

Abstract
The bacterial adrenergic sensor kinases QseC and QseE respond to epinephrine and/or norepinephrine to initiate a complex phosphorelay regulatory cascade that modulates virulence gene expression in several pathogens. We have previously shown that QseC activates virulence gene expression in Salmonella enterica serovar Typhimurium. Here we report the role of QseE in S. Typhimurium pathogenesis as well as the interplay between these two histidine sensor kinases in gene regulation. An S. Typhimurium qseE mutant is hampered in the invasion of epithelial cells and intramacrophage replication. The ΔqseC strain is highly attenuated for intramacrophage survival but has only a minor defect in invasion. However, the ΔqseEC strain has only a slight attenuation in invasion, mirroring the ΔqseC strain, and has an intermediary intramacrophage replication defect in comparison to the ΔqseE and ΔqseC strains. The expressions of the sipA and sopB genes, involved in the invasion of epithelial cells, are activated by epinephrine via QseE. The expression levels of these genes are still decreased in the ΔqseEC double mutant, albeit to a lesser extent, congruent with the invasion phenotype of this mutant. The expression level of the sifA gene, important for intramacrophage replication, is decreased in the qseE mutant and the ΔqseEC double mutant grown in vitro. However, as previously reported by us, the epinephrine-dependent activation of this gene occurs via QseC. In the systemic model of S. Typhimurium infection of BALB/c mice, the qseC and qseE mutants are highly attenuated, while the double mutant has an intermediary phenotype. Altogether, these data suggest that both adrenergic sensors play an important role in modulating several aspects of S. Typhimurium pathogenesis.
INTRODUCTION
The QseC and QseE adrenergic histidine sensor kinases sense the host hormones epinephrine (Epi) and/or norepinephrine (NE). Both of these sensors were first described as regulating virulence in enterohemorrhagic Escherichia coli (EHEC) (11, 48, 54). However, Epi and NE and/or QseC regulation of virulence gene expression is not exclusive to EHEC, since these hormones were also reported to play a role in the pathogenesis of other bacterial species, such as enteropathogenic Escherichia coli (EPEC) (52), Francisella tularensis (46), Vibrio parahaemolyticus (39), as well as Salmonella enterica serovar Typhimurium (4, 46, 54). Moreover, QseC has been reported to be a central virulence factor in other pathogens, such as uropathogenic Escherichia coli (UPEC) (24, 34). In addition to sensing Epi/NE, QseC also senses bacterial autoinducer-3 (AI-3) (11), and QseE also senses phosphate and sulfate sources (48).
The QseC and QseE sensor kinases are part of a two-component system, where their respective cognate response regulators are QseB and QseF. QseE phosphorylates QseF only, while QseC, in addition to phosphorylating QseB, also phosphorylates QseF and KdpE (31, 61). Next, these response regulators bind to their target genes, promoting changes in gene expression.
The role of QseC in S. Typhimurium pathogenesis has been under investigation (4, 37, 38, 45, 46); however, a more complete understanding of this regulatory cascade in this gastrointestinal pathogen is still missing. We have shown previously that QseC plays an important role in S. Typhimurium systemic infection in mice (38, 46). It was also reported previously that NE induced motility and augmented swine infection via QseC in S. Typhimurium (4). However, QseC and QseE do not seem to play a role in S. Typhimurium colonization in a bovine ligated ileal loop model (45). Using recessive homozygous Dbh (dopamine beta hydroxylase) mice, which lack Epi/NE, we have previously shown that the kinetics of infection of S. Typhimurium in these animals differed from those of infection in wild-type (WT) (Epi/NE-producing) animals and that QseC played an important role in the recognition of these two hormones in vivo. Moreover, the qseC mutant presented decreased motility, had a mild defect in the invasion of epithelial HeLa cells, and had a striking decrease in survival within J774 macrophages. QseC regulates the transcription of Salmonella pathogenicity island 1 (SPI-1) genes, the SPI-2 effector sifA, and flagellar genes during pathogenesis in vivo and in vitro (38).
S. Typhimurium pathogenesis is a truly complex and orchestrated process. Classically, the main islands involved in S. Typhimurium infection are SPI-1 and SPI-2, both of which encode type III secretion systems (T3SSs) essential for S. Typhimurium virulence (20, 21, 23, 41, 51). The SPI-1-encoded T3SS is required for the efficient invasion of the intestinal epithelium (21), while the SPI-2-encoded T3SS is essential for S. Typhimurium replication and survival within macrophages and systemic infection in mice (10, 28, 41).
The SPI-1 locus also contains genes that encode effectors and regulators. Among these effectors, SopE, SopE2, and SopB, on the pathogen side, as well as the Rho GTPases Cdc42, Rac1, and RhoG, on the host side, are required for a coordinated and efficient invasion. Together, they lead to actin cytoskeletal reorganization, membrane ruffling, and bacterial internalization by macropinocytosis. SopB, together with the above-mentioned effectors, leads to chloride secretion through lipid dephosphorylation (62) and indirectly stimulates Cdc42 and RhoG through its phosphoinositide phosphatase activity (19, 27, 43, 62). SopB also promotes intestinal disease by increasing the intracellular concentration of d-myo-inositol 1,4,5,6-tetrakisphosphate, which stimulates cellular chloride secretion (62). Although SipA is not exclusively required for cell invasion, it helps to initiate the actin polymerization at the site of S. Typhimurium entry by decreasing the critical concentration and increasing the stability of actin filaments (29, 36, 64).
Encoded outside SPI-2, the effector SifA is secreted through the SPI-2 T3SS and is crucial for inducing the tubulation of the S. Typhimurium phagosome. SifA binds to the mammalian kinesin-binding protein SKIP. SifA coexpressed with SseJ induces the tubulation of mammalian cell endosomes, indicating that SifA likely mimics or activates the RhoA family of GTPases (42).
Here we describe the contribution of QseE and its interplay with QseC to S. Typhimurium pathogenesis. Our data suggest that QseE is also important for S. Typhimurium virulence both in vitro and in vivo and that QseC and QseE have a complex relationship in virulence gene regulation in S. Typhimurium. These results further highlight the multifaceted role of virulence chemical cues and their cascades in bacterial pathogenesis.
MATERIALS AND METHODS
Strains and plasmids.
All strains and plasmids used in this study are listed in Table 1. Strains were grown aerobically in LB or N-minimal medium at pH 4.5 or pH 7.0, where indicated (7, 10, 13, 35), at 37°C. Recombinant DNA and molecular biology techniques were performed as previously described (50). Most of the oligonucleotides used in this study were previously reported (38). Otherwise, they are listed in Table 2.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or description | Reference or source |
|---|---|---|
| Strains | ||
| SL1344 | Salmonella enterica serovar Typhimurium prototype | 30 |
| CGM220 | SL1344 qseC mutant | 46 |
| CGM224 | SL1344 qseC-complemented strain | This study |
| CGM225 | SL1344 qseE mutant | This study |
| CGM226 | SL1344 qseE-complemented strain | This study |
| CGM227 | SL1344 qseE and qseC double mutant | This study |
| CGM228 | SL1344 qseC (pBADMycHisA) and qseE (pACYC184) complemented | This study |
| TOP10 | E. coli F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 deoR recA1 araD139 Δ(ara-leu)7697 galU galK rpsL (Strr) endA1 nupG | Invitrogen |
| DH5α | E. coli supE44 ΔlacU169 (80dlacZΔM15) hsdR17 | Stratagene |
| Plasmids | ||
| pBADMycHisA | Cloning vector and C-terminal Myc-His expression vector | Invitrogen |
| pACYC184 | Cloning vector | NEB |
| TOPO PCR Blunt | PCR blunt cloning vector with topoisomerase | Invitrogen |
| pKD3 | λ Red template plasmid | 12 |
| pKD46 | λ Red helper plasmid (recombinase) | 12 |
| pKD20 | λ Red resolvase plasmid | 9 |
Table 2.
Oligonucleotides used in this study
| Primer | Sequence |
|---|---|
| qseC-F | GGTACCAAATTGACGCAACGTCTCAG |
| qseC-R | GAATTCGCCCAACTTACTACGGCCTC |
| qseE-F | GGTACCAGCGACACGTTGAAGCGC |
| qseE-R | GAATTCGCGTGTTTGTCAGATGCAGG |
| qseE-F Lambda Red | TTGTCAGATGCAGGCAATGGCAGCGAGATGCGAAAACAAACTTCCTGCGCGTGTAGGCTGGGAGCTGCTTC |
| qseE-R Lambda Red | GACACGTTGAAGCGCTGGTCTGTTTTCCCCCGTTCTTTACGACAATTGGTCATATGAATATCCTCCTTA |
Construction of the qseE and qseEC mutants.
The construction of the isogenic nonpolar S. Typhimurium SL1344 qseE mutant and qseEC double knockout mutant were achieved by using λ Red mutagenesis (12). The qseC mutant (CGM220) (46) was complemented with the qseC gene cloned into the pBADMycHisA (KpnI and EcoRI) vector (Invitrogen), generating strain CGM224. Similarly, the qseE mutant (CGM225) was complemented with the qseE gene cloned into the pBADMycHisA (KpnI and EcoRI) vector (Invitrogen), generating strain CGM226. The qseEC double mutant (CGM227) was also complemented with qseC cloned into pBADMycHisA (Invitrogen) and qseE cloned into the pACYC184 vector (EcoRI site into the Cmr cassette), generating strain CGM228.
Quantitative real-time RT-PCR.
Cultures were grown aerobically overnight in LB or N-minimal medium at 250 rpm to the late-exponential growth phase (optical density at 600 nm [OD600] of 1.0) for the in vitro assays, in the absence or presence of 50 μM Epi. RNA from three biological samples was extracted by using the RiboPure-Bacteria RNA isolation kit (Ambion), according to the manufacturer's instructions. The primers used for the real-time assays were designed by using Primer Express v1.5 (Applied Biosystems) (Table 2). Real-time reverse transcription (RT)-PCR was performed in a one-step reaction using an ABI 7500 sequence detection system (Applied Biosystems). For each 20-μl reaction mixture, 10 μl 2× SYBR master mix, 0.1 μl Multi-Scribe reverse transcriptase (Applied Biosystems), and 0.1 μl RNase inhibitor (Applied Biosystems) were added. The amplification efficiency of each primer pair was verified by using standard curves of known RNA concentrations. The rpoA (RNA polymerase subunit A) gene was used as the endogenous control. Data collection was performed by using ABI Sequence Detection 1.3 software (Applied Biosystems). Data were normalized to levels of rpoA and analyzed by using the comparative critical threshold (CT) method, as previously described (60). The expression levels of the target genes under different growth conditions were compared by using the relative quantification method (60). Real-time data are presented as fold changes compared to WT levels. Error bars represent the standard deviations of the ΔΔCT values (60). Statistical significances were determined by one-way analysis of variance (ANOVA) and posttested with the Bonferroni correction. An ANOVA P value of <0.0001 and a posttest P value of <0.01 were considered highly significant.
HeLa cell invasion and adhesion assays.
Epithelial HeLa cells were infected with S. Typhimurium at a multiplicity of infection (MOI) of 100:1 for 90 min at 37°C in 5% CO2, as previously described (17, 18, 44). To ensure the uniform inoculation of all strains in these assays, the inocula were normalized to the same OD600 value and plated onto LB, and CFU were enumerated through serial dilutions. These cells were treated with 40 μg/ml of gentamicin for 1 h to kill extracellular bacteria and lysed with 1% Triton X. Bacteria were diluted and plated onto LB plates for CFU determinations (17, 18, 44). All the assays in the presence of Epi-bitartrate (50 μM) were performed by preconditioning the cells with 10% dialyzed fetal bovine serum (FBS) to avoid Epi/NE traces from bovine sera and light protection to avoid Epi degradation during the assay. HeLa cell adhesion control assays were performed by employing cytochalasin D, as previously reported (15). Statistical significances were determined by one-way ANOVA compared to WT CFU. P values of <0.05 were considered significant.
Macrophage infection.
J774 murine macrophages were infected with opsonized S. Typhimurium cells with normal mouse serum at 37°C for 15 min and washed. These macrophages were infected at an MOI of 100:1 for 30 min of bacterium/cell interactions at 37°C in 5% CO2. To ensure the uniform inoculation of all strains in these assays, the inocula were normalized to the same OD600 and plated onto LB, and CFU were enumerated through serial dilutions. These cells were treated with 40 μg/ml of gentamicin for 1 h to kill extracellular bacteria and lysed with 1% Triton X. Bacteria were diluted and plated onto LB plates for CFU determinations (14, 17, 44). All the assays in the presence of Epi-bitartrate (50 μM) were performed by preconditioning the cells with 10% dialyzed FBS to avoid Epi/NE traces from bovine sera and light protection to avoid Epi degradation during the assay. Statistical significances were determined by one-way ANOVA compared to WT CFU. P values of <0.05 were considered significant.
Mouse survival experiments with Salmonella Typhimurium.
Mice (BALB/c, 7 to 9 weeks old, and female) were infected i.p. (intraperitoneally) with a predetermined lethal dose of 1 × 106 CFU of S. Typhimurium WT strain SL1344 or the same CFU of the innocuous E. coli K-12 strain DH5α (the K-12 strain was used as a negative infectivity control to ensure that there were no issues with organ perforations during i.p. injection and that the death of mice was not due to endotoxic effects) or specific S. Typhimurium mutants, as indicated. We employed 10 mice per strain using the same i.p. infection with 1 × 106 CFU, and these experiments were repeated at least twice to ensure reproducibility. Mice were returned to their cages and monitored daily for signs of morbidity (anorexia, rapid shallow breathing, scruffy fur, decreased muscle tone, and lethargy) and death. After 9 days postinfection, the remaining animals were euthanized by CO2 asphyxiation. Systemic infection (i.p. route) for replication within tissues was performed as previously described (14). For CFU enumeration in spleens and livers after 20 h of i.p. infection, the mice were sacrificed to remove the spleens and livers. Those organs were harvested, homogenized, and plated onto LB agar plates for bacterial cell counting to determine tissue colonization (CFU) (14, 41).
RESULTS
QseE plays a role in invasion of epithelial cells.
Invasion is the first step in infection, since the gastrointestinal epithelium is the first barrier to be surpassed (22–24). Here we investigated the role of the sensor kinases QseC and QseE in invasion. Previously, we reported that QseC has a minor, although significant, role in the invasion of epithelial cells (38). Here we performed gentamicin protection assays to measure epithelial cell invasion by the WT and the qseC, qseE, and qseEC mutants as well as their respective complemented strains. As most commercially available fetal bovine serum (FBS) formulations used to supplement HeLa epithelial cell culture media contain traces of Epi/NE, we used dialyzed FBS (Gibco, Invitrogen), which has all molecules with a molecular mass of less than 10,000 Da removed. In the absence of Epi/NE, the qseC mutant had a significant (P = 0.034) 1-order-of-magnitude decrease in HeLa cell invasion compared to the WT, and a similar reduction was observed for the qseEC double mutant (P = 0.024) (Fig. 1A). In contrast, the qseE mutant had a striking decrease in invasion of approximately 5 orders of magnitude compared to the WT (P = 0.018) (Fig. 1A). All of these phenotypes were restored upon complementation in trans. These results suggested that although QseC plays a minor role in the invasion of epithelial cells, QseE has an important role in this process, and the phenotype of the double kinase mutant mirrors the ΔqseC phenotype (Fig. 1A).
Fig 1.

(A) Invasion assay of HeLa epithelial cells by the WT; qseE, qseC, and qseEC mutant; and complemented strains in the absence and presence of 50 μM Epi. Statistical differences were considered significant compared to WT levels at a P value of <0.05, determined by ANOVA. *, P = 0.041; **, P = 0.018; ***, P = 0.018; ****, not significant (P = 0.293); *****, P = 0.014; +, P = 0.024; ++, P = 0.025; +++, P = 0.044; ++++, P = 0.048; +++++, P = 0.034; #, not significant (P = 0.055); ##, P = 0.029; ###, P = 0.015. (B) Growth curves of the WT, mutants, and the respective complemented strains. No statistically significant differences were observed, according to one-way ANOVA (P = 1.00). (C) HeLa cell adhesion control assay with cytochalasin D (Sigma).
The initial assays were performed in the absence of Epi, whereas QseC would have only the S. Typhimurium self-produced AI-3 signal and QseE SO4 and PO4 sources present in the media to sense. Because both QseC and QseE sense Epi, we also performed these assays in the presence of Epi. Epi increased the invasiveness of WT S. Typhimurium by 1.5 orders of magnitude compared to invasion in the absence of Epi (P = 0.04). Epi also increased the invasiveness of the qseE mutant by 2 orders of magnitude, although it never reached the levels of invasiveness of the WT strain either in the presence or in the absence of Epi (P = 0.018). These data suggest that the qseE mutant can still respond to Epi, probably because QseC is still intact in this strain, and that other factors sensed or regulated exclusively through QseE also play an important role in invasion. The complementation of the qseE mutant restored its ability to sense Epi (P = 0.014). The qseC mutant did not increase its invasiveness in the presence of Epi, and the complementation of qseC restored its ability to increase its invasiveness in the presence of this hormone (P = 0.015). These results may reflect the fact that QseC acts upstream of QseE, with the transcription of qseE being activated by QseC (49). The phenotype of the qseEC mutant again mirrored the qseC phenotype (Fig. 1A). The differences in the invasive abilities among these strains cannot be attributed to either differences in growth or differential adhesion to HeLa cells, given that the growth rates of all strains were similar to that of the WT (Fig. 1B) and cytochalasin D adhesion assays with HeLa cells (15) showed no significant differences in adhesion among these strains (Fig. 1C).
These differences in the invasion assays prompted us to investigate whether the expression levels of SPI-1 genes were influenced by QseC and QseE, given that the SPI-1 T3SS is responsible for the invasion of epithelial cells (22–24). To test this hypothesis, we performed quantitative RT-PCR (qRT-PCR) to measure the expression levels of SPI-1 genes under in vitro conditions conducive to SPI-1 expression (LB). We previously reported that the expression levels of sipA and sopB were significantly decreased, by 5- and 2.5-fold, respectively, in a qseC mutant (38). Here we show that the expression levels of both sipA and sopB were also significantly decreased in the qseE mutant and the qseEC double mutant albeit to a lesser extent in the double mutant than in the single mutant (Fig. 2). The qseE mutant had a 4-fold decrease in sopB and a 10-fold decrease in sipA expression levels, while the qseEC double mutant had a 2-fold decrease in the expression levels of both sopB and sipA compared to the WT. These decreases in expression levels were rescued upon complementation (Fig. 2). These qRT-PCR data are congruent with the invasion phenotypes observed for the qseE and qseEC mutants. Therefore, the interplay between QseE and QseC has a significant role in SPI-1-mediated epithelial cell invasion.
Fig 2.

QseE and QseEC regulate the transcriptional expression of SPI-1 genes in the absence and presence of 50 μM Epi. (A) Transcriptional expression of sopB in LB in the WT (wild type), the mutants, and the respective complemented strains. (B) Transcriptional expression of sipA in LB in the WT, the mutant, and the respective complemented strains. *, statistical differences are considered highly significant compared to WT expression levels (P < 0.0001 by ANOVA and P < 0.01 by posttest).
Role of QseE and QseC in S. Typhimurium survival within macrophages.
S. Typhimurium intracellular survival and replication are critical steps for the progression of S. Typhimurium infection, and they are SPI-2-mediated processes (42). Next, we investigated whether QseE and QseC influence S. Typhimurium survival within murine J774 macrophages. Here again, we used dialyzed FBS (Gibco, Invitrogen), which has all molecules with a molecular mass of less than 10,000 Da removed to avoid traces of Epi/NE present in normal undialyzed FBS. We previously reported that QseC plays an important role in intramacrophage survival (38), with a decrease of over 4 orders of magnitude, as further confirmed in Fig. 3. The other two mutants, the qseE and qseEC mutants, also presented significant decreases in survival within macrophages. The qseE mutant presented a decreased survival of approximately 4 orders of magnitude within macrophages compared to the WT (P = 0.046), while the qseC mutant had a reduction of 6 orders of magnitude (P = 0.0001). The qseEC double mutant showed an intermediate phenotype, with a reduction of survival of 5 orders of magnitude within macrophages compared to the WT (P = 0.0001). These differences were rescued upon their respective complementations in trans (Fig. 3). Epi was also added to this assay mixture, similarly to the HeLa cell invasion assay described above. The addition of Epi significantly increased macrophage replication for the WT and qseE mutant strains (P = 0.046 and 0.001, respectively) but did not change the ability of either the qseC or the qseEC mutant to replicate within these cells, suggesting again that QseC may be the primary, most upstream sensor of Epi in this signaling cascade. Epi could still be sensed by the qseE mutant but not by the qseC and double mutants in this assay (Fig. 3). The complemented strains' ability to replicate within macrophages was not changed, supposedly because the overexpression of these sensors, albeit in low-copy-number vectors, may be sufficient to bypass the need for the sensing of these signals in certain assays.
Fig 3.
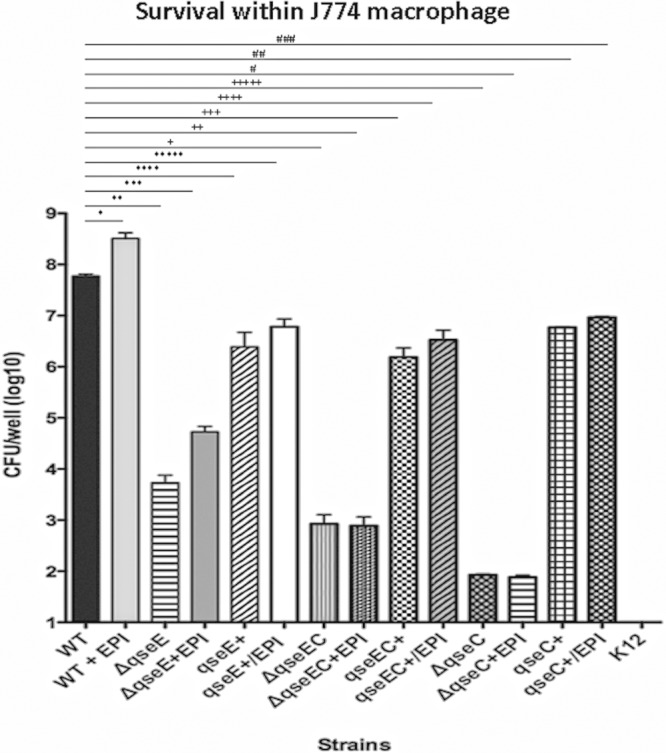
Intramacrophage survival assays with J774 cells infected with the WT; qseE, qseC, and qseEC mutant; and complemented strains in the absence and presence of 50 μM Epi. Statistical differences were considered significant compared to WT levels at a P value of <0.05, determined by ANOVA. *, P = 0.046; **, P = 0.0001; ***, P = 0.0001; ****, P = 0.001; *****, P = 0.001; +, P = 0.0001; ++, P = 0.0001; +++, P = 0.0001; ++++, P = 0.001; +++++, P = 0.0001; #, P = 0.0001; ##, P = 0.001; ###, P = 0.001.
The SPI-2 T3SS effector SifA is critical for intramacrophage survival and replication. The transcription of the sifA gene is optimal under conditions of SPI-2 expression in N-minimal medium, which has low concentrations of phosphate and magnesium (7). S. Typhimurium also encounters an acidic pH within the vacuole (5). To assess whether pH influences sifA expression, we assessed sifA transcription under conditions of acidic and neutral pHs. The expression level of sifA was increased 10-fold at an acidic pH (4.5) compared to a neutral pH (7.0) (Fig. 4A). Consequently, from here on, we assessed sifA expression levels only under acidic conditions (pH 4.5). As previously observed for a qseC mutant (38), the transcription of sifA was decreased approximately 100-fold in both the qseE and qseEC mutants compared to the WT, and these differences were rescued upon complementation (Fig. 4B). The expression level of sifA was also measured, via qRT-PCR, within J774 macrophages by employing the same conditions as those used for the intramacrophage survival assays (Fig. 3). The expression level of sifA within macrophages was increased 100-fold compared to its expression level under in vitro conditions (minimal medium at pH 4.5) in WT S. Typhimurium (Fig. 4C), and the sifA expression level was decreased over 10-fold in the qseC and qseEC mutants but not in the qseE mutant (Fig. 4C). These data suggest that QseC plays an important role in sifA expression both during growth in vitro and during intramacrophage replication and that although QseE is important for the expression of this gene in vitro, it is dispensable within macrophages. This pattern of sifA expression level decreases is congruent with the diminished intramacrophage survival of these mutants (Fig. 3), indicating that the QseC and QseE sensor kinases play a pleiotropic role in S. Typhimurium pathogenesis.
Fig 4.

QseE and QseEC regulate the transcriptional expression of sifA (SPI-2) in the absence and presence of 50 μM Epi. (A) Transcriptional expression of sifA in N-minimal medium (N-Min) in acidic and neutral milieus in the WT (wild type), the mutants, and the respective complemented strains. (B) Transcriptional expression of sifA in N-minimal medium in an acidic milieu in the WT, the mutants, and the respective complemented strains. (C) Assay of intramacrophage transcriptional expression of sifA within J774 cells of the WT and mutants, also compared to in vitro conditions (N-minimal medium). *, statistical differences considered highly significant compared to N-minimal medium at pH 7.0 (A) and WT expression levels (B and C) (P < 0.0001 by ANOVA and P < 0.01 by posttest); **, not statistically significantly changed (P = 0.085 by ANOVA and posttest [Bonferroni correction]).
Epinephrine increases S. Typhimurium virulence gene expression.
Since Epi is a common cue that can be sensed by both QseC and QseE (11, 48, 54), we assessed gene expression in the presence of Epi. Epi increased the expression levels of sopB, sipA, and sifA (Fig. 2 and 4) (38). We previously reported that the Epi/NE-dependent increase in the sopB expression level is not dependent on QseC, leading us to propose that the sensor of Epi toward the activation of the expression of this gene was QseE. In agreement with our initial hypothesis, the Epi activation of sopB expression is dependent on QseE (Fig. 2B), given that a qseE mutant does not respond to Epi and cannot activate sopB expression. Interestingly, the qseCE double mutant presented a decrease in the sopB expression level in the presence of Epi, suggesting that in the absence of both of these sensors, another Epi-dependent signaling pathway represses sopB expression (Fig. 2B). In support of these data, a third adrenergic receptor in S. Typhimurium was previously proposed (53). The expression of sipA follows the same trend as that of sopB, with the Epi-dependent regulation of this gene also occurring through QseE (Fig. 2B).
Next, we assessed Epi-dependent sifA expression (Fig. 4B). We previously reported that the qseC mutant could not respond to Epi/NE to activate sifA expression, leading us to suggest that the Epi activation of sifA expression occurred through QseC (38). Here again, congruent with our previously reported hypothesis, the qseE mutant still responds to Epi to activate sifA expression (Fig. 4B), given that QseC is present in this strain and the double kinase mutant is irresponsive. These data suggest that during in vitro growth, the Epi activation of SPI-1 effectors occurs primarily through QseE, while the expression of the sifA SPI-2 effector occurs primarily through QseC.
Role of QseE and QseC in vivo.
Using 129x1/SvJ mice, which are more resistant to S. Typhimurium infection, we previously showed that a qseC mutant was attenuated for infection. We used this resistant mouse strain in the past, because dopamine hydroxylase knockout mice (these mice do not produce either Epi or NE) had this genetic background, and in these initial studies, we also employed these animals to show that QseC was involved in the sensing of Epi/NE during murine infection. In the animal studies performed here, we employed systemic (intraperitoneal [i.p.]) infection to reproduce the typhoid-like model using BALB/c mice, which are Nramp1−/− or Nramp1 and are more susceptible to S. Typhimurium infection (8). i.p. infections were also performed by using E. coli K-12 as a negative control, to ensure that endotoxic effects were not responsible for the death of these animals. After 1 day postinfection with WT S. Typhimurium, 85% of the mice were alive, while 100% of the qseC and qseE mutant-infected mice were also alive at this time (Fig. 5A). On day 2, 100% of the WT strain-infected mice were dead, while 100% of qseC, 80% of qseE, and 30% of qseEC mutant-infected mice were alive. By day 3, the survival rate of the qseC mutant-infected mice dropped slightly, to approximately 85%. Mice infected with the qseE mutant succumbed to death only at day 7. The survival rate of qseEC mutant-infected mice also dropped by day 4, and none survived by day 5 postinfection. Meanwhile, the survival rate of the qseC mutant-infected mice was constant at 85% throughout the experiment, and the K-12-infected negative controls had no deaths, as expected (Fig. 5A).
Fig 5.

QseE is important for murine infection. (A) Survival plots of BALB/c mice infected (i.p.) with the WT and mutants, as indicated, using lethal doses of S. Typhimurium. (B) Quantification of replication within tissues as loads in spleens and livers harvested from BALB/c mice after 20 h postinfection. The infection route was oral gavage with lethal doses of the WT or the qseE mutant. *, P < 0.001.
Next, to better understand the role of QseE during systemic infection, we harvested spleens and livers of mice infected with either the WT or the qseE mutant (20 h postinfection), similarly to experiments that we previously performed using mice infected with the qseC mutant (38). S. Typhimurium replication in the spleens and livers of mice during early stages (at 20 h postinfection) was significantly reduced for the qseE mutant, with an approximate decrease of 5 orders of magnitude in spleens and livers compared to WT-infected animals (Fig. 5B). These data indicate that QseE also has an important role during in vivo colonization within spleen and liver replication in a systemic S. Typhimurium infection model.
DISCUSSION
Bacterial cell-to-cell chemical communication is a complex process, which can aid pathogens to sense their surroundings in order to successfully colonize specific niches, survive host defenses, and outcompete indigenous microbiota (3). Many histidine sensor kinases were implicated in this regulation to fine-tune the expression of virulence factors (54). Therefore, cell communication in S. Typhimurium has been the subject of various studies (33, 55–58), although only recently has a role for QseC and QseE been investigated (4, 37, 38, 45, 46). QseC has been shown to increase virulence by the upregulation of SPI-1 gene expression; however, the most striking QseC-dependent gene regulation pertains to the expression of SifA (38), which is an SPI-2 T3SS effector essential for Salmonella-induced filament formation, i.e., intracellular survival and replication (6, 46). Prolonged survival within macrophages and increased motility are QseC-mediated phenotypes (38). However, the role of QseE as well as the interplay between these two kinases in S. Typhimurium pathogenesis remained undefined. Considering that QseE plays a role in EHEC virulence and interfaces with QseC in this regulatory cascade, (11, 48, 54), our initial hypothesis was that QseE might also have a role in the regulation of S. Typhimurium virulence.
The invasion of epithelial cells is one of the initial steps in S. Typhimurium pathogenesis. Here we showed that the qseE mutant has a striking reduction in HeLa cell invasion (Fig. 1). This is in contrast to the qseC mutant, where a mild 1-log reduction was observed. These results were consistent with the transcription profiles of these mutants: while QseC mildly regulates the expression levels of SPI-1 genes and effectors (38) primarily involved in invasion, QseE has a more pronounced effect (Fig. 2). Even the Epi-dependent activation of these genes (sipA and sopB) seems to occur primarily through QseE rather than QseC (Fig. 2) (38). SipA facilitates actin stability, while SopB is directly involved in the stimulation of cellular chloride secretion (26, 62). Although both are not required for invasion, their presence is essential for efficient invasion, an essential initial step in S. Typhimurium pathogenesis (16, 22, 25, 29, 59, 63, 64). The invasion difference in the qseE mutant was supported by the reductions in the expression levels of sopB and sipA. Recently, SopB was also described to be important to vacuole maturation; i.e., it helps S. Typhimurium to evade lysosomal fusion and host defenses (2). The phenotype of the qseEC double mutant mirrored the qseC mutant phenotype (Fig. 1), and congruent with the invasion phenotype, the levels of transcription of SPI-1 genes were also decreased less in the double mutant than in the qseE single mutant (Fig. 2). These data suggest a novel role for QseE during S. Typhimurium pathogenesis, with emphasis on epithelial cell invasion, while QseC seems to play a more important role in systemic disease and intramacrophage replication. Furthermore, there seems to be an intricate interplay between these two sensor kinases in the timing of the extensive repertoire of S. Typhimurium virulence gene expression.
Following invasion, intramacrophage survival and replication are an essential second step in S. Typhimurium pathogenesis (6, 19). Hence, we investigated survival within J774 macrophages to assess the role of QseC and QseE and their interplay during intracellular survival. We observed that the qseE mutant presented a significant decrease in survival, similarly to the double knockout in intramacrophage survival/replication, while the qseC mutant presented a striking reduction (Fig. 3) (38). These results are again congruent with the transcription of virulence factors, where sifA transcriptional regulation is dependent mostly on QseC, especially within macrophages (Fig. 4) (38).
Given that both kinases regulated many aspects of S. Typhimurium virulence in vitro, we next assessed the role of these sensors during murine infection. To examine the role of QseC and QseE in vivo, we assessed mouse survival using lethal systemic infection with S. Typhimurium. Previously, we reported that QseC was important during systemic murine infection, using a resistant murine strain (38, 46). Here, using an S. Typhimurium-susceptible murine strain (BALB/c), we observed that the contribution of QseC to systemic infection is even more profound in this model (Fig. 5A). Additionally, we also report that QseE plays a significant role in systemic infection (Fig. 5) albeit to a lesser degree than QseC. It is, however, puzzling that each individual mutant has a more profound effect during murine infection than the double mutant, which, although significantly attenuated compared to the WT, is much less attenuated than the single mutants (Fig. 5).
There is extensive interplay at the genetic and biochemical levels between the QseBC and the QseEF systems as well as with other two-component systems. QseBC activates the transcription of qseEF, and this activation is enhanced in the presence of Epi, suggesting a hierarchal relationship between these two systems, with QseBC acting upstream of QseEF (49). There is also cross talk between these systems as well as with other two-component systems at the biochemical level, with QseC phosphorylating not only its cognate response regulator, QseB, but also the noncognate response regulators QseF and KdpE (31) (Fig. 6). The cognate sensor kinase for KdpE is KdpD, which senses potassium, and the KdpD sensor kinase is known to be important for S. Typhimurium pathogenesis in Caenorhabditis elegans (1). Additional layers of complexity also exist, with the QseEF system repressing the expression of rcsB (40, 47) and with QseF, in addition to being phosphorylated by QseE and QseC, also being phosphorylated by UhpB, BaeS, EnvZ, and RstB (61). Finally, one also has to take into consideration that QseC and QseE, in addition to being kinases, are also phosphatases, and response regulators such as QseB can bind to different sites depending on their phosphorylation state (31). It was also reported previously that QseC acts through QseB not only by phosphorylating this response regulator but also by dephosphorylating it (34). Combinatorial sensing by several two-component systems was proposed previously to enable the integration of multiple stimuli and the amplification of signals. This is especially relevant given that bacteria are exposed to different gradients of environmental signals and/or cues during host infection (32).
Fig 6.

Model for AI-3/epinephrine/norepinephrine (Nore) regulation in Salmonella enterica serovar Typhimurium via QseEF and QseBC cross talk during pathogenesis. QseC senses AI-3, Epi, and NE to augment its autophosphorylation, while QseE senses PO4, SO4, Epi, and NE. QseE exclusively phosphorylates QseF, while QseC phosphorylates QseB, QseF, and KdpE, whose cognate sensor kinase, KdpD, senses potassium. Additionally, QseF is phosphorylated by UhpB, BaeS, EnvZ, and RstB. An additional level of cross talk involves the QseEF repression of rcsB transcription. Besides phosphorylating QseB, QseC also dephosphorylates this response regulator to exert its control in gene expression. Finally, the concerted and integrated action of QseBC and QseEF with several two-component systems, forming a network, leads to the optimal activation of SPI-1 and sifA expression and the enhancement of S. Typhimurium pathogenesis. However, which of the response regulators is directly activating the transcription of SPI-1 and sifA remains to be determined. OM, outer membrane; IM, inner membrane.
These data further highlight the importance of the cross talk among different two-component systems in bacterial pathogens. The data also support the concept that histidine sensor kinases and response regulators are network components rather than stand-alone systems. They are organized in intricate networks that confer plasticity and rapid responses to change gene expression toward adaptation to complex and dynamic environments (32). A more complete understanding of two-component system regulation in bacterial pathogenesis is essential for the future development of novel antibacterial therapies.
ACKNOWLEDGMENTS
This work was supported by NIH grant UO1-AI053067 and the Burroughs Wellcome Fund.
Footnotes
Published ahead of print 1 October 2012
REFERENCES
- 1. Alegado RA, Chin CY, Monack DM, Tan MW. 2011. The two-component sensor kinase KdpD is required for Salmonella typhimurium colonization of Caenorhabditis elegans and survival in macrophages. Cell. Microbiol. 13:1618–1637 [DOI] [PubMed] [Google Scholar]
- 2. Bakowski MA, et al. 2010. The phosphoinositide phosphatase SopB manipulates membrane surface charge and trafficking of the Salmonella-containing vacuole. Cell Host Microbe 7:453–462 [DOI] [PubMed] [Google Scholar]
- 3. Bassler BL, Losick R. 2006. Bacterially speaking. Cell 125:237–246 [DOI] [PubMed] [Google Scholar]
- 4. Bearson BL, Bearson SM. 2008. The role of the QseC quorum-sensing sensor kinase in colonization and norepinephrine-enhanced motility of Salmonella enterica serovar Typhimurium. Microb. Pathog. 44:271–278 [DOI] [PubMed] [Google Scholar]
- 5. Beuzon CR, Banks G, Deiwick J, Hensel M, Holden DW. 1999. pH-dependent secretion of SseB, a product of the SPI-2 type III secretion system of Salmonella typhimurium. Mol. Microbiol. 33:806–816 [DOI] [PubMed] [Google Scholar]
- 6. Beuzon CR, et al. 2000. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 19:3235–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bustamante VH, et al. 2008. HilD-mediated transcriptional cross-talk between SPI-1 and SPI-2. Proc. Natl. Acad. Sci. U. S. A. 105:14591–14596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Canonne-Hergaux F, Gruenheid S, Govoni G, Gros P. 1999. The Nramp1 protein and its role in resistance to infection and macrophage function. Proc. Assoc. Am. Physicians 111:283–289 [DOI] [PubMed] [Google Scholar]
- 9. Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14 [DOI] [PubMed] [Google Scholar]
- 10. Cirillo DM, Valdivia RH, Monack DM, Falkow S. 1998. Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol. Microbiol. 30:175–188 [DOI] [PubMed] [Google Scholar]
- 11. Clarke MB, Hughes DT, Zhu C, Boedeker EC, Sperandio V. 2006. The QseC sensor kinase: a bacterial adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A. 103:10420–10425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deiwick J, Nikolaus T, Erdogan S, Hensel M. 1999. Environmental regulation of Salmonella pathogenicity island 2 gene expression. Mol. Microbiol. 31:1759–1773 [DOI] [PubMed] [Google Scholar]
- 14. Detweiler CS, Monack DM, Brodsky IE, Mathew H, Falkow S. 2003. virK, somA and rcsC are important for systemic Salmonella enterica serovar Typhimurium infection and cationic peptide resistance. Mol. Microbiol. 48:385–400 [DOI] [PubMed] [Google Scholar]
- 15. Eaves-Pyles T, Szabo C, Salzman AL. 1999. Bacterial invasion is not required for activation of NF-kappaB in enterocytes. Infect. Immun. 67:800–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feng Y, Wente SR, Majerus PW. 2001. Overexpression of the inositol phosphatase SopB in human 293 cells stimulates cellular chloride influx and inhibits nuclear mRNA export. Proc. Natl. Acad. Sci. U. S. A. 98:875–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fierer J, et al. 1993. Expression of the Salmonella virulence plasmid gene spvB in cultured macrophages and nonphagocytic cells. Infect. Immun. 61:5231–5236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Finlay BB, Ruschkowski S, Dedhar S. 1991. Cytoskeletal rearrangements accompanying Salmonella entry into epithelial cells. J. Cell Sci. 99(Pt 2):283–296 [DOI] [PubMed] [Google Scholar]
- 19. Friebel A, et al. 2001. SopE and SopE2 from Salmonella typhimurium activate different sets of RhoGTPases of the host cell. J. Biol. Chem. 276:34035–34040 [DOI] [PubMed] [Google Scholar]
- 20. Galan JE. 1996. Molecular genetic bases of Salmonella entry into host cells. Mol. Microbiol. 20:263–271 [DOI] [PubMed] [Google Scholar]
- 21. Galan JE, Curtiss R., III 1989. Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sci. U. S. A. 86:6383–6387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Galkin VE, et al. 2002. The bacterial protein SipA polymerizes G-actin and mimics muscle nebulin. Nat. Struct. Biol. 9:518–521 [DOI] [PubMed] [Google Scholar]
- 23. Groisman EA, Ochman H. 1993. Cognate gene clusters govern invasion of host epithelial cells by Salmonella typhimurium and Shigella flexneri. EMBO J. 12:3779–3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hadjifrangiskou M, et al. 2011. A central metabolic circuit controlled by QseC in pathogenic Escherichia coli. Mol. Microbiol. 80:1516–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hapfelmeier S, et al. 2004. Role of the Salmonella pathogenicity island 1 effector proteins SipA, SopB, SopE, and SopE2 in Salmonella enterica subspecies 1 serovar Typhimurium colitis in streptomycin-pretreated mice. Infect. Immun. 72:795–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haraga A, Ohlson MB, Miller SI. 2008. Salmonellae interplay with host cells. Nat. Rev. Microbiol. 6:53–66 [DOI] [PubMed] [Google Scholar]
- 27. Hardt WD, Chen LM, Schuebel KE, Bustelo XR, Galan JE. 1998. S. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 93:815–826 [DOI] [PubMed] [Google Scholar]
- 28. Hensel M, et al. 1998. Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol. Microbiol. 30:163–174 [DOI] [PubMed] [Google Scholar]
- 29. Higashide W, Dai S, Hombs VP, Zhou D. 2002. Involvement of SipA in modulating actin dynamics during Salmonella invasion into cultured epithelial cells. Cell. Microbiol. 4:357–365 [DOI] [PubMed] [Google Scholar]
- 30. Hoiseth SK, Stocker BA. 1981. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291:238–239 [DOI] [PubMed] [Google Scholar]
- 31. Hughes DT, Clarke MB, Yamamoto K, Rasko DA, Sperandio V. 2009. The QseC adrenergic signaling cascade in enterohemorrhagic E. coli (EHEC). PLoS Pathog. 5:e1000553 doi:10.1371/journal.ppat.1000553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jung K, Fried L, Behr S, Heermann R. 2012. Histidine kinases and response regulators in networks. Curr. Opin. Microbiol. 15:118–124 [DOI] [PubMed] [Google Scholar]
- 33. Kaper JB, Sperandio V. 2005. Bacterial cell-to-cell signaling in the gastrointestinal tract. Infect. Immun. 73:3197–3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kostakioti M, Hadjifrangiskou M, Pinkner JS, Hultgren SJ. 2009. QseC-mediated dephosphorylation of QseB is required for expression of genes associated with virulence in uropathogenic Escherichia coli. Mol. Microbiol. 73:1020–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lundberg U, Vinatzer U, Berdnik D, von Gabain A, Baccarini M. 1999. Growth phase-regulated induction of Salmonella-induced macrophage apoptosis correlates with transient expression of SPI-1 genes. J. Bacteriol. 181:3433–3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McGhie EJ, Hayward RD, Koronakis V. 2001. Cooperation between actin-binding proteins of invasive Salmonella: SipA potentiates SipC nucleation and bundling of actin. EMBO J. 20:2131–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Merighi M, et al. 2009. Genome-wide analysis of the PreA/PreB (QseB/QseC) regulon of Salmonella enterica serovar Typhimurium. BMC Microbiol. 9:42 doi:10.1186/1471-2180-9-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moreira CG, Weinshenker D, Sperandio V. 2010. QseC mediates Salmonella enterica serovar Typhimurium virulence in vitro and in vivo. Infect. Immun. 78:914–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakano M, Takahashi A, Sakai Y, Nakaya Y. 2007. Modulation of pathogenicity with norepinephrine related to the type III secretion system of Vibrio parahaemolyticus. J. Infect. Dis. 195:1353–1360 [DOI] [PubMed] [Google Scholar]
- 40. Njoroge J, Sperandio V. 2012. Enterohemorrhagic Escherichia coli virulence regulation by two bacterial adrenergic kinases, QseC and QseE. Infect. Immun. 80:688–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ochman H, Soncini FC, Solomon F, Groisman EA. 1996. Identification of a pathogenicity island required for Salmonella survival in host cells. Proc. Natl. Acad. Sci. U. S. A. 93:7800–7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ohlson MB, et al. 2008. Structure and function of Salmonella SifA indicate that its interactions with SKIP, SseJ, and RhoA family GTPases induce endosomal tubulation. Cell Host Microbe 4:434–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Patel JC, Galan JE. 2006. Differential activation and function of Rho GTPases during Salmonella-host cell interactions. J. Cell Biol. 175:453–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pfeifer CG, Marcus SL, Steele-Mortimer O, Knodler LA, Finlay BB. 1999. Salmonella typhimurium virulence genes are induced upon bacterial invasion into phagocytic and nonphagocytic cells. Infect. Immun. 67:5690–5698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pullinger GD, et al. 2010. Norepinephrine augments Salmonella enterica-induced enteritis in a manner associated with increased net replication but independent of the putative adrenergic sensor kinases QseC and QseE. Infect. Immun. 78:372–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rasko DA, et al. 2008. Targeting QseC signaling and virulence for antibiotic development. Science 321:1078–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reading NC, Rasko D, Torres AG, Sperandio V. 2010. A transcriptome study of the QseEF two-component system and the QseG membrane protein in enterohaemorrhagic Escherichia coli O157:H7. Microbiology 156:1167–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reading NC, Rasko DA, Torres AG, Sperandio V. 2009. The two-component system QseEF and the membrane protein QseG link adrenergic and stress sensing to bacterial pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 106:5889–5894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reading NC, et al. 2007. A novel two-component signaling system that activates transcription of an enterohemorrhagic Escherichia coli effector involved in remodeling of host actin. J. Bacteriol. 189:2468–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 51. Shea JE, Hensel M, Gleeson C, Holden DW. 1996. Identification of a virulence locus encoding a second type III secretion system in Salmonella typhimurium. Proc. Natl. Acad. Sci. U. S. A. 93:2593–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sircili MP, Walters M, Trabulsi LR, Sperandio V. 2004. Modulation of enteropathogenic Escherichia coli virulence by quorum sensing. Infect. Immun. 72:2329–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spencer H, et al. 2010. Genome-wide transposon mutagenesis identifies a role for host neuroendocrine stress hormones in regulating the expression of virulence genes in Salmonella. J. Bacteriol. 192:714–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sperandio V, Torres AG, Jarvis B, Nataro JP, Kaper JB. 2003. Bacteria-host communication: the language of hormones. Proc. Natl. Acad. Sci. U. S. A. 100:8951–8956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Surette MG, Bassler BL. 1998. Quorum sensing in Escherichia coli and Salmonella typhimurium. Proc. Natl. Acad. Sci. U. S. A. 95:7046–7050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Surette MG, Bassler BL. 1999. Regulation of autoinducer production in Salmonella typhimurium. Mol. Microbiol. 31:585–595 [DOI] [PubMed] [Google Scholar]
- 57. Surette MG, Miller MB, Bassler BL. 1999. Quorum sensing in Escherichia coli, Salmonella typhimurium, and Vibrio harveyi: a new family of genes responsible for autoinducer production. Proc. Natl. Acad. Sci. U. S. A. 96:1639–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Taga ME, Semmelhack JL, Bassler BL. 2001. The LuxS-dependent autoinducer AI-2 controls the expression of an ABC transporter that functions in AI-2 uptake in Salmonella typhimurium. Mol. Microbiol. 42:777–793 [DOI] [PubMed] [Google Scholar]
- 59. Terebiznik MR, et al. 2002. Elimination of host cell PtdIns(4,5)P(2) by bacterial SigD promotes membrane fission during invasion by Salmonella. Nat. Cell Biol. 4:766–773 [DOI] [PubMed] [Google Scholar]
- 60. Walters M, Sperandio V. 2006. Autoinducer 3 and epinephrine signaling in the kinetics of locus of enterocyte effacement gene expression in enterohemorrhagic Escherichia coli. Infect. Immun. 74:5445–5455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yamamoto K, et al. 2005. Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli. J. Biol. Chem. 280:1448–1456 [DOI] [PubMed] [Google Scholar]
- 62. Zhou D, Chen LM, Hernandez L, Shears SB, Galan JE. 2001. A Salmonella inositol polyphosphatase acts in conjunction with other bacterial effectors to promote host cell actin cytoskeleton rearrangements and bacterial internalization. Mol. Microbiol. 39:248–259 [DOI] [PubMed] [Google Scholar]
- 63. Zhou D, Mooseker MS, Galan JE. 1999. An invasion-associated Salmonella protein modulates the actin-bundling activity of plastin. Proc. Natl. Acad. Sci. U. S. A. 96:10176–10181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhou D, Mooseker MS, Galan JE. 1999. Role of the S. typhimurium actin-binding protein SipA in bacterial internalization. Science 283:2092–2095 [DOI] [PubMed] [Google Scholar]