

Abstract
Clostridium difficile toxins A (TcdA) and B (TcdB) induce a pronounced systemic and intestinal inflammatory response. A2B adenosine receptors (A2BARs) are the predominant adenosine receptors in the intestinal epithelium. We investigated whether A2BARs are upregulated in human intestinal cells by TcdA or TcdB and whether blockade of A2BARs can ameliorate C. difficile TcdA-induced enteritis and alter the outcome of C. difficile infection (CDI). Adenosine receptor subtype (A1, A2A, A2B, and A3) mRNAs were assayed in HCT-8 cells. Ileal loops from wild-type rabbits and mice and A2BAR−/− mice were treated with TcdA, with or without the selective A2BAR antagonist ATL692 or PSB1115. A murine model of CDI was used to determine the effect of A2BAR deletion or blockade with the orally available agent ATL801, on clinical outcome, histopathology and intestinal interleukin-6 (IL-6) expression from infection. TcdA and TcdB upregulated A2BAR gene expression in HCT-8 cells. ATL692 decreased TcdA-induced secretion and epithelial injury in rabbit ileum. Deletion of A2BARs reduced secretion and histopathology in TcdA-challenged mouse ileum. Deletion or blockade of A2BARs reduced histopathology, IL-6 expression, weight loss, diarrhea, and mortality in C. difficile-infected mice. A2BARs mediate C. difficile toxin-induced enteritis and disease. Inhibition of A2BAR activation may be a potential strategy to limit morbidity and mortality from CDI.
INTRODUCTION
Adenosine receptors—A1, A2A, A2B, and A3—are G protein-coupled receptors expressed in a wide variety of tissues. A2B adenosine receptors (A2BARs) are the predominant adenosine receptors in intestinal epithelial cells and are increased in the presence of inflammation (36). A2BAR mRNA had been shown to be upregulated in the presence of colitis in both murine and human tissues (23). Adenosine, through its activity on A2BAR, mediates chloride secretion (4, 23, 36) and secretion of inflammatory cytokines, interleukin-6 (IL-6), and keratinocyte-derived chemokine (KC) (33). In murine models of inflammatory bowel disease (IBD), the effects of blocking or deleting A2BARs have been inconsistent. Reduced tissue levels of inflammatory mediators, clinical disease activity score, and histopathology were reported by Kolachala et al. (24, 25), while increased pathology was reported by Frick et al. (13).
Clostridium difficile infection (CDI) induces a pronounced systemic and intestinal inflammatory response. Intestinal disease is induced by large clostridial toxins A (TcdA) and B (TcdB) (39). These toxins glucosylate the small G protein family of Rho, Rac, and Cdc42 leading to actin depolymerization, cytoskeleton disruption, and intestinal barrier dysfunction (20, 21). Although the exact mechanisms involved are still unclear, infiltration of inflammatory cells and secretion of proinflammatory mediators occur (32). Inflammatory diarrhea ensues, and a systemic inflammatory response is observed in severe cases. Severe infection presents as pseudomembranous colitis, toxic megacolon, severe sepsis, or septic shock. Although primarily considered antibiotic-precipitated disease, treatment of CDI still relies on antimicrobial agents active against C. difficile. Unfortunately, antimicrobial therapy is associated with recurrent disease in 20% of the initial cases, with increasing recurrence with increasing numbers of prior CDIs (22). Treatment of severe disease and recurrences is challenging.
This study aims to determine whether A2BARs are involved in C. difficile toxin-induced intestinal injury, secretion, and inflammation. Furthermore, we examine the effects of inhibition of A2BAR activity during CDI in the mouse model of the disease.
(This study was partially presented at the 48th Annual Meeting of the Infectious Disease Society of America, Vancouver, Canada, 21 to 24 October 2010.)
MATERIALS AND METHODS
Adenosine receptor subtype assay.
Adenosine receptor subtype (A1, A2A, A2B, and A3) mRNAs were assayed by quantitative PCR (qPCR) in a human colonic cell line, HCT-8, with or without TcdA or TcdB. (Toxins were kindly provided by David Lyerly.) HCT-8 cells were grown in filtered RPMI 1640 medium in the presence of 10% fetal bovine serum, 1 mM sodium pyruvate, and 0.1 U of Pen Strep (penicillin-streptomycin; Gibco catalog no. 15140) in 5% CO2 at 37°C. A 96-well plate was seeded with trypsin-EDTA-dissociated cells in 200 μl of the medium. Upon 80% confluence, the cells were treated with TcdA (0.01, 0.1, 1, 10, and 100 ng/ml) and were incubated from 0 to 4 h. The Invitrogen Fast SYBR green cells-to-CT one-step kit was used under the manufacturer's instructions. For qPCR, 2 μl cDNA sample was mixed with 5 μl Fast SYBR green PCR master mix, 0.2 μl each of 10 μM forward and reverse primers (Table 1), and 2.6 μl DNase-free distilled water (dH2O) in a 96-well plate for DNA amplification performed with the CFX96 real-time PCR detection system (Bio-Rad). The PCR parameters were sequentially set for 3 stages: 1 cycle for 20 s at 95.0°C, 40 cycles for 3 s at 95.0°C and 30 s at 60.0°C, and 1 cycle of a hold at 25.0°C. The relative gene expression was determined using 18S rRNA as the housekeeping gene.
Table 1.
List of primer sequences for reverse transcription-qPCR analyses
| Gene type | Accession no. | Locus positions | Primer | Sequence |
|---|---|---|---|---|
| A1AR | NM_000674 | 530–675 | Forward | GCGGTGAAGGTGAAC |
| Reverse | AGGCAGGTGTGGAAG | |||
| A2AAR | NM_000675 | 1340–1460 | Forward | AGTTCCGCCAGACCTTCC |
| Reverse | ACCTGCTCTCCGTCACTG | |||
| A2BAR | NM_000676 | 704–824 | Forward | GGTCATTGCTGTCCTCTG |
| Reverse | TTCATTCGTGGTTCCATCC | |||
| A3AR | NM_000677 | 621–737 | Forward | AGGGTAGGAATGAGCAAGTTG |
| Reverse | CAGGTGAGCCAGCAAGATC | |||
| 18S rRNA | NR_003286 | 1373–1561 | Forward | CCGATAACGAACGAGACTCTGG |
| Reverse | TAGGGTAGGCACACGCTGAGCC |
Radioligand binding assay of A2BAR antagonists.
Radioligand binding assays were performed with ATL692, ATL801, and PSB1115 as described previously (37). Recombinant mouse ARs were stably expressed in HEK-293 cells (A2AAR, A2BAR, and A3AR), or CHO-K1 cells (A1AR) or transiently expressed along with G protein subunits by baculoviral infection of Sf9 cells (A2AAR, high-affinity assay). The radioligands were 125I-ABA for A1AR and A3AR, 125I-ZM241385 for A2AAR, 125I-ABOPX for A2B AR, or 125I-APE for the A2AAR/high-affinity assay, and radioligand was counted in a Wallac Wizard 1470 gamma counter (Perkin Elmer, Boston, MA). Nonselective binding was measured with the nonselective agonist NECA. Each assay was performed at least in triplicate. Competition binding curves (n = 3 to 4) were constructed and 50% inhibitory concentrations (IC50s) were calculated using a 4-parameter logistic fit (PRISM 5.0; GraphPad Software, San Diego, CA). Ki values were calculated from IC50s (10).
Rabbit ileal loop model.
All animal experiment protocols were approved by the Center for Comparative Medicine of the University of Virginia and Animal Care and Use Committee. New Zealand White rabbits (Burlington) weighing approximately 2 kg were used in the rabbit ileal loop model as described previously (1, 2). Control loops were injected with either phosphate-buffered saline (PBS) (1 ml/loop) alone or TcdA (10 to 20 μg/loop dissolved in 1 ml of PBS). A2BARs in rabbit ileal loops, with or without TcdA (10 μg), were blocked with either ATL692 (0.5 or 5 μM; Dogwood Pharmaceuticals) or PSB1115 (5 μM; Tocris Bioscience), which are highly and moderately A2BAR selective at mouse and human receptors, respectively (3). In one experiment, ileal loops were treated with TcdA (10 μg) alone, PBS alone, and ATL692 (5 μM) alone (no TcdA challenge). Ileal loops were harvested after 4 h of incubation and assessed for intraluminal secretion (expressed as the ratio of the volume of fluid to the length of the loop). A small section of the ileum was snap-frozen for the later myeloperoxidase (MPO) assay. Ileal tissues were fixed with 10% formalin overnight. Fixed tissues were sent in 70% ethanol (EtOH) for further processing and staining with hematoxylin-eosin (H&E) by the UVA Research Histology Core. Each slide was read blindly by at least 2 investigators (C.A.W. and G.M.C.). A grading scale, previously formulated and published (1, 2), was used to grade each slide according to mucosal disruption, cellularity, and vascular congestion.
Murine ileal loop model.
To confirm results from the rabbit experiments and determine the effects of deletion of A2BAR, mouse ileal loop experiments were also performed. C57BL/6 wild-type (WT) and A2BAR −/− mice, weighing 23 to 25 g each, were used for the mouse ileal loop model as previously described (8). Ileal loops were injected with either PBS (100 μl/loop) alone or TcdA (50 μg/loop dissolved in 100 μl of PBS). The ligated ileal loops were then returned to the abdomen, and the abdominal wall was sutured closed. After 4 h of incubation, intraluminal fluid and histopathology were assessed in the harvested ileal specimens as described above. In addition, ileal weight was measured and the weight/length (W/L) ratio was reported in milligrams per centimeter.
MPO assay.
Neutrophilic infiltration was estimated by measuring myeloperoxidase (MPO) activity. Rabbit ileal tissue (50 mg) was homogenized in 1 ml hexadecyltrimethylammonium bromide (Sigma) buffer (1 mg/50 ml) for 15 s, placed in a −70°C freezer for 10 min, and again homogenized. The homogenate was centrifuged at 4,500 rpm for 20 min at 4°C. An aliquot of 7 μl from each sample was pipetted into a 96-well plate in duplicate. Two hundred microliters of o-dianisidine dihydrochloride (Sigma) and 1% hydrogen peroxide were then added. Absorbance was read immediately (time zero, basal level) and at 1 min using an enzyme-linked immunosorbent assay (ELISA) reader at 450 nm. The results were reported as MPO units per mg tissue. One unit of activity was defined as that degrading 1 μmol of hydrogen peroxide per min at 22°C.
Preparation of C. difficile inocula.
C. difficile VPI10463 was purchased from the American Type Culture Collection (Manassas, VA). This strain was maintained in anaerobic chopped meat broth (CMB; Anaerobe System) and incubated for 24 h at 37°C. After incubation, the culture was centrifuged (10,000 × g, 4°C, 10 min) and pelleted. The pellet was washed 2 additional times with brain heart infusion (BHI) prior to resuspension in fresh reduced CMB and quantified by spectrophotometer. An optical density (OD) reading of 1.0 was calculated to be equivalent to 5 × 107 CFU C. difficile/ml. The final concentration of C. difficile in CFU/ml was validated by counting the bacteria using a hemocytometer under a light microscope. All visualized cells were rod shaped, with the minority containing spores.
Murine model of C. difficile infection.
The infection model is a modification of the published protocol by Chen et al. (9). This protocol has been approved by the Center for Comparative Medicine at the University of Virginia. From 6 to 4 days prior to infection, mice were given an antibiotic cocktail containing vancomycin (0.0045 mg/g), colistin (0.0042 mg/g), gentamicin (0.0035 mg/g), and metronidazole (0.0215 mg/g) in drinking water. One day prior to infection, clindamycin (32 mg/kg) was injected subcutaneously. Infection was performed with VPI10463 at an inoculum of 105 cells administered by oral gavage. For knockout infection studies, A2BAR−/− mice (originally from Katya Ravid and bred in-house) with age-matched (6- to 8-week-old) and sex-matched (male or female) wild-type littermates, were used. Mice were divided into the following groups: uninfected A2BAR−/−, infected A2BAR−/−, uninfected A2BAR WT littermates, and infected A2BAR WT littermates. For the wild-type treatment studies, C57BL/6 (The Jackson Laboratory) male 8-week-old mice were used. The mice were divided into the following groups: uninfected wild-type (WT), infected WT, and infected WT treated with A2BAR. Treated mice were given either A2BAR antagonist ATL801 (10 mg/kg/day) or vehicle by chow given daily for 5 days, beginning 1 day postinfection. A clinical scoring system was developed on the basis of weight loss, diarrhea, activity level, and appearance of eyes and hair (with each parameter scored from 0 to 3, where 0 is considered normal and 3 as the worst, with a maximum score of 20). Mice judged moribund by the clinical score (score of ≥14) on any day and all surviving mice at the end of the experiment were sacrificed by cervical dislocation under sedation (ketamine-xylazine). Stool specimens were collected daily. Diarrhea was scored as follows: 1 for soft or color change (yellow), 2 for wet tail or mucoid, and 3 for liquid or no stool (ileus). Mice were observed for either 1 week (knockout studies) or 2 weeks (wild-type treatment studies) after infection. Upon euthanasia, cecal and colonic tissues were fixed in 10% zinc formalin overnight and then placed in 10% ethanol before being sent for paraffin embedding and H&E staining at the UVA Research Histology Core. Histopathologic scoring was performed blindly (C.A.W.) as we have previously described (31).
IHC.
Immunohistochemistry (IHC) was performed on 5-μm formalin-fixed, paraffin-embedded (FFPE) sections of cecum and colon. After deparaffinization through xylene and gradients of EtOH, tissue slides were retrieved in IHC-Tek proteinase solution (catalog no. IW-1101) for 20 min at 37°C. The slides were then processed with the primary antibody anti-IL-6 (1:200 rabbit polyclonal ab6672; Abcam, Cambridge, United Kingdom) diluted in antibody diluent (catalog no. IW-1000 or IW-1001) to reduce background and nonspecific staining for 60 min at room temperature. A serum blocking step was not needed. The anti-rabbit (Dako, Carpinteria, CA) secondary antibody was applied for 30 min. IL-6 was detected by using a high-sensitivity peroxidase visualization kit from Dako (Carpinteria, CA) and 3,3′-diaminobenzidine (DAB) chromogen (Vector, Burlingame, CA). For counterstaining, Mayer's hematoxylin was used (30 s). Slides were washed and rehydrated through gradients of EtOH and xylene. Staining was performed at the Tissue Research Core Facility at the University of Virginia Medical School. Images were taken using an Olympus DP71 microscope and Microsuite Pathology Edition software. IL-6-stained tissue sections were scored blindly (C.A.W. and S.Z.M.) based on the presence and intensity of staining in the epithelial cells, lamina propria, and submucosa. Each section was scored from 0 (absence of staining) to 3 (most intense staining), and the sum of all scores was considered the final IHC score of the tissue section. The maximum score was 9.
Toxin A/B ELISA.
TcdA or TcdB was detected using a Toxin A/B II ELISA kit (Tech Lab, Blacksburg, VA). Each stool sample was weighed, and the amount of diluent per sample was normalized to provide the same mass stool/diluent ratio for each sample. The diluent-sample mixtures were homogenized by grinding and vortexing, and 1:10, 1:100, and 1:1,000 serial dilutions of the sample were made. One hundred fifty microliters of the 1:1,000 dilution of each sample was added to a precoated well provided in the kit. The rest of the procedure was performed as recommended by the manufacturer. The absorbance was read at 450 nm on a Bio Tek Gen5 plate reader (Bio Tek Instruments, Highland Park, VT).
Quantification of C. difficile in stool.
Genomic DNA was extracted from the stool or cecal samples using a modified protocol for the QIAamp DNA stool minikit (Qiagen). All qPCRs were performed on the Bio-Rad iCycler using primers to detect the toxin B (tcdB) gene (forward, GGAGAGTCATCCAACTTATATG; reverse, CCACCAATTTCTTTTAATGCAG). qPCRs, in a final volume of 20 μl, consisted of 10 μl QuantiFast SYBR green Supermix (Qiagen), 1 μl each of forward and reverse primers, 4 μl sterile water, and 4 μl sample DNA. PCR assays were performed in duplicate under the following cycling conditions: 3 min at 95°C, 10 s at 95°C, and 30 s at 57°C for 40 repeats and with a melt curve between 65°C and 95°C of 5 s at 0.5°C intervals.
Toxin assay of C. difficile VPI10463.
ATL801 (stock in dimethyl sulfoxide [DMSO]) was diluted into BHI at final concentrations of 1, 8, and 64 μg/ml using 24-well plates. C. difficile (VPI10463; 5.5 log CFU) was added to each well and incubated in an anaerobic chamber for a total of 24 h. The presence of toxin was assessed in supernatants from each well using the TcdA/B II kit from TechLab according to the manufacturer's instructions as described above.
Statistical analysis.
Results of measurements of gene expression, secretion and inflammation, histopathology, percentage of weight change, clinical score, and diarrhea were expressed as means ± standard errors of the mean (SEM), as generated by GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA). The differences between experimental groups were compared using one-way analysis of variance (ANOVA) with Bonferroni's multiple comparison test. Student's t test was performed to analyze differences between 2 groups. To compare experimental groups across a time period, two-way ANOVA with Bonferroni's correction was performed. Survival curves were subjected to the log-rank (Mantel-Cox) test. Statistical significance was set at P ≤ 0.05. The mortality of wild-type C57BL/6 mice (The Jackson Laboratory) infected with 105 VPI10463 cells was expected to be 80 to 100% in dose-ranging studies. To detect a 50% difference between control untreated and treated infected mice, a sample size of 6 to 11 per group was calculated to be sufficient to attain a power of 80% with a significance level of 5%.
RESULTS
TcdA or TcdB upregulates A2BAR gene expression in human ileocolonic cells.
To determine whether C. difficile toxins affect expression of adenosine receptors, quantitative real-time PCR was performed on intoxicated HCT-8 cells. In the absence of either TcdA or TcdB, the mean A2BAR mRNA expression was 2- to 4-fold higher than that of A2AAR and ≥20-fold higher than those of A1AR and A3AR transcripts in HCT8 cells at baseline. A2BAR mRNA increased by over 50-fold after 4 h of exposure to 0.1 ng/ml TcdA and to a lesser extent by TcdB (Fig. 1). A2AAR mRNA was also induced, to a lesser extent, while A1AR and A3AR were not induced. Of note, we have previously shown that both toxins induce apoptosis in HCT-8 cells (11), and thus, the decrease in adenosine receptor expression at higher toxin doses may be explained by cell loss. However, even at the highest dose of toxins (100 ng/ml at 4 h of incubation), A2BAR mRNA levels were still high relative to those of the other adenosine receptor subtypes. These results demonstrate that intestinal epithelial cells upregulate A2BAR mRNA expression in response to C. difficile toxins.
Fig 1.
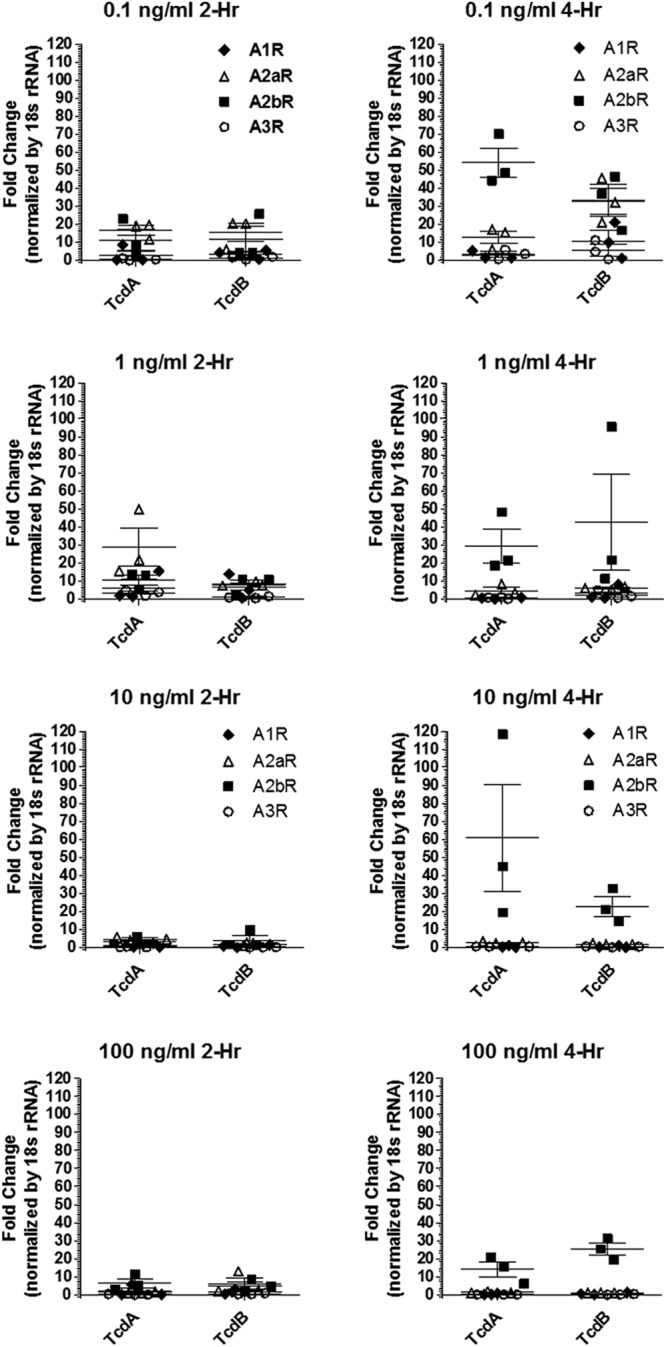
Adenosine receptor (A1, A2A, A2B, and A3) mRNA expression in HCT-8 cells treated with C. difficile toxin A (TcdA) or B (TcdB). Assays were performed 2 and 4 h postintoxication with either TcdA or TcdB at concentrations from 0.1 to 100 μg/ml. Each treatment had 3 replicates per time point.
The A2BAR antagonists ATL692 and PSB1115 decrease TcdA-induced secretion or epithelial injury in rabbit ileum.
To test whether inhibition of A2BARs affects toxin-induced intestinal secretion and histopathology, rabbit ileal loops were treated with or without the selective A2BAR blocker ATL692 or PSB1115. Ileal loops challenged with TcdA had markedly increased secretion compared with PBS control loops (P < 0.0001) (Fig. 2). ATL692 (0.5 and 5 μM) significantly reduced TcdA-induced secretion (by 39% and 61%, with P not significant and P < 0.001, respectively). PSB1115 (5 μM) also significantly decreased intestinal secretion by 56% (P < 0.001). Upon microscopy, intestinal tissues from TcdA-challenged mice had pronounced mucosal disruption, cellularity, and vascular congestion. By blinded histopathologic scoring, ATL692 (0.5 and 5 μM) significantly reduced TcdA-induced epithelial injury (by 24% and 44%, with P = 0.004 and P = 0.001, respectively) in rabbits. In contrast, PSB1115 (5 μM) did not alter TcdA's effect on histopathology.
Fig 2.
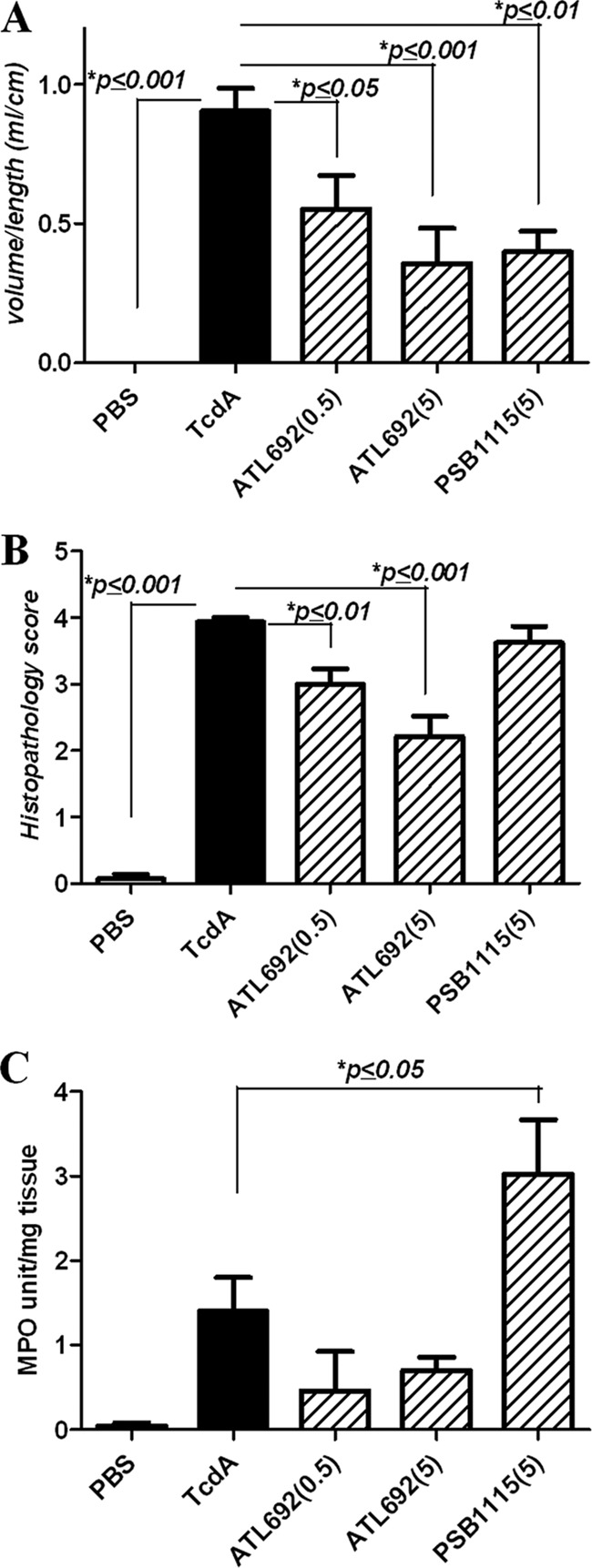
Effects of A2BAR inhibition in C. difficile toxin A (TcdA)-stimulated rabbit ligated ileal loops. Ileal loops were treated with TcdA (10 μg/loop) with or without the A2BAR antagonist ATL692 (0.5 or 5 μM) or PSB1115 (5 μM) and incubated for 4 h. (A) Intestinal secretion measured by the volume of fluid in the lumen over the length of the ileal segment (P < 0.001 by ANOVA with Bonferroni's multiple comparison test). (B) Ileal histopathology scores (P < 0.001 by ANOVA with Bonferroni's multiple comparison test). (C) Myeloperoxidase (MPO) activity (P < 0.0001 by ANOVA with Bonferroni's multiple comparison test). n = 7 or 8/group).
In an experiment in which loops were treated with only PBS and ATL692 (no TcdA challenge), intestinal secretion and histopathology scores were not different between the 2 treatment groups (n = 5 per group) (see Fig. S1 in the supplemental material). As expected, loops treated with TcdA alone had significantly elevated secretion and damage (histopathology) compared to either of the unchallenged groups.
In addition to histopathology, we assessed MPO activity as a measure of intestinal tissue inflammation. MPO activity was reduced in ATL692-treated ileal tissues by at least 50%. PSB1115-treated tissues, which had reduced secretion but displayed increased histopathology, had the most elevated MPO activity (116% increase) compared with TcdA control loops, possibly due to the relatively less selectivity by this agent. Results of radioligand binding assays are presented in Table 2. ATL692 was more selective for mouse A2BARs than PSB1115 and ATL801. Binding affinities to A1ARs and A3ARs were uniformly poor for all 3 A2BAR antagonists assayed.
Table 2.
Binding affinity of A2BAR antagonists to mouse adenosine receptors
| A2BAR antagonist |
Ki (nM) for: |
|||
|---|---|---|---|---|
| A1 | A2A | A2B | A3 | |
| ATL692 | 3,810 | 8,365 | 5 | >10,000 |
| PSB1115 | 2,210 | 7,700 | 332 | >10,000 |
| ATL801 | 7,550 | 8,340 | 187 | >10,000 |
Deletion of A2BAR protects against TcdA-induced enteritis in mice.
We next examined whether the deletion of the A2BARs abrogates TcdA effects in ileal loops. As expected, intestinal secretion and histopathology scores were significantly increased in ligated ileal loops from wild-type (WT) mice as observed in rabbits. A2BAR−/− mice challenged with TcdA had less secretion (by 52%; P < 0.01) and a lower histopathology score (by 67%; P < 0.001) than WT TcdA control mice (Fig. 3A and B). A2BAR−/− mice treated with saline alone also displayed decreased secretion (by 45%), but this was not a statistically significant difference compared with saline-treated WT mice. Representative ileal tissues are shown in Fig. 3C.
Fig 3.

Effects of C. difficile toxin A (TcdA) in C57BL/6 wild-type (WT) and A2BAR knockout (KO) mouse ligated ileal loops. Ileal loops (1 loop/mouse, 4 mice/group) were treated with TcdA (50 μg/loop) and incubated for 4 h. (A) Intestinal secretion measured by weight over the length of the ileal loop (P = 0.0006 by ANOVA with Bonferroni's multiple comparison test). (B) Ileal histopathology scores (P < 0.0001 by ANOVA with Bonferroni's multiple comparison test). (C) Representative H&E-stained tissue sections of ileal loops from WT saline-treated, WT TcdA-treated, A2BAR KO saline-treated, and A2BAR KO TcdA-treated mice.
Deletion of the A2BAR protects mice from CDI-induced morbidity and mortality.
To investigate the relevance of the A2BAR in the development of disease from C. difficile, we infected A2BAR−/− and littermate WT mice (bred in-house) with VPI10463. Of 10 A2BAR WT mice, 3 (30%) died within 2 to 3 days postinfection (Fig. 4). Diarrhea and weight loss occurred in A2BAR−/− mice to a lesser extent than in controls, and no A2BAR−/− mice died from the disease. The surviving infected WT mice had marked weight loss compared to infected A2BAR−/− mice, and starting on day 2, the latter displayed weight loss that was reversed by day 6. Fecal C. difficile toxin levels were higher in WT than A2BAR−/− mice, although there was no difference observed in clostridial shedding. Histopathology scores in A2BAR−/− mice were lower, suggesting a greater ability to control infection and reduced toxin-induced intestinal tissue injury in the absence of A2BAR activation (Fig. 5).
Fig 4.

Clinical course of A2BAR knockout (KO) and littermate C57BL/6 wild-type (WT) mice infected with C. difficile VPI 10463. n = 7 to 9 in the control group, and n = 10/group in the infected groups. (A) Survival curve (P = 0.067 for WT infected versus KO infected, by Mantel-Cox test). (B) Percentage of weight change from day of inoculation (day 0). For statistical analyses, the latest weights of dead mice were carried through the end of observation (*, P < 0.05 on days 4 to 7 for WT infected versus KO infected, by two-way ANOVA with Bonferroni's correction). (C) Diarrhea scores. For statistical analyses, the latest scores of dead mice were carried through the end of observation. *, P < 0.01 for WT infected versus KO infected on days 3 to 5 by two-way ANOVA with Bonferroni's correction). (D) C. difficile toxin positivity in cecal contents by ELISA (P < 0.0001 by ANOVA with Bonferroni's multiple comparison test). (E) Clostridial shedding, detected by PCR of the C. difficile TcdB gene (tcdB) in available fecal specimens from infected KO and WT mice. The lower the cycle threshold value, the higher the degree of clostridial shedding. Uninfected controls did not have detectable tcdB. Histopathology is presented in Fig. 5.
Fig 5.

Histopathology in C. difficile (VPI 10463)-infected A2BAR knockout (KO) and littermate C57BL/6 wild-type (WT) mice. (A) Representative H&E-stained tissue sections from cecal specimens obtained during sacrifice of moribund mice or of surviving mice at the end of the observation period. (B) Cecal histopathology scores (P = 0.0047 by ANOVA with Bonferroni's multiple comparison test). n = 7 to 9 in the control group, and n = 10/group in infected groups. Survival, clinical and diarrhea scores, C. difficile shedding, and toxin burden are presented in Fig. 4.
Treatment by A2BAR blockade increases survival in mice with CDI.
To test whether A2BAR blockade alters the outcome of infection, we treated infected mice (The Jackson Laboratory) with either the A2BAR antagonist ATL801 or vehicle starting 1 day after inoculation. ATL801 was used in the mouse model of infection because this compound has better oral availability than ATL692. Moreover, ATL801 has also been shown previously to decrease disease and histopathology scores in the mouse models of inflammatory bowel disease (24). While infected mice receiving vehicle alone developed severe disease, infected mice treated with ATL801 developed only mild diarrhea and disease that peaked at day 3 and was followed by progressive improvement (Fig. 6). All untreated infected mice died by day 4, while only 2 of 8 (25%) of the ATL801-treated mice succumbed to infection (with deaths occurring on days 4 and 6) indicating that A2BAR blockade attenuates severe disease and death. Similar to what was observed in infected A2BAR−/− mice, only a few ATL801-treated infected mice were toxin positive at sacrifice (2 weeks after infection) despite there being no significant differences in fecal clostridial shedding between infected controls and treated infected mice. Given that these mice developed clinical disease, it is possible that treatment contributed to amelioration of symptoms and recovery rather than prevention of infection.
Fig 6.

Effects of A2BAR inhibition on C. difficile infection in mice. C57BL/6 wild-type mice (8 mice/group) were infected with C. difficile VPI 10463 (105 CFU) and treated with the A2BAR antagonist ATL801 (10 mg/kg/day for 5 days) 24 h after infection. (A) Survival curve (P < 0.0001, by Mantel-Cox test). (B) Percentage of weight change from day of inoculation (day 0). No significant differences between untreated infected versus ATL801-treated mice were observed when weights of dead mice where carried through the end of observation for statistical analysis. (C) Diarrhea scores. For statistical analyses, the latest scores of dead mice were carried through the end of observation. (The only significant difference was the diarrhea score at day 5 for untreated infected versus ATL801-treated infected mice [P < 0.05 by two-way ANOVA with Bonferroni's correction[.) (D) Cecal histopathology scores (P < 0.0001 by ANOVA with Bonferroni's multiple comparison test). (E) C. difficile toxin positivity in cecal contents by ELISA (P < 0.0001 by ANOVA with Bonferroni's multiple comparison test). (F) Clostridial shedding, detected by PCR of the C. difficile TcdB gene (tcdB) in available fecal specimens from uninfected and infected mice. The lower the cycle threshold value, the higher the degree of clostridial shedding.
Of note, A2BAR−/− and littermate WT mice (Fig. 4) were bred in-house. We have noted that mice bred in-house are less susceptible to severe infection than mice directly bred by The Jackson Laboratory, such as those used for the treatment studies with ATL801 (Fig. 6). Moreover, a higher degree of bacterial shedding in infected mice (The Jackson Laboratory) was associated with increased contamination or colonization of uninfected controls housed in the same sash, an observation not noted in mice bred in-house (Fig. 4E and 6F). Differences in microbiota among mice housed in different environments may explain the variation in susceptibility to infection.
C. difficile toxin production in vitro is not affected by A2BAR blockade.
Because TcdA/B levels were decreased in stools from mice treated with ATL801, we asked whether the A2BAR blocker alters clostridial toxin production in vitro. ATL801, at various doses, did not affect toxin levels in pure C. difficile culture supernatant, indicating that ATL801 does not have any direct activity against clostridial toxin production.
Intestinal tissue IL-6 expression is augmented during CDI and depressed with A2BAR blockade or deletion.
IL-6 secretion in intestinal epithelial cells has been shown by others to be mediated by activation of A2BAR by apical or basolateral adenosine (33). Therefore, IL-6 production in both wild-type and A2BAR−/− mice during infection was measured by IHC. As shown in Fig. 7A, cecal tissues from infected C57BL/6 mice had greater IL-6 immunoreactivity in the mucosa as well as in the submucosa than tissues from uninfected mice. Infected mice treated with the A2BAR antagonist ATL801 had significantly less IL-6 immunoreactivity in intestinal tissues than untreated infected mice. In studies with A2BAR−/− mice, although there were no significant differences in total IHC scores between all groups, wild-type littermates exhibited enhanced IL-6 expression in the enterocytes and submucosa during infection compared to infected A2BAR−/− mice (Fig. 7B), suggesting possible compartmentalization of the A2BAR effect. Overall, these findings provide evidence that C. difficile-induced disease may be mediated by IL-6 secreted as a consequence of A2BAR activation.
Fig 7.

IL-6 intestinal tissue expression during C. difficile infection in mice. (A1) IL-6 immunohistochemistry (IHC) in uninfected, infected, and ATL801-treated infected C57BL/6 mice. (A2) Mean IHC scores were from combined enterocyte, lamina propria, and submucosa scores from 3 representative mice per group (P < 0.0007 by ANOVA with Bonferroni's multiple comparison test). (B1) IL-6 IHC in infected wild-type (WT; n = 7) littermate and infected A2BAR knockout (KO; n = 5) littermate. (B2) Enterocyte IHC scores (unpaired two-tailed t test). (B3) Submucosa IHC scores (unpaired two-tailed t test).
DISCUSSION
Clostridium difficle infection is characterized by intense intestinal inflammation and injury and, in severe cases, a systemic inflammatory response. We have demonstrated that intestinal epithelial A2BAR expression is upregulated by TcdA or TcdB and that A2BAR deletion or blockade ameliorates C. difficile toxin-induced intestinal epithelial injury, secretion, and inflammation. Furthermore, A2BAR deletion or blockade improves outcome from infection in the mouse model.
Although adenosine has been recognized to be produced during inflammatory conditions, expression of adenosine receptors had never been studied during CDI. The A2BAR is the predominant adenosine receptor subtype expressed in the cecum, colon, and intestinal epithelial cells (34, 35). In the study, we found that upregulation of A2BARs occurs in TcdA- or TcdB-challenged human intestinal epithelial cells. Others have shown that both A2AAR and A2BAR are expressed during dextran sodium sulfate (DSS)-induced colitis (13), while A2BAR is selectively induced in ischemia-reperfusion injury in mice (17). A2AAR mRNA is not as prominently expressed as A2BAR in epithelial cells; A2AAR may be more localized in immune rather than epithelial cells in the intestinal tract (12, 18). Although it was previously shown that A2AAR agonists decreased intestinal secretion and epithelial injury in TcdA-induced enteritis in both rabbit and mouse ileal loop models (8, 41), we recently found that A2AAR activation provides only a modest benefit against CDI in the murine model (C. A. Warren et al., personal communication). Limited benefit may occur because during infection, the initial insult from the bacteria is localized in the intestinal epithelium where A2BARs, and not A2AARs, predominate.
How the C. difficile toxins affected adenosine receptor expressions was unclear. Tumor necrosis factor alpha (TNF-α), a cytokine that is expressed during colitis, has been shown to upregulate A2BAR transcript and protein (23). TcdA and TcdB are known to cause apoptosis (5, 7, 14). Adenosine released or generated from adenine nucleotides as a consequence of cell death could bind stimulatory G protein (Gs)-coupled A2BARs to stimulate cyclic AMP (cAMP) generation, thereby stimulating CREB-mediated release of cytokines, such as IL-6 (33). IL-8 also is increased in response to TcdA or TcdB stimulation of intestinal epithelial cell lines (19, 29), but whether this cytokine, or IL-6, also upregulates A2BARs is unclear. Moreover, protein kinase A activates the apical cystic fibrosis conductance regulator, causing net Cl secretion (15, 36), which may explain increased ileal secretion in TcdA-challenged tissues. Indeed, in addition to increasing intestinal permeability by its effect on epithelial tight junctions, TcdA has been shown to increase Cl secretion in guinea pig ileum (30).
TcdA causes mucosal disruption and increased intestinal secretion in both rabbit and mouse ileal loops (27, 38, 40). Treatment with anti-inflammatory agents, such as COX-2 inhibitors (1), other nonsteroidal anti-inflammatory drugs (NSAIDs) (28), or angiotensin II subtype I receptor blockers (2), ameliorates epithelial injury and intestinal fluid accumulation in the ileal lumen. In the present study, we show that reduction of inflammation by A2BAR blockade also controls the inflammatory response and prevents toxin-induced intestinal epithelial damage and secretion. Interestingly, another A2BAR inhibitor, PSB1115, blocked secretion but unlike A2BAR deletion did not alter the histopathology score and even increased myeloperoxidase activity, suggesting differences in the selectivity of these compounds and possible off-target effects by PSB1115. Our findings on blockade or deletion of A2BAR in both TcdA-induced enteritis and C. difficile infection in mice further confirm the role of A2BAR in mediating epithelial injury and secretion. These findings mirror what others have found in murine models of colitis in which A2BAR−/− mice were reported to be less susceptible to chemicals (DSS or 2,4,6-trinitrobenzene sulfonic acid [TNBS]) or infection with Salmonella enterica serovar Typhimurium (given to streptomycin-pretreated mice) than WT controls (25). However, in the latter study, A2BAR−/− mice were observed to be more susceptible to overwhelming sepsis and death from exposure to live S. Typhimurium cells (given without pretreatment with streptomycin). In contrast to the invasive nature of S. Typhimurium, disease from C. difficile is primarily mediated by its toxins (39). Although a systemic inflammatory reaction is observed in CDI, bacteremia is rarely observed (26). Control of toxin-induced intestinal inflammation locally by inhibition or deletion of A2BAR appears to prevent progression of disease and death. Indeed, when WT mice (from The Jackson Laboratory and which are highly susceptible to severe infection) were infected but treated with ATL801, protection was conferred to 75% of the mice. While fecal toxin levels in ATL801-treated mice were reduced, we showed that the A2BAR antagonist itself did not alter clostridial toxin production in vitro. Since fecal toxin also decreased in A2BAR−/− infected mice, this effect appears to be indirect. We speculate that modulation of the epithelial inflammatory response has beneficial effects to the intestinal microbiota, which in concert with the host, is critical for the control of C. difficile growth and possibly secretion of toxin (6). Indeed, despite being bred in the same facility (and therefore presumably sharing the same microbiome) and infected with the same amount of C. difficile, A2BAR−/− mice were still less susceptible to severe disease and had lower toxin levels than their littermate wild-type controls.
Interestingly, other investigators have demonstrated more of an anti-inflammatory and protective role for A2BAR receptors in ischemia-reperfusion injury (16, 17) and murine colitis (13). Differences in experimental protocols, animal strains, and environmental conditions have been proposed as possible reasons for the discrepancies in the findings (13). In addition, it is possible that the A2BARs may actually have different effects, depending on the cell types involved and the localization and type of tissue insult. Global A2BAR deletion may also produce changes in the development of epithelial and immune cells. Studies using targeted gene deletions in immune and epithelial cells and the use of more selective pharmacologic agents will be critical to elucidate the roles of the different adenosine receptors in various cells in various disease states.
In conclusion, we have shown evidence that A2BARs are upregulated by C. difficile toxin in intestinal epithelial cells. Blockade or deletion of A2BARs decreased toxin-induced intestinal tissue injury and secretion and improved outcomes in the murine model of infection. A2BAR inhibition may potentially limit damage from toxigenic C. difficile infection.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health, National Institute of Allergy and Infectious Diseases (U01 AI075526).
We acknowledge Edna Zaenker and Tara Bracken for technical assistance. We are grateful to Patcharin Pramoonjago, University of Virginia Biorepository and Tissue Research Facility and UVA Histology Research Core, for expertise and assistance in processing intestinal tissues.
R.A.F. was previously partially funded by Dogwood Pharmaceuticals, Inc. J.L. was a consultant for Dogwood Pharmaceuticals, Inc. C.A.W., Y.L., G.M.C., E.V.O., R.S.F., S.Z.-M., and R.L.G. do not have any conflicts of interest.
Footnotes
Published ahead of print 8 October 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Alcantara C, Stenson WF, Steiner TS, Guerrant RL. 2001. Role of inducible cyclooxygenase and prostaglandins in Clostridium difficile toxin A-induced secretion and inflammation in an animal model. J. Infect. Dis. 184:648–652 [DOI] [PubMed] [Google Scholar]
- 2. Alcantara CS, et al. 2005. Angiotensin II subtype 1 receptor blockade inhibits Clostridium difficile toxin A-induced intestinal secretion in a rabbit model. J. Infect. Dis. 191:2090–2096 [DOI] [PubMed] [Google Scholar]
- 3. Auchampach JA, et al. 2009. Characterization of the A2B adenosine receptor from mouse, rabbit, and dog. J. Pharmacol. Exp. Ther. 329:2–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barrett KE, Cohn JA, Huott PA, Wasserman SI, Dharmsathaphorn K. 1990. Immune-related intestinal chloride secretion. II. Effect of adenosine on T84 cell line. Am. J. Physiol. 258:C902–C912 [DOI] [PubMed] [Google Scholar]
- 5. Brito GA, et al. 2002. Mechanism of Clostridium difficile toxin A-induced apoptosis in T84 cells. J. Infect. Dis. 186:1438–1447 [DOI] [PubMed] [Google Scholar]
- 6. Britton RA, Young VB. 2012. Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol. 20:313–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carneiro BA, et al. 2006. Caspase and Bid involvement in Clostridium difficile toxin A-induced apoptosis and modulation of toxin A effects by glutamine and alanyl-glutamine in vivo and in vitro. Infect. Immun. 74:81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cavalcante IC, et al. 2006. Effect of novel A2A adenosine receptor agonist ATL 313 on Clostridium difficile toxin A-induced murine ileal enteritis. Infect. Immun. 74:2606–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen X, et al. 2008. A mouse model of Clostridium difficile-associated disease. Gastroenterology 135:1984–1992 [DOI] [PubMed] [Google Scholar]
- 10. Cheng Y, Prusoff WH. 1973. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22:3099–3108 [DOI] [PubMed] [Google Scholar]
- 11. D'Auria KM, et al. 2012. Systems analysis of the transcriptional response of human ileocecal epithelial cells to Clostridium difficile toxins and effects on cell cycle control. BMC Syst. Biol. 6:2 doi:10.1186/1752-0509-6-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. 2001. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 53:527–552 [PMC free article] [PubMed] [Google Scholar]
- 13. Frick JS, et al. 2009. Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J. Immunol. 182:4957–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gerhard R, et al. 2008. Glucosylation of Rho GTPases by Clostridium difficile toxin A triggers apoptosis in intestinal epithelial cells. J. Med. Microbiol. 57:765–770 [DOI] [PubMed] [Google Scholar]
- 15. Greger R. 2000. Role of CFTR in the colon. Annu. Rev. Physiol. 62:467–491 [DOI] [PubMed] [Google Scholar]
- 16. Hart ML, et al. 2011. Hypoxia-inducible factor-1alpha-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5′-nucleotidase (CD73) and the A2B adenosine receptor. J. Immunol. 186:4367–4374 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17. Hart ML, Jacobi B, Schittenhelm J, Henn M, Eltzschig HK. 2009. Cutting edge: A2B adenosine receptor signaling provides potent protection during intestinal ischemia/reperfusion injury. J. Immunol. 182:3965–3968 [DOI] [PubMed] [Google Scholar]
- 18. Hasko G, Pacher P. 2008. A2A receptors in inflammation and injury: lessons learned from transgenic animals. J. Leukoc. Biol. 83:447–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He D, et al. 2002. Clostridium difficile toxin A triggers human colonocyte IL-8 release via mitochondrial oxygen radical generation. Gastroenterology 122:1048–1057 [DOI] [PubMed] [Google Scholar]
- 20. Just I, et al. 1994. Clostridium difficile toxin B acts on the GTP-binding protein Rho. J. Biol. Chem. 269:10706–10712 [PubMed] [Google Scholar]
- 21. Just I, Selzer J, von Eichel-Streiber C, Aktories K. 1995. The low molecular mass GTP-binding protein Rho is affected by toxin A from Clostridium difficile. J. Clin. Invest. 95:1026–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kelly CP, Lamont JT. 2008. Clostridium difficile—more difficult than ever. N. Engl. J. Med. 359:1932–1940 [DOI] [PubMed] [Google Scholar]
- 23. Kolachala V, et al. 2005. TNF-alpha upregulates adenosine 2b (A2b) receptor expression and signaling in intestinal epithelial cells: a basis for A2bR overexpression in colitis. Cell. Mol. Life Sci. 62:2647–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kolachala VL, et al. 2008. Blockade of adenosine A(2B) receptors ameliorates murine colitis. Br. J. Pharmacol. 155:127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kolachala VL, et al. 2008. A2B adenosine receptor gene deletion attenuates murine colitis. Gastroenterology 135:861–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Libby DB, Bearman G. 2009. Bacteremia due to Clostridium difficile—review of the literature. Int. J. Infect. Dis. 13:e305–e309 [DOI] [PubMed] [Google Scholar]
- 27. Lima AA, Lyerly DM, Wilkins TD, Innes DJ, Guerrant RL. 1988. Effects of Clostridium difficile toxins A and B in rabbit small and large intestine in vivo and on cultured cells in vitro. Infect. Immun. 56:582–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lima AA, et al. 2008. Role of phospholipase A2 and tyrosine kinase in Clostridium difficile toxin A-induced disruption of epithelial integrity, histologic inflammatory damage and intestinal secretion. J. Appl. Toxicol. 28:849–857 [DOI] [PubMed] [Google Scholar]
- 29. Mahida YR, Makh S, Hyde S, Gray T, Borriello SP. 1996. Effect of Clostridium difficile toxin A on human intestinal epithelial cells: induction of interleukin 8 production and apoptosis after cell detachment. Gut 38:337–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moore R, Pothoulakis C, Lamont JT, Carlson S, Madara JL. 1990. C. difficile toxin A increases intestinal permeability and induces Cl-secretion. Am. J. Physiol. 259:G165–G172 [DOI] [PubMed] [Google Scholar]
- 31. Pawlowski SW, et al. 2010. Murine model of Clostridium difficile infection using gnotobiotic aged C57Bl/6 mice and a BI strain. J. Infect. Dis. 202:1708–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pothoulakis C, Lamont JT. 2001. Microbes and microbial toxins: paradigms for microbial-mucosal interactions. II. The integrated response of the intestine to Clostridium difficile toxins. Am. J. Physiol. Gastrointest. Liver Physiol. 280:G178–G183 [DOI] [PubMed] [Google Scholar]
- 33. Sitaraman SV, et al. 2001. Neutrophil-epithelial crosstalk at the intestinal lumenal surface mediated by reciprocal secretion of adenosine and IL-6. J. Clin. Invest. 107:861–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stehle JH, et al. 1992. Molecular cloning and expression of the cDNA for a novel A2-adenosine receptor subtype. Mol. Endocrinol. 6:384–393 [DOI] [PubMed] [Google Scholar]
- 35. Strohmeier GR, et al. 1997. Surface expression, polarization, and functional significance of CD73 in human intestinal epithelia. J. Clin. Invest. 99:2588–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Strohmeier GR, Reppert SM, Lencer WI, Madara JL. 1995. The A2b adenosine receptor mediates cAMP responses to adenosine receptor agonists in human intestinal epithelia. J. Biol. Chem. 270:2387–2394 [DOI] [PubMed] [Google Scholar]
- 37. Sullivan GW, Linden J, Buster BL, Scheld WM. 1999. Neutrophil A2A adenosine receptor inhibits inflammation in a rat model of meningitis: synergy with the type IV phosphodiesterase inhibitor, rolipram. J. Infect. Dis. 180:1550–1560 [DOI] [PubMed] [Google Scholar]
- 38. Triadafilopoulos G, Pothoulakis C, O'Brien MJ, Lamont JT. 1987. Differential effects of Clostridium difficile toxins A and B on rabbit ileum. Gastroenterology 93:273–279 [DOI] [PubMed] [Google Scholar]
- 39. Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 18:247–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Warny M, et al. 2000. p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL-8 production, and enteritis. J. Clin. Invest. 105:1147–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Warren CA, et al. 2012. Effects of adenosine A(2)A receptor activation and alanyl-glutamine in Clostridium difficile toxin-induced ileitis in rabbits and cecitis in mice. BMC Infect. Dis. 12:13 doi:10.1186/1471-2334-12-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.