.
INTRODUCTION
The intestinal lumen is host to a diverse population of microbes, which provide benefits by conferring protection against pathogens. While the presence of these beneficial microbes in the intestinal lumen does not evoke overt host responses, microbial invasion of host tissue needs to be met by a rapid activation of host defenses through innate immune mechanisms. One mechanism by which the innate immune system senses microbial entry into tissue is the detection of conserved microbial structures, known as pathogen-associated molecular patterns (PAMPs), by host cell receptors, such as Toll-like receptors (TLRs) (40). However, while this concept is well established through tissue culture models, less is known about the identity of signals that contribute to inflammation in the intestine, a site exposed to large microbial communities.
Amyloid fibrils are a PAMP that is commonly present in biofilm material produced by members of the phyla Firmicutes, Bacteroides, and Proteobacteria (41, 44). The best-characterized member of this family of surface structures is the curli (55), which are amyloid fibrils (13) produced by Salmonella enterica serovar Typhimurium, Escherichia coli, and other closely related members of the Enterobacteriaceae (65, 85). Curli amyloid fibrils of E. coli and S. Typhimurium stimulate innate immune responses (4, 5), which requires both TLR2 (79, 80) and TLR1 (78). Although other TLR2/TLR1 ligands, such as lipoproteins, can be purified from the bacterial cell wall, curli amyloid fibrils are a prominent TLR2/TLR1 agonist recognized on intact cells of E. coli (78). TLR2 is expressed by various cell types in the gastrointestinal tract, including epithelial cells and antigen-presenting cells (10–12, 35, 59). Collectively, these observations raise the question of whether amyloid fibrils induce TLR2-dependent responses when bacteria transit from the gut lumen into the intestinal mucosa.
To address this question, we studied a pathogen, S. Typhimurium, which actively invades the intestinal mucosa, thereby evoking a rapid and robust activation of host defenses. S. Typhimurium causes acute cecal inflammation in mice preconditioned by treatment with streptomycin (mouse colitis model), in part by using its invasion-associated type III secretion system (T3SS-1) to enter the intestinal mucosa (2). One challenge of detecting the contribution of TLR2-dependent responses in vivo is the fact that the presence of S. Typhimurium in tissue is sensed by a multitude of additional pathways, including the detection of flagellin by TLR5 (26), the detection of the lipid A moiety of lipopolysaccharide (LPS) by TLR4 (81), the detection of cell wall fragments by nucleotide-binding oligomerization domain 1 (NOD1) and NOD2 (24, 25, 46), the detection of cytosolic access by T3SS-1 through NLRC4 (NOD-like receptor [NLR] family caspase-associated recruitment domain [CARD]-containing protein 4) (21, 52, 53) and the activation of NLRP3 (NLR family pyrin domain-containing protein 3) by an unknown mechanism (8).
Due to the multitude of mechanisms contributing to S. Typhimurium-induced intestinal inflammation, inactivation of a single pathway does not reduce the overall severity of pathological changes in the mucosa. For example, activation of NLRC4 and NLRP3 results in proteolytic activation of interleukin 1β (IL-1β), an important proinflammatory cytokine. However, IL-1 receptor deficiency has little effect on the development of intestinal pathology in the mouse colitis model of S. Typhimurium infection (43). Similarly, mice deficient for TLR2, TLR4, or TLR5 do not exhibit significant changes in the severity of intestinal pathology in this model (33). In contrast, mice deficient for myeloid differentiation primary response protein 88 (MyD88), an adaptor protein required for signaling through TLR2, TLR4, TLR5, and the receptor for IL-1β, exhibit a marked reduction in the severity of intestinal lesions in the mouse colitis model (43). These data suggest that pathology scoring does not provide sufficient sensitivity to resolve the contribution of an individual pathway to S. Typhimurium-induced inflammation.
A more sensitive approach to monitoring inflammation is the detection of cytokine responses, particularly those that become amplified in tissue. Two types of immune responses operational during S. Typhimurium-induced colitis are generated by the activation of the CD4+ T helper cells, specifically the T helper 1 (Th1) immune response and the activation of T helper 17 (Th17) immune responses (67). The Th17-type immune response consists of antigen-presenting cells that, in response to microbial stimuli, release IL-23, IL-6, and/or IL-1β, cytokines which in turn act on naïve T cells to induce the production of IL-17A and IL-22 (27, 28, 43). Recently, a novel innate lymphoid cell population, lymphoid tissue inducer (LTi) cells, has also been shown to produce IL-17A as well as IL-22 upon inflammation in the gut (39, 75). The Th1 type response consists of antigen-presenting cells that, in response to microbial stimuli, produce tumor necrosis factor alpha (TNF-α) as well as IL-12 and/or IL-18, two cytokines that act on T cells and NK cells to induce gamma interferon (IFN-γ) production (20, 27, 34, 49–51). The amplification products IL-17A, IL-22, and IFN-γ are among the cytokines whose expression is most prominently induced in the intestinal mucosa early after S. Typhimurium infection (27, 63). Expression levels of IL-17A, IL-22, and IFN-γ are therefore sensitive indicators of inflammation elicited during S. Typhimurium infection.
Here, we addressed the in vivo role of TLR2 activation by curli fibrils during S. Typhimurium-induced acute intestinal inflammation by monitoring the expression of cytokines that are involved in the Th1- and Th17-type cell responses in the mouse colitis model.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
S. Typhimurium strain IR715 is a fully virulent, spontaneous nalidixic acid-resistant derivative of strain ATCC 14028 (73). SF15 (82), a derivative of IR715 which carries a marked mutation in the csgBA genes, was used to generate an unmarked deletion of the csgBA genes. A cellulose mutant that has a kanamycin cassette insertion in the bcsE gene was kindly provided by John Gunn at Ohio State University. To confer streptomycin resistance, plasmid pHP45Ω was introduced into all strains used in animal experiments (60). Bacteria were grown in Luria-Bertani (LB) broth or LB agar containing the following antibiotics as appropriate: carbenicillin (100 μg/ml), nalidixic acid (50 μg/ml), and kanamycin (50 μg/ml).
Generation of a curli mutant.
To generate an unmarked deletion of csgBA, the upstream and downstream regions of csgBA genes were amplified with primer pairs Agf7-Agf8 and Agf3-Agf4 (Table 1), respectively. These PCR products were ligated to each other after digestion with PstI. Another PCR product was amplified from this DNA using primers Agf9 and Agf10. The resulting PCR product was cloned into pCR2.1 vector and transformed into Top10 cells (Invitrogen). The insert in pCR2.1 was digested with EcoRI and ligated into vector pRDH10, which was previously digested with the same enzyme, giving rise to plasmid pSF24, and this plasmid was transformed into E. coli S17 λpir (70). The strain carrying the unmarked csgBA deletion was produced by introducing plasmid pSF24 into S. Typhimurium SF15 by conjugation. An exconjugant with an unmarked deletion of csgBA was selected by sacB counterselection and designated CT16.
Table 1.
Primers used in this studya
| Primer | Sequence (5′-3′) |
|---|---|
| Agf3 | AACTGCAGTTTACCACGACTCAGCAAGG |
| Agf4 | AAATGGATAACGCCGCCC |
| Agf7 | GTAGGGATAATCGTCAGGGG |
| Agf8 | AACTGCAGTTGTCTTTGTGCTGTCCAGG |
| Agf9 | GTTTTTGCGGCTTAAATTATCCTG |
| Agf10 | GAGGGTTCGGCGTTACCTTATAG |
All primers were developed as part of the present study.
Characterization of bacterial strains.
To detect curli formation by Western blotting, bacteria grown on T-medium plates containing 0.1% tryptone (pH 7.2) at 30°C for 48 h were recovered from plates, and curli fibrils were depolymerized by 90% formic acid treatment as described previously (15). Next, formic acid-treated extracts were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), and proteins were transferred to Immobilon-P (Millipore) membranes using a Trans-Blot semidry transfer cell (Bio-Rad). The presence of CsgA was detected by using rabbit anti-CsgA serum as described previously (37).
Curli expression was further detected by flow cytometry in bacterial strains grown on T-medium plates at 30°C for 48 h. Bacterial cells were recovered in phosphate-buffered saline (PBS), and curli fibril expression was detected by labeling cells with rabbit anti-CsgA serum (1:250 dilution) and goat anti-rabbit fluorescein isothiocyanate (FITC) conjugate (1:250 dilution; Jackson Immunolabs). The FITC fluorescence (CsgA expression) intensity was determined for each particle (LSRII; Becton, Dickinson).
Cellulose production was monitored by binding of colonies grown on LB agar supplemented with 200 μg/ml calcofluor (fluorescent brightener 28) at room temperature for 48 h. Calcofluor binding was observed under a 366-nm UV light source (71). Motility of bacteria was tested as described previously (84).
Invasion assay.
The human colon carcinoma cell line HT-29 was obtained from the American Type Culture Collection and maintained in McCoy's 5a medium with 1.5 mM l-glutamine (Gibco) supplemented with 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 atmosphere. HT-29 cells were seeded at a density of 5 × 105 cells per well in 24-well tissue culture plates 24 h prior to the experiment. Bacteria were grown on T-medium plates at 30°C for 48 h to induce curli formation (15). S. Typhimurium cells were recovered from the plates in PBS and added to the HT-29 monolayers at a multiplicity of infection (MOI) of 5 for 1 h. Cells were washed five times with Dulbecco's phosphate-buffered saline (DPBS) (Gibco), and the medium was replaced with medium containing 0.1 mg/ml gentamicin (Gibco). After 90 min, cells were washed three times with DPBS and lysed with 0.5 ml of 1% Triton X-100 (Sigma). Intracellular bacteria were quantified by spreading serial 10-fold dilutions of cell lysates on LB agar plates to determine the number of CFU. The experiment was repeated three times.
Experimental infections of mice.
Four- to six-week-old female C57BL/6 mice and mice deficient in TLR2 (B6.129-Tlr2tm1Kir/J) were obtained from The Jackson Laboratories. The streptomycin-pretreated mouse model was described previously (2). Briefly, mice were inoculated intragastrically with 20 mg of streptomycin (0.1 ml of a 200 mg/ml solution in PBS) 24 h before bacterial inoculation. Bacteria were grown with shaking in LB broth containing carbenicillin at 37°C overnight. For infection, groups of five mice were inoculated intragastrically with either 0.1 ml of sterile LB broth (mock infection) or 1 × 109 CFU of S. Typhimurium strains carrying plasmid pHP45W. Mice were sacrificed at indicated time points after infection. To determine the number of viable S. Typhimurium, samples of cecum (proximal section), liver, spleen, Peyer's patches, mesenteric lymph nodes, and colon contents were collected from each mouse and homogenized in PBS, and 10-fold serial dilutions were plated on LB agar plates containing antibiotic. The tip of the cecum was collected for histopathological analysis. The center section of the cecum was immediately snap-frozen in liquid nitrogen and stored at −80°C for RNA isolation. For intracellular staining and flow cytometry analysis, animals were injected with 250 μg of brefeldin A (in PBS) intravenously 4 h prior to the appropriate time points, 48 and 72 h after infection, as described previously (48). All animal experiments were at least repeated twice with identical results.
Real-time PCR.
RNA was extracted from snap-frozen tissues with 1 ml TriReagent (Molecular Research Center) according to the manufacturer's protocol. Reverse transcription of total RNA (1 μg) was performed in 50 μl volume according to manufacturer's instructions (TaqMan reverse transcription reagents; Applied Biosystems). Real-time PCR was performed using the SYBR green method (Applied Biosystems) according to the manufacturer's instructions. Real-time PCR was performed for each cDNA sample (5 μl per reaction) in duplicate using the 7900HT fast real-time PCR system. For the mouse experiments, primers for mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (66), mouse IL-17A (57), mouse IL-22 (27), mouse gamma interferon (IFN-γ) (57), mouse tumor necrosis factor alpha (TNF-α) (83), mouse lipocalin 2 (62), regenerating islet-derived 3 gamma (Reg3γ) (27), and Reg3β (77) were used. Results were analyzed using the comparative Ct method and by normalizing the data to GAPDH. Fold increases in gene expression in infected mice were calculated relative to the average level of the respective cytokine in the mock-infected mice.
Isolation of cells from the cecum.
Ceca of mice were removed and cleared of feces with ice-cold Ca2+- and Mg2+-free Hanks balanced salt solution (Mediatech, Inc.) with 2% fetal bovine serum (FBS) (Gibco) and 15 mM HEPES (Fisher Scientific). Cecal tissue was cut into 5-mm sections and placed in a buffer containing Ca2+- and Mg2+-free Hanks balanced salt solution (Mediatech, Inc.) with 10% FBS (Gibco), 2 mM EDTA, and 15 mM HEPES (Fisher Scientific). Tissue sections were incubated at 37°C with shaking (100 rpm) for 20 min. To extract intraepithelial lymphocytes (IELs), the mixture was put through a 100-μm cell strainer. IELs were contained within the buffer, and tissue was placed in fresh buffer for a second extraction. IEL-depleted tissue sections were then washed free of EDTA with ice-cold Ca2+- and Mg2+-free Hanks balanced salt solution (Mediatech, Inc.) with 2% FBS (Gibco) and 15 mM HEPES (Fisher Scientific). Tissue was subsequently digested with 400 μg/ml of Liberase (Roche) and 500 μg/ml of DNase (Sigma) in Ca2+- and Mg2+-free Hanks balanced salt solution (Mediatech, Inc.) including 2% bovine serum albumin for 30 min at 37°C. Digested tissue was subjected to homogenization with a gentleMACS dissociator (Miltenyi Biotech). Tissue- and IEL-derived cell suspensions were subjected to enrichment via a Percoll (GE) density gradient (35/80%) as described previously (47).
Flow cytometry.
Cecal IELs and tissue-derived single-cell suspensions were treated with LEAF (low endotoxin azide-free)-purified anti-mouse CD16/32 antibody (Biolegend) for 5 min on ice at a concentration of 1.0 μg per 106 cells in 100 μl. To identify T cells and B cells, the following antibodies were used: Alexa Fluor 700-conjugated anti-mouse CD4 antibody (Biolegend), Pacific Blue-conjugated anti-mouse CD45R/B220 antibody (Biolegend), FITC-conjugated anti-mouse CD314 (NKG2D) antibody (Biolegend), allophycocyanin (APC)-Cy7-conjugated anti-mouse CD8a antibody (Biolegend), phycoerythrin (PE)-Cy7-conjugated anti-mouse CD3 antibody (Biolegend), and APC-conjugated anti-mouse γδ-T-cell receptor (γδ-TCR) (Ebioscience). Cells were incubated with these antibodies (0.5 μg per 106 cells in a 100-μl volume) for 30 min on ice in the dark. Cells were fixed with 4% paraformaldehyde (PFA) and permeabilized with BD Perm/Wash buffer (BD Bioscience) according to the manufacturer's instructions. Cells were then stained for intracellular IL-17 using PE-conjugated anti-mouse IL-17A antibody (Biolegend) for 30 min in the dark at a concentration of 0.5 μg per 106 cells in a 100-μl volume.
To identify monocytes and granulocytes, the following antibodies were used: Alexa Fluor 488-conjugated anti-mouse F4/80 antibody (Biolegend), PE-Cy7-conjugated anti-mouse CD205 (DEC-205) antibody (Biolegend), PerCP-Cy5.5-conjugated anti-mouse CD11b antibody (Biolegend), APC-Cy7-conjugated anti-mouse Ly-6G/Ly-6C (Gr-1) antibody (Biolegend), and Pacific Blue-conjugated anti-mouse CD45R/B220 antibody (Biolegend). Cells were incubated with these antibodies at a concentration of 0.5 μg per 106 cells for 30 min on ice in the dark. Next, cells were fixed with 4% PFA and permeabilized with BD Perm/Wash buffer (BD Bioscience) according to the manufacturer's suggested protocol. Cells were then stained for intracellular IL-23 and IL-6 using a PE-conjugated anti-mouse IL-12/IL-23 p40 antibody (Biolegend) and APC-conjugated anti-mouse IL-6 antibody (Biolegend) for 30 min in the dark.
To identify LTi cells, we used a dump gate system using the following antibodies: Alexa Fluor 700-conjugated anti-mouse CD4 antibody (Biolegend), Pacific Blue-conjugated anti-mouse CD45R/B220 antibody (Biolegend), PerCP-Cy5.5-conjugated anti-mouse CD11b antibody, APC-Cy7-conjugated anti-mouse Ly-6G/Ly-6C (Gr-1) antibody, APC-conjugated anti-mouse CD11c (eBioscience), PE-Cy7-conjugated anti-mouse NK-1.1 antibody (Biolegend), and FITC-conjugated anti-CD3 antibody (BD Biosciences). Cells were stained for IL-17A as described above.
Cells were analyzed using a LSRII FACScan flow cytometer (BD Biosciences), and data were processed using FlowJo software.
Dendritic-cell cultures.
BMDCs were cultured as described previously (23). Briefly, femurs were removed and flushed with complete medium (Iscove's modified Dulbecco's medium, 10% FBS, 2 mM glutamine, ß-mercaptoethanol, and antibiotic). Cells were then seeded at a concentration of 1 × 106/ml in complete medium supplemented with 3 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) (Pharmingen) in 24-well plates. Half the medium was removed and replaced with fresh medium containing GM-CSF every other day. On day 6, cells were treated with purified curli fibrils (10 μg/ml, 2.5 μg/ml, 1.25 μg/ml, and 0.625 μg/ml) for 6 h, and supernatants were removed to be analyzed for IL-6 and IL-23 production by enzyme-linked immunosorbent assay (ELISA) (eBioscience) and to stimulate T-cell cultures.
In vitro T-cell culture.
Naïve CD4+ T cells were purified from spleens of C57BL/6 mice using Automacs (Miltenyi). T cells were seeded in 24-well plates pretreated with 5 μg/ml anti-CD3 antibody (BD Biosciences) at 2 × 105/well. Cells were then cultured in the presence of 1 μg/ml anti-CD28 antibody (BD Biosciences), and supernatants were collected from bone marrow-derived dendritic cells. Supernatants were removed to analyze IL-17 production by ELISA after 72 h. The experiment was repeated twice, with similar results.
Statistical analysis.
A parametric test (Student's t test) was used to determine whether differences were statistically significant (P < 0.05) for all experiments except flow cytometry. For tissue culture experiments, percentage values were transformed logarithmically prior to statistical analysis using Student's t test. For analysis of bacterial numbers and cytokine expression in vivo, values were converted logarithmically to calculate geometric means. For flow cytometry experiments, a one-way analysis of variance (ANOVA) was used to calculate statistically significant differences (P < 0.05).
RESULTS
Characterization of the curli mutant.
To investigate the role of curli fibrils during intestinal inflammation, we constructed a mutant which lacks the ability to produce curli fibrils (CT16, csgBA) by introducing an unmarked deletion of the csgBA curli biosynthesis genes into an S. Typhimurium wild-type strain (IR715). Production of curli fibrils was monitored by flow cytometry (Fig. 1A) as well as Western blotting in whole-cell extracts from the S. Typhimurium wild type and its isogenic csgBA mutant grown on T-medium plates (Fig. 1B). As expected, curli fibrils were detected only on the S. Typhimurium wild-type strain. Cellulose is coexpressed with curli fibrils, and production of cellulose has recently been reported to reduce the inflammatory responses in uroepithelial cells generated by curli fibrils of a uropathogenic E. coli strain (42). Therefore, we determined if the deletion of csgBA genes affected this strain's ability to produce cellulose. We used plates containing calcofluor and a strain which carries a mutation in the bcsE cellulose biosynthesis gene as a negative control. Both wild-type S. Typhimurium and the csgBA mutant displayed bright fluorescence under UV light, indicating normal production of cellulose (Fig. 1C), whereas no fluorescence was detected in the negative control. Motility contributes to the ability of S. Typhimurium to invade the intestinal mucosa and trigger inflammation (72). We determined that the motility of the csgBA mutant was similar to that of wild-type S. Typhimurium (Fig. 1D). Next, we assessed the invasiveness of both the wild-type and mutant strains using a colonic epithelial cell line, HT-29 (Fig. 1E). Invasiveness of the csgBA mutant was not significantly different (P > 0.05) from that of the S. Typhimurium wild type. Finally, we measured the expression of the lipoprotein-encoding genes lppA and lppB. We did not see any differences in the gene expression profiles between the S. Typhimurium wild type and the csgBA mutant (data not shown). We concluded that comparison of the wild type and the csgBA mutant was well suited to specifically investigate the role of curli fibrils in the intestinal phase of S. Typhimurium infection.
Fig 1.

Characterization of an S. Typhimurium mutant carrying an unmarked deletion of csgBA genes. Expression of curli fibrils was detected by flow cytometry (A) and Western blotting (B). Cellulose production was monitored on plates containing calcofluor (C). Motility was determined on plates containing 0.3% agar (D). Invasiveness of strains was tested using human colonic epithelial (HT-29) cells (E). Each experiment was repeated three times independently, with similar outcomes, and a representative example is shown.
Curli fibrils contribute to induction of IL-22 and IL-17A transcripts in the cecal mucosa.
The goal of this study was to determine whether curli fibrils, a potent TLR2/TLR1 ligand (78), contribute to cytokine responses in the intestinal mucosa during S. Typhimurium infection. To test this hypothesis, we performed a time course experiment in which wild-type mice (C57BL/6) were pretreated with streptomycin and orally infected with the S. Typhimurium wild-type strain, an isogenic csgBA mutant, or sterile medium (mock infection). Groups of mice were euthanized at 24, 48, 72, and 96 h after infection. Consistent with the invasion data, no differences in bacterial numbers recovered from the cecum, colon contents, and mesenteric lymph nodes were observed between the S. Typhimurium wild type and the csgBA mutant at any of the time points investigated (Fig. 2).
Fig 2.

Bacterial numbers in the organs of streptomycin-pretreated mice after infection with wild-type S. Typhimurium and a csgBA mutant (curli deficient). The numbers of CFU in the colon contents (A), cecum (B), and mesenteric lymph nodes (C) were determined at the indicated time points.
To compare the Th1 and Th17 inflammatory responses, we extracted RNA from the cecal mucosa and determined the relative expression of genes encoding TNF-α (Tnfa), IFN-γ (Ifng), IL-22 (Il22), and IL-17A (Il17a) by quantitative real-time PCR. In addition, we monitored expression of several genes that are highly induced during S. Typhimurium infection (27): Reg3g, encoding regenerating islet-derived 3 gamma (REGIIIγ); Reg3b, encoding REGIIIβ; and Lcn2, encoding lipocalin 2. Compared to mock-infected mice, the development of acute intestinal inflammation in animals infected with the S. Typhimurium wild type was accompanied by a marked increase in the expression of Tnfa, Ifng, Il22, Il17a, Lcn2 (Fig. 3), Reg3g, and Reg3b (data not shown). Expression of Tnfa, Ifng, Il22, and Il17a peaked at 48 h after infection, while expression Lcn2 peaked at 96 h after infection. There were no significant differences in the mRNA levels of Tnfa, Ifng (Fig. 3A and B), Reg3g, and Reg3b (data not shown) induced by the csgBA mutant and those induced by the S. Typhimurium wild type. However, mRNA levels of Il22 were significantly lower (P < 0.05) 24 h, 48 h, and 96 h after infection with the csgBA mutant than after infection with the wild type (Fig. 3C). Similarly, reduced transcript levels of Il17a were detected at 48 h after infection with the csgBA mutant compared to infection with the wild type (Fig. 3D). Notably, Lcn2 expression was significantly reduced at 72 h in the animals infected with the csgBA mutant compared to animals infected with the wild type (Fig. 3E).
Fig 3.

Immune response elicited in the ceca of streptomycin-pretreated mice after infection with wild-type S. Typhimurium and a csgBA mutant (curli deficient). Transcript levels of Tnfa (A), Ifng (B), Il17a (C), Il22 (D), and Lcn2 (E) were measured by quantitative real-time PCR at the indicated time points after infection. Bars represent changes in mRNA levels compared to a group of mock-infected mice from the same time point (geometric means and standard errors). ns, not significant.
TLR2 activation contributes to expression of IL-17A and IL-22 in the cecal mucosa.
Responses to curli fibrils are mediated though the TLR2/TLR1 complex and lead to the activation of nuclear factor kappa–light-chain enhancer of activated B cells (NF-κB) (78). To determine whether decreased induction of Il17a and Il22 mRNA 48 h after infection with the csgBA mutant was due to the reduced activation of TLR2, we repeated the experiment with TLR2-deficient mice. No significant differences in bacterial numbers were detected in the colon contents, cecum, or mesenteric lymph nodes between wild-type (C57BL/6) and TLR2-deficient mice 48 h after infection with the S. Typhimurium wild type or the curli mutant (data not shown).
Interestingly, expression of Il22 and Il17a was significantly lower in TLR2-deficient mice than in wild-type mice (C57BL/6) infected with the S. Typhimurium wild type. Remarkably, the expression levels of Il22 and Il17a induced by the S. Typhimurium wild type in TLR2-deficient mice were similar to those elicited by the csgBA mutant in wild-type (C57BL/6) mice. We did not detect any differences in the expression of Ifng between wild-type mice (C57BL/6) and TLR2-deficient mice (Fig. 4). We next evaluated the contribution of TLR2 to the inflammatory pathology in the intestine. There were no differences in the overall cecal pathology score between wild-type mice (C57BL/6) and TLR2-deficient mice infected with the S. Typhimurium wild type or the csgBA mutant (data not shown). Collectively, these data suggested that TLR2 contributes to the induction of Il22 and Il17a expression in the cecal mucosa during S. Typhimurium infection and that this response was at least in part dependent on the ability of S. Typhimurium to produce curli fibrils.
Fig 4.

Immune response elicited in the cecum of streptomycin-pretreated C57BL/6 and TLR2-deficient (TLR2-/-) mice after infection with wild-type S. Typhimurium and a csgBA mutant (curli deficient). Transcript levels of Il17a (A), Il22 (B), and Ifng (C) were measured by quantitative real-time PCR at 48 h after infection. Bars represent changes in mRNA levels compared to a group of mock-infected mice from the same time point (geometric means and standard errors). ns, not significant.
Curli fibrils induce IL-17A production in vitro.
Cytokines released by antigen-presenting cells can stimulate T cells to release IL-17A and IL-22, which is a mechanism to amplify cytokine responses in tissue (67). One T-cell subset involved in this process in the intestinal mucosa is CD4+ T helper cells, also known as Th17 cells (43), and IL-17A and IL-22 are therefore often referred to as Th17 cytokines. One possible mechanism by which curli amyloid fibrils might induce Il22 and Il17a expression in the intestinal mucosa is a TLR2-dependent activation of antigen-presenting cells to release cytokines, such as IL-6 and IL-23, which in turn act on T cells (27, 28, 43). Interestingly, addition of recombinant serum amyloid A to cocultures of lamina propria dendritic cells (DCs) and naïve CD4+ T cells results in the differentiation of cells and production of Th17 cytokines (38). This observation was consistent with our hypothesis, because serum amyloid A and curli amyloid fibrils both induce host responses through TLR2 (14, 36, 79), presumably because amyloids of host and microbial origins share structural features (80). We therefore investigated the effect of TLR2 activation by curli fibrils on the differentiation of Th17 cells and production of Th17 cytokines in vitro.
To this end, curli fibrils were purified from a S. Typhimurium msbB mutant, a strain expressing tetra-acylated lipid A that does not signal through TLR4. We isolated bone marrow-derived DCs (BMDCs) from wild-type (C57BL/6) and TLR2-deficient mice and incubated them for 6 h with different concentrations of purified curli fibrils. CD4+ T cells isolated from spleens of wild-type C57BL/6 mice were incubated with the BMDC supernatants for 72 h. Remarkably, T cells incubated with supernatants of BMDC derived from wild-type mice produced significantly larger amounts of IL-17A (P < 0.05) than T cells incubated with supernatants from BMDCs derived from TLR2-deficient mice (Fig. 5A). These data suggested that curli fibrils induced BMDCs to produce cytokines that drove release of IL-17A by T cells in a TLR2-dependent manner. Consistent with this idea, supernatants from wild-type BMDCs stimulated with curli fibrils contained significantly larger amounts of IL-6 and IL-23 (P < 0.05) than supernatants from BMDCs derived from TLR2-deficient mice (Fig. 5B and C). Collectively, these data support the model that curli fibrils signal through TLR2 on antigen-presenting cells to induce the release of cytokines that in turn stimulate T cells to release IL-17A.
Fig 5.
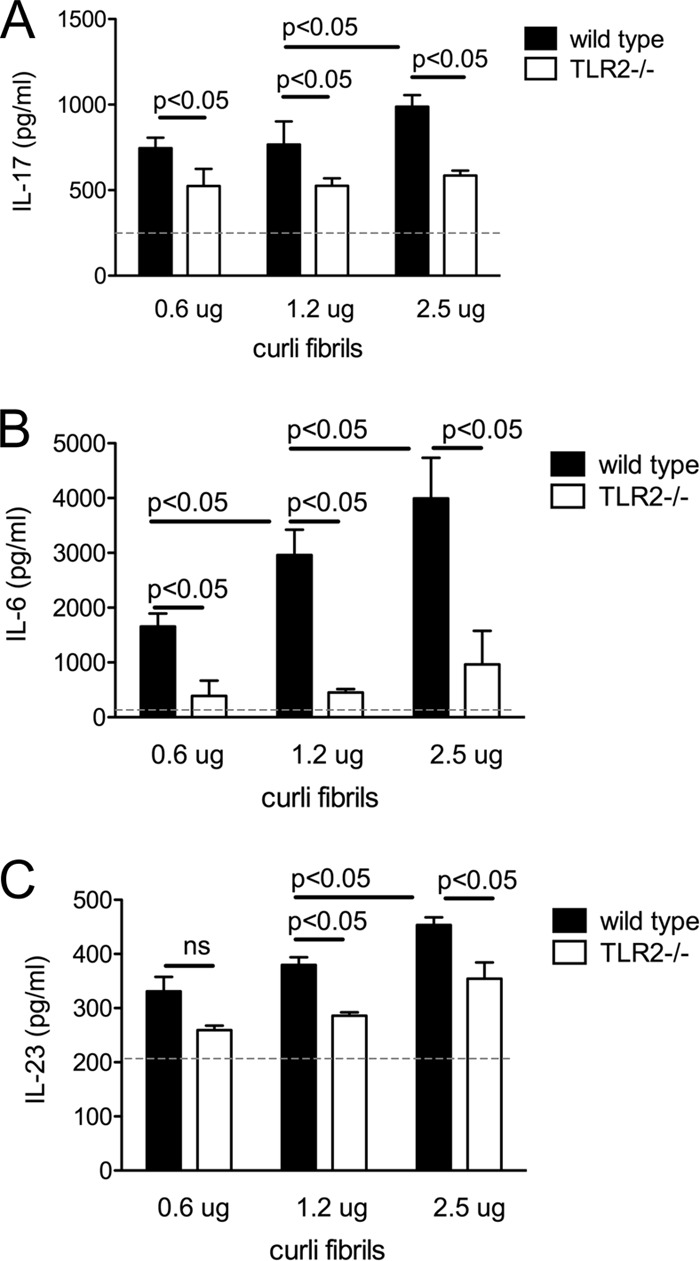
Splenic naïve CD4+ T cells were treated with supernatants collected from bone marrow-derived dendritic cells, which were previously incubated with increasing concentrations of curli fibrils (0.6 μg/ml, 1.2 μg/ml, and 2.5 μg/ml), for 6 h in the presence of anti-CD3 and anti-CD28 antibodies. IL-17 production was determined in the T-cell supernatants after 72 h (A) by ELISA. IL-6 (B) and IL-23 (C) production was determined in the bone marrow-derived dendritic cell supernatants from wild-type and TLR2-deficient mice, which were treated with curli fibrils (0.6 μg/ml, 1.2 μg/ml, and 2.5 μg/ml). Background cytokines levels in the untreated supernatants were marked with a dashed line in the graphs. ns, not significant.
Curli fibrils activate CD4+ T helper cells, CD8+ cytotoxic T cells, and γδ T cells to produce IL-17A in vivo.
In addition to CD4+ T cells, other cell populations have recently been shown to produce IL-17 by either direct or indirect stimulation by microbes or their stimuli during infection. CD8+ cytotoxic T cells, γδ T cells, NK cells, NKT cells, and LTi cells are among the populations that are significant producers of IL-17A upon stimulation (16, 27, 29, 54, 56, 58, 68, 75). To confirm our in vitro findings with CD4+ T cells and further determine which of the above populations contribute to IL-17A production in vivo during S. Typhimurium infection, we employed a multicolor flow cytometry approach. We infected C57BL/6 mice with either the wild type or the csgBA mutant following streptomycin pretreatment. At 48 or 72 h postinfection, IELs and tissue-associated lymphocytes were collected from ceca of mice. By using antibodies specific for various cell types and flow cytometry, we found that IL-17A production was significantly higher in CD3+ cell populations, suggesting that T cells were involved in the production of this cytokine (data not shown). When we investigated the various cell populations among CD3+ cells, we did not see any difference in the percentages of CD3+ CD4+, CD3+ CD8+, and γδ T cells between animals infected with S. Typhimurium and those infected with its csgBA mutant in either IEL fractions (data not shown) or tissue fractions (Fig. 6A) at 48 h after infection. However, there was a marked increase in the mean fluorescence intensity of R-phycoerythrin (PE), which was indicative of IL-17A production, in the tissue fractions obtained from animals infected with wild-type S. Typhimurium compared to the fluorescence intensity in animals infected with the csgBA mutant among CD3+ CD4+, CD3+ CD8+, and γδ T cells at this time point (Fig. 6A and B). At 72 h after infection, we observed a significant increase in the number of γδ T cells in the animals infected with wild-type S. Typhimurium while the numbers of γδ T cells stayed the same in the animals infected with the csgBA mutant. Notably, there was a significant increase in the percentage of CD3+ CD8+ T cells in the tissue fractions of animals infected with the csgBA mutant compared to animals infected with wild-type S. Typhimurium at 72 h postinfection (Fig. 6C). Nonetheless, IL-17A production by all the cell populations was lower, as determined by lower mean fluorescence intensity of PE, at 72 h than at 48 h. Moreover, there were no differences in IL-17A production by the cell populations investigated between the animal groups infected with wild-type S. Typhimurium and those infected with its csgBA mutant (Fig. 6C and D). When we investigated the LTi cells at 48 h postinfection, we determined that although the abundance of LTi cells were very small at this time point, the production of IL-17A by these cells was immense (Fig. 6E and F). NK cells as well as NKT cells were also contributors of IL-17A production independent of curli production at both time points investigated. At both 48 and 72 h, we did not observe any significant changes in the production of IL-17A in IELs in the animals infected with either wild-type S. Typhimurium or the csgBA mutant (data not shown).
Fig 6.

Immune cell infiltration and IL-17 production in C57BL/6 mice infected with wild-type S. Typhimurium and csgBA mutant (curli deficient) bacteria. Cecal tissue-derived cells, depleted of intraepithelial lymphocytes (IELs), were stained with antibodies for CD3, CD4, CD8, B220, γδ-TCR, NKG2D, and IL-17A and analyzed using flow cytometry. The proportion of CD3+ CD4+, CD3+ CD8+, or γδ T cells in total tissue-associated lymphocytes at 48 (A) and 72 (C) hours postinfection was determined. The mean fluorescence intensity (MFI) of PE-IL-17A was determined among CD3+ CD4+, CD3+ CD8+, and γδ T cells at 48 (B) and 72 (D) hours postinfection. The proportion of LTi cells in tissue-associated lymphocytes (E) and their IL-17A-producing capacity (F) were determined 48 h postinfection by analyzing CD4+, CD3−, CD8−, B220−, GR1−, CD11b−, CD11c−, and NK1.1− cells.
To determine the role of antigen-presenting cells in the process of IL-17A production and confirm our in vitro findings, we used the flow cytometry approach and measured IL-6 and IL-23 production by dendritic cells and macrophages in the cecal tissue. There was no observed difference in the percentage of dendritic cells or macrophages at 48 h between the two groups of animals infected with wild-type S. Typhimurium or the csgBA mutant. However, IL-6 production among dendritic cells but not macrophages was significantly higher in the animals infected with wild-type S. Typhimurium than in the animals infected with the csgBA mutant. Interestingly, IL-23 production in both dendritic cells and macrophages was significantly higher in the tissue of mice infected with wild-type S. Typhimurium than in that of mice infected with the csgBA mutant (Fig. 7).
Fig 7.
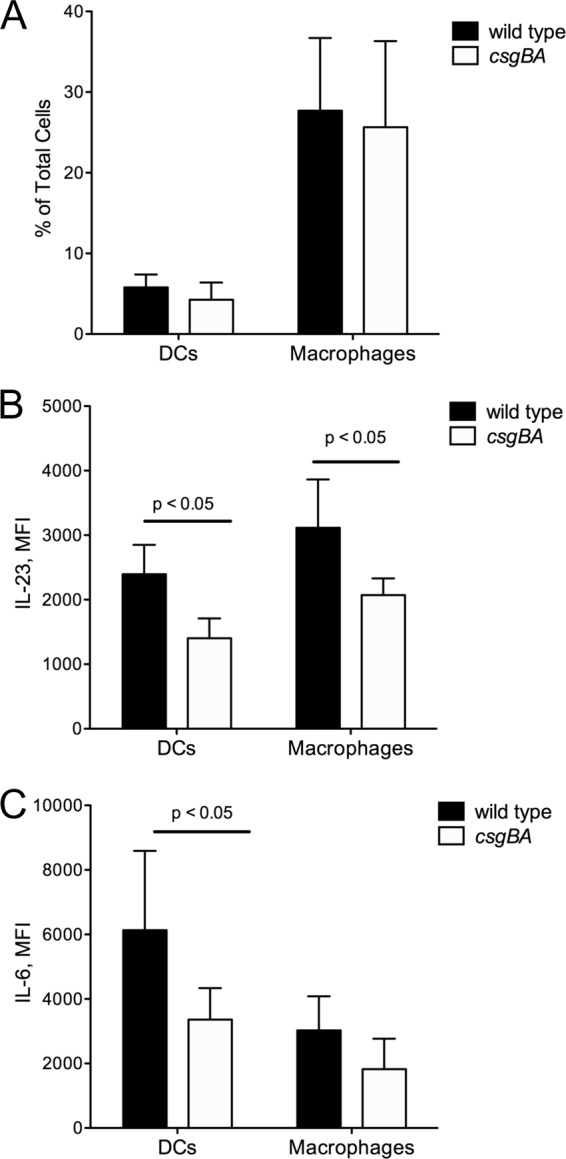
IL-6 and IL-23 production in dendritic cells and macrophages in C57BL/6 mice infected with wild-type S. Typhimurium and csgBA mutant (curli-deficient) bacteria 48 h postinfection. (A) The proportion of dendritic cells (CD11b+ and DEC-205+) and macrophages (F4/80+) in cecal tissue-derived cells was analyzed via flow cytometry. The mean fluorescence intensity of IL-23 (B) and IL-6 (C) among dendritic cells and macrophages was also determined. Significant statistical differences are indicated (P < 0.05).
DISCUSSION
The intestinal immune system must avoid producing excessive inflammatory responses against the microbial communities colonizing the gut lumen, while at the same time orchestrating appropriate inflammatory responses against microbes that breach the epithelial barrier. One pathogen recognition receptor implicated in innate immune surveillance in the intestinal mucosa is TLR2 (9, 10). Epithelial cells and antigen-presenting cells in the intestinal mucosa express TLR2 (10–12, 35, 59). Expression of TLR2 is elevated in individuals with inflammatory bowel disease (22) and the receptor has been shown to control intestinal epithelial barrier function by augmenting tight junction formation (10). These data suggest that detection of conserved microbial structures by TLR2 contributes to the maintenance of intestinal homeostasis.
Triacylated lipoproteins are an important component of the Gram-negative bacterial cell wall (32) and stimulate innate immune responses through TLR2 and TLR1 in vitro (1, 6, 76). It is generally assumed that lipoproteins are largely responsible for TLR2-dependent responses induced by Gram-negative bacterial cells in vivo. However, this idea has never been convincingly demonstrated through mutant analysis, because bacterial genomes encode a multitude of lipoproteins, some of which are essential for cell wall-related functions, thereby complicating mutant analysis. For example, the E. coli genome encodes 96 different lipoproteins, some of which are highly abundant in the cell envelope (7). Inactivation of the abundant Braun lipoproteins in S. Typhimurium produces a pleiotropic phenotype, including inhibition of motility, reduced invasiveness, and avirulence, in mice (17, 19, 69).
The idea that lipoproteins are the predominant TLR2-ligand detected on intact bacterial cells has recently been called into question by tissue culture experiments with intact E. coli cells. These experiments reveal that NF-κB activation is much greater when HeLa cells expressing TLR2 and TLR1 are stimulated with wild-type E. coli expressing curli amyloid fibrils than with an isogenic E. coli csgBA mutant that cannot elaborate this surface structure (78). Curli amyloid fibrils might serve as an important TLR2-ligand detected on intact bacterial cells because these structures are secreted, thereby being more readily accessible to detection by TLR2 than lipoproteins, which are buried in the cell envelope. Our data suggest that detection of amyloid fibrils did contribute to TLR-2-dependent cytokine expression in the intestinal mucosa in response to infection with an invasive enteric pathogen, S. Typhimurium.
TLR2 was required to induce maximum expression of Il22 and Il17a, but not of Tnfa and Ifng, in the cecal mucosa during S. Typhimurium infection. IL-17A is produced by a number of immune cells, including (but not limited to) CD4+ T cells (Th17 cells), γδ T cells, CD8+ T cells, and LTi cells, in the intestinal mucosa of mice (28, 43, 68). However, the mechanism through which S. Typhimurium infection induces IL-17A production by these cell types in vivo has not been fully resolved. Both NOD1/NOD2-dependent and MyD88-dependent mechanisms contribute to IL-17A production in the cecal mucosa, and the relative importance of each mechanism changes over time (25, 43). The TLR2/curli-dependent induction of Il17a expression in the cecal mucosa during S. Typhimurium infection described here is likely to be a mechanism that contributes to the MyD88-dependent induction of Il17a expression observed previously (43). Interestingly, although the overall expression patterns of Il17a and IL22 were blunted in TLR2 deficient animals, wild-type S. Typhimurium elicited higher levels of expression of these cytokines than the csgBA mutant, suggesting that other signaling pathways or receptors may also contribute to the recognition of curli fibrils. Consistent with this, recent studies determined an intracellular innate receptor, NALP3, for amyloid-beta of Alzheimer's disease, which also signals via TLR2 as a consequence of its fibrillar amyloidogenic quaternary structure (31, 80). Interestingly, NALP3 is involved in the generation of IL-1β, a cytokine involved in promoting the differentiation of Th17 cells as well as direct stimulation of γδ T cells to produce IL-17A (30, 74). Thus, it is intriguing to speculate that the expression of Il17a may be affected by IL-1β production through the stimulation of NALP3 by curli fibrils even in the absence of TLR2. However, further analysis is required to confirm this hypothesis. We are investigating the mechanism underlying this interesting phenotype.
In vitro stimulation with cytokines produced by antigen-presenting cells, such as IL-23 or IL-6, induces T-cell differentiation that biases T cells toward a Th17 phenotype and subsequent IL-17 production (3, 30, 61, 74). In a coculture experiment, curli amyloid fibrils signaled through TLR2 on dendritic cells to induce the release of cytokines, which in turn induced release of IL-17A by T cells. This scenario is compatible with previous observations, indicating that T cells are important for IL-17A production in the cecal mucosa during S. Typhimurium infection (27). Correspondingly, when we used multicolor flow cytometry approach to investigate the cell types that contribute to the production of IL-17A, we found that the csgBA mutant elicited lower levels of IL-23 and IL-6, cytokines that drive the differentiation of CD4+ T cells, in antigen-presenting cells in the cecal mucosa of mice. Furthermore, IL-17A was produced not only by CD4+ T cells but also by γδ T cells, CD8+ T cells, and LTi cells in the cecal tissue during infection. However, the expression of curli by S. Typhimurium affected IL-17A production only by CD4+ T cells, γδ T cells, and CD8+ T cells. Although LTi cells produced IL-17A in large amounts, the percentage of this population was small (approximately 1% of total cells) at the time of the investigation (48 h), and curli expression affected neither the population percentage nor the IL–17A production by LTi cells. This could be due to the fact that LTi cells are innate cells that are active during initial stages of infection, and thus, 48 h postinfection could be too late to investigate the features of this population.
Our in vitro data suggest that curli fibrils drive the differentiation of naïve CD4+ cells into Th17 cells. Although the details of the Th17 cell differentiation in vivo are not fully understood, IL–17A production by CD4+ cells does not occur till 3 days after in vitro stimulation, suggesting that differentiated Th17 cells do not arise at early times during the infection. Thus, the IL–17A production that we observed by CD4 + and CD8+ T cells in vivo at 48 h was most likely due to memory responses generated by immune cells during previous bacterial or amyloid encounters. Despite the fact that curli are produced only by a number of commensal and pathogenic enteric bacteria, amyloids are present in biofilms of representatives from the phyla Bacteroidetes and Firmicutes, which dominate the microbiota of the gastrointestinal tract (13, 18, 41, 44, 45, 64). Moreover, expression of various serum amyloid A (SAA) proteins in the gastrointestinal tract has been demonstrated (38). Since TLR2 recognizes both curli fibrils and host amyloid fibrils, including amyloid-beta and SAA, it is possible that T cells and antigen-presenting cells become exposed to various amyloids through out their life span, which could generate a memory response. However, to support this hypothesis and determine the involvement of TLR2 in such immune responses, further investigation is needed.
To our knowledge, our study is the first to demonstrate that TLR2 contributes to IL-17A and IL-22 responses in the mucosa via the recognition of bacterial amyloids, specifically, curli fibrils in the gut mucosa. Furthermore, our data point to TLR2 as a sentinel receptor that enables cells in the intestinal mucosa to monitor bacterial translocation from the gut.
ACKNOWLEDGMENTS
Work in C.T.'s laboratory was supported by Scientist Development Grant 0835248N from the American Heart Association and Mid-Atlantic Regional Center for Excellence for Biodefense and Emerging Infectious Diseases Research grant U54 AI57168 from the National Institutes of Health. Work in D.G.'s laboratory was supported by Public Health Service Grants AI047325, AI084065, AI052306. Work in R.M.T.'s laboratory was supported by Public Health Service Grants AI082320 and AI050553. G.J.R. was supported by a grant from the Pennsylvania Department of Health.
The Pennsylvania Department of Health specifically disclaims responsibilities for any analyses, interpretations, and conclusions.
Footnotes
Published ahead of print 1 October 2012
REFERENCES
- 1. Aliprantis AO, et al. 1999. Cell activation and apoptosis by bacterial lipoproteins through Toll-like receptor-2. Science 285:736–739 [DOI] [PubMed] [Google Scholar]
- 2. Barthel M, et al. 2003. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect. Immun. 71:2839–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bettelli E, et al. 2006. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441:235–238 [DOI] [PubMed] [Google Scholar]
- 4. Bian Z, Brauner A, Li Y, Normark S. 2000. Expression of and cytokine activation by Escherichia coli curli fibers in human sepsis. J. Infect. Dis. 181:602–612 [DOI] [PubMed] [Google Scholar]
- 5. Bian Z, Yan ZQ, Hansson GK, Thoren P, Normark S. 2001. Activation of inducible nitric oxide synthase/nitric oxide by curli fibers leads to a fall in blood pressure during systemic Escherichia coli infection in mice. J. Infect. Dis. 183:612–619 [DOI] [PubMed] [Google Scholar]
- 6. Brightbill HD, et al. 1999. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science 285:732–736 [DOI] [PubMed] [Google Scholar]
- 7. Brokx SJ, et al. 2004. Genome-wide analysis of lipoprotein expression in Escherichia coli MG1655. J. Bacteriol. 186:3254–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Broz P, et al. 2010. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med. 207:1745–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cario E, et al. 2002. Commensal-associated molecular patterns induce selective toll-like receptor-trafficking from apical membrane to cytoplasmic compartments in polarized intestinal epithelium. Am. J. Pathol. 160:165–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cario E, Gerken G, Podolsky DK. 2007. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology 132:1359–1374 [DOI] [PubMed] [Google Scholar]
- 11. Cario E, Gerken G, Podolsky DK. 2004. Toll-like receptor 2 enhances ZO-1-associated intestinal epithelial barrier integrity via protein kinase C. Gastroenterology 127:224–238 [DOI] [PubMed] [Google Scholar]
- 12. Chabot S, Wagner JS, Farrant S, Neutra MR. 2006. TLRs regulate the gatekeeping functions of the intestinal follicle-associated epithelium. J. Immunol. 176:4275–4283 [DOI] [PubMed] [Google Scholar]
- 13. Chapman MR, et al. 2002. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295:851–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheng N, He R, Tian J, Ye PP, Ye RD. 2008. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J. Immunol. 181:22–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collinson SK, Emody L, Muller KH, Trust TJ, Kay WW. 1991. Purification and characterization of thin, aggregative fimbriae from Salmonella enteritidis. J. Bacteriol. 173:4773–4781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cua DJ, Tato CM. 2010. Innate IL-17-producing cells: the sentinels of the immune system. Nat. Rev. Immunol. 10:479–489 [DOI] [PubMed] [Google Scholar]
- 17. Dailey FE, Macnab RM. 2002. Effects of lipoprotein biogenesis mutations on flagellar assembly in Salmonella. J. Bacteriol. 184:771–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dueholm MS, et al. 2010. Functional amyloid in Pseudomonas. Mol. Microbiol. 77:1009–1020 [DOI] [PubMed] [Google Scholar]
- 19. Fadl AA, et al. 2005. Murein lipoprotein is a critical outer membrane component involved in Salmonella enterica serovar typhimurium systemic infection. Infect. Immun. 73:1081–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Flores-Langarica A, et al. 2011. T-zone localized monocyte-derived dendritic cells promote Th1 priming to Salmonella. Eur. J. Immunol. 41:2654–2665 [DOI] [PubMed] [Google Scholar]
- 21. Franchi L, et al. 2006. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 7:576–582 [DOI] [PubMed] [Google Scholar]
- 22. Frolova L, Drastich P, Rossmann P, Klimesova K, Tlaskalova-Hogenova H. 2008. Expression of Toll-like receptor 2 (TLR2), TLR4, and CD14 in biopsy samples of patients with inflammatory bowel diseases: upregulated expression of TLR2 in terminal ileum of patients with ulcerative colitis. J. Histochem. Cytochem. 56:267–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gallucci S, Lolkema M, Matzinger P. 1999. Natural adjuvants: endogenous activators of dendritic cells. Nat. Med. 5:1249–1255 [DOI] [PubMed] [Google Scholar]
- 24. Geddes K, et al. 2010. Nod1 and Nod2 regulation of inflammation in the Salmonella colitis model. Infect. Immun. 78:5107–5115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Geddes K, et al. 2011. Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat. Med. 17:837–844 [DOI] [PubMed] [Google Scholar]
- 26. Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. 2001. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J. Immunol. 167:1882–1885 [DOI] [PubMed] [Google Scholar]
- 27. Godinez I, et al. 2008. T cells help to amplify inflammatory responses induced by Salmonella enterica serotype Typhimurium in the intestinal mucosa. Infect. Immun. 76:2008–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Godinez I, et al. 2009. Interleukin-23 orchestrates mucosal responses to Salmonella enterica serotype Typhimurium in the intestine. Infect. Immun. 77:387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goto M, et al. 2009. Murine NKT cells produce Th17 cytokine interleukin-22. Cell Immunol. 254:81–84 [DOI] [PubMed] [Google Scholar]
- 30. Guo L, et al. 2009. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc. Natl. Acad. Sci. U. S. A. 106:13463–13468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Halle A, et al. 2008. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9:857–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hantke K, Braun V. 1973. Covalent binding of lipid to protein. Diglyceride and amide-linked fatty acid at the N-terminal end of the murein-lipoprotein of the Escherichia coli outer membrane. Eur. J. Biochem. 34:284–296 [DOI] [PubMed] [Google Scholar]
- 33. Hapfelmeier S, et al. 2008. Microbe sampling by mucosal dendritic cells is a discrete, MyD88-independent step in ΔinvG S. Typhimurium colitis. J. Exp. Med. 205:437–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harrington L, Srikanth CV, Antony R, Shi HN, Cherayil BJ. 2007. A role for natural killer cells in intestinal inflammation caused by infection with Salmonella enterica serovar Typhimurium. FEMS Immunol. Med. Microbiol. 51:372–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hart AL, et al. 2005. Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology 129:50–65 [DOI] [PubMed] [Google Scholar]
- 36. He RL, et al. 2009. Serum amyloid A induces G-CSF expression and neutrophilia via Toll-like receptor 2. Blood 113:429–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Humphries AD, et al. 2003. The use of flow cytometry to detect expression of subunits encoded by 11 Salmonella enterica serotype Typhimurium fimbrial operons. Mol. Microbiol. 48:1357–1376 [DOI] [PubMed] [Google Scholar]
- 38. Ivanov II, et al. 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139:485–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ivanov II, Diehl GE, Littman DR. 2006. Lymphoid tissue inducer cells in intestinal immunity. Curr. Top. Microbiol. Immunol. 308:59–82 [DOI] [PubMed] [Google Scholar]
- 40. Janeway CA., Jr 1989. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp. Quant Biol. 54:1–13 [DOI] [PubMed] [Google Scholar]
- 41. Jordal PB, et al. 2009. Widespread abundance of functional bacterial amyloid in mycolata and other gram-positive bacteria. Appl. Environ. Microbiol. 75:4101–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kai-Larsen Y, et al. 2010. Uropathogenic Escherichia coli modulates immune responses and its curli fimbriae interact with the antimicrobial peptide LL-37. PLoS Pathog. 6:e1001010 doi:10.1371/journal.ppat.1001010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Keestra AM, et al. 2011. Early, MyD88-dependent induction of interleukin-17A expression during Salmonella colitis. Infect. Immun. 79:3131–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Larsen P, et al. 2007. Amyloid adhesins are abundant in natural biofilms. Environ. Microbiol. 9:3077–3090 [DOI] [PubMed] [Google Scholar]
- 45. Larsen P, Nielsen JL, Otzen D, Nielsen PH. 2008. Amyloid-like adhesins produced by floc-forming and filamentous bacteria in activated sludge. Appl. Environ. Microbiol. 74:1517–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Le Bourhis L, et al. 2009. Role of Nod1 in mucosal dendritic cells during Salmonella pathogenicity island 1-independent Salmonella enterica serovar Typhimurium infection. Infect. Immun. 77:4480–4486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lefrancois L, Lycke N. 2001. Isolation of mouse small intestinal intraepithelial lymphocytes, Peyer's patch, and lamina propria cells. Curr. Protoc. Immunol. 17:3.19.1–3.19.16 [DOI] [PubMed] [Google Scholar]
- 48. Liu F, Whitton JL. 2005. Cutting edge: re-evaluating the in vivo cytokine responses of CD8+ T cells during primary and secondary viral infections. J. Immunol. 174:5936–5940 [DOI] [PubMed] [Google Scholar]
- 49. Mastroeni P, et al. 1999. Interleukin 18 contributes to host resistance and gamma interferon production in mice infected with virulent Salmonella typhimurium. Infect. Immun. 67:478–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mastroeni P, Harrison JA, Chabalgoity JA, Hormaeche CE. 1996. Effect of interleukin 12 neutralization on host resistance and gamma interferon production in mouse typhoid. Infect. Immun. 64:189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mastroeni P, et al. 1998. Interleukin-12 is required for control of the growth of attenuated aromatic-compound-dependent salmonellae in BALB/c mice: role of gamma interferon and macrophage activation. Infect. Immun. 66:4767–4776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Miao EA, et al. 2006. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 7:569–575 [DOI] [PubMed] [Google Scholar]
- 53. Miao EA, et al. 2010. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U. S. A. 107:3076–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Michel ML, et al. 2007. Identification of an IL-17-producing NK1.1− iNKT cell population involved in airway neutrophilia. J. Exp. Med. 204:995–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Olsen A, Jonsson A, Normark S. 1989. Fibronectin binding mediated by a novel class of surface organelles on Escherichia coli. Nature 338:652–655 [DOI] [PubMed] [Google Scholar]
- 56. Ono Y, et al. 2012. T-helper 17 and interleukin-17-producing lymphoid tissue inducer-like T cells make different contributions to colitis in mice. Gastroenterology. doi:10.1053/j.gastro.2012.07.108 [DOI] [PubMed] [Google Scholar]
- 57. Overbergh L, et al. 2003. The use of real-time reverse transcriptase PCR for the quantification of cytokine gene expression. J. Biomol. Tech. 14:33–43 [PMC free article] [PubMed] [Google Scholar]
- 58. Passos ST, et al. 2010. IL-6 promotes NK cell production of IL-17 during toxoplasmosis. J. Immunol. 184:1776–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Podolsky DK, Gerken G, Eyking A, Cario E. 2009. Colitis-associated variant of TLR2 causes impaired mucosal repair because of TFF3 deficiency. Gastroenterology 137:209–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Prentki P, Krisch HM. 1984. In vitro insertional mutagenesis with a selectable DNA fragment. Gene 29:303–313 [DOI] [PubMed] [Google Scholar]
- 61. Rachitskaya AV, et al. 2008. Cutting edge: NKT cells constitutively express IL-23 receptor and RORgammat and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J. Immunol. 180:5167–5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Raffatellu M, et al. 2009. Lipocalin-2 resistance confers an advantage to Salmonella enterica serotype Typhimurium for growth and survival in the inflamed intestine. Cell Host Microbe 5:476–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Raffatellu M, et al. 2008. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat. Med. 14:421–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Romero D, Aguilar C, Losick R, Kolter R. 2010. Amyloid fibers provide structural integrity to Bacillus subtilis biofilms. Proc. Natl. Acad. Sci. U. S. A. 107:2230–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Romling U, Bian Z, Hammar M, Sierralta WD, Normark S. 1998. Curli fibers are highly conserved between Salmonella typhimurium and Escherichia coli with respect to operon structure and regulation. J. Bacteriol. 180:722–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Roux CM, et al. 2007. Brucella requires a functional type IV secretion system to elicit innate immune responses in mice. Cell Microbiol. 9:1851–1869 [DOI] [PubMed] [Google Scholar]
- 67. Santos RL, et al. 2009. Life in the inflamed intestine, Salmonella style. Trends Microbiol. 17:498–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schulz SM, Kohler G, Holscher C, Iwakura Y, Alber G. 2008. IL-17A is produced by Th17, gammadelta T cells and other CD4- lymphocytes during infection with Salmonella enterica serovar Enteritidis and has a mild effect in bacterial clearance. Int. Immunol. 20:1129–1138 [DOI] [PubMed] [Google Scholar]
- 69. Sha J, et al. 2004. The two murein lipoproteins of Salmonella enterica serovar Typhimurium contribute to the virulence of the organism. Infect. Immun. 72:3987–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Simon R, Priefer U, Puhler A. 1983. Vector plasmids for in vivo and in vitro manipulations of gram negative bacteria, p 98–106 In Puhler A. (ed), Molecular genetics of the bacteria-plant interaction. Springer-Verlag, Berlin, Germany [Google Scholar]
- 71. Solano C, et al. 2002. Genetic analysis of Salmonella enteritidis biofilm formation: critical role of cellulose. Mol. Microbiol. 43:793–808 [DOI] [PubMed] [Google Scholar]
- 72. Stecher B, et al. 2004. Flagella and chemotaxis are required for efficient induction of Salmonella enterica serovar Typhimurium colitis in streptomycin-pretreated mice. Infect. Immun. 72:4138–4150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Stojiljkovic I, Baumler AJ, Heffron F. 1995. Ethanolamine utilization in Salmonella typhimurium: nucleotide sequence, protein expression, and mutational analysis of the cchA cchB eutE eutJ eutG eutH gene cluster. J. Bacteriol. 177:1357–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sutton CE, et al. 2009. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31:331–341 [DOI] [PubMed] [Google Scholar]
- 75. Takatori H, et al. 2009. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 206:35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Takeuchi O, et al. 2002. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol. 169:10–14 [DOI] [PubMed] [Google Scholar]
- 77. Tebar LA, et al. 2008. Deletion of the mouse RegIIIbeta (Reg2) gene disrupts ciliary neurotrophic factor signaling and delays myelination of mouse cranial motor neurons. Proc. Natl. Acad. Sci. U. S. A. 105:11400–11405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tukel C, et al. 2010. Toll-like receptors 1 and 2 cooperatively mediate immune responses to curli, a common amyloid from enterobacterial biofilms. Cell Microbiol. 12:1495–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tukel C, et al. 2005. CsgA is a pathogen-associated molecular pattern of Salmonella enterica serotype Typhimurium that is recognized by Toll-like receptor 2. Mol. Microbiol. 58:289–304 [DOI] [PubMed] [Google Scholar]
- 80. Tukel C, et al. 2009. Responses to amyloids of microbial and host origin are mediated through Toll-like receptor 2. Cell Host Microbe 6:45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Vazquez-Torres A, et al. 2004. Toll-like receptor 4 dependence of innate and adaptive immunity to Salmonella: importance of the Kupffer cell network. J. Immunol. 172:6202–6208 [DOI] [PubMed] [Google Scholar]
- 82. Weening EH, et al. 2005. The Salmonella enterica serotype Typhimurium lpf, bcf, stb, stc, std, and sth fimbrial operons are required for intestinal persistence in mice. Infect. Immun. 73:3358–3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wilson RP, et al. 2008. The Vi-capsule prevents Toll-like receptor 4 recognition of Salmonella. Cell Microbiol. 10:876–890 [DOI] [PubMed] [Google Scholar]
- 84. Winter SE, et al. 2009. Contribution of flagellin pattern recognition to intestinal inflammation during Salmonella enterica serotype typhimurium infection. Infect. Immun. 77:1904–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zogaj X, Bokranz W, Nimtz M, Romling U. 2003. Production of cellulose and curli fimbriae by members of the family Enterobacteriaceae isolated from the human gastrointestinal tract. Infect. Immun. 71:4151–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]