

Abstract
The human pathogens enteropathogenic Escherichia coli (EPEC) and vaccinia virus trigger actin assembly in host cells by activating the host adaptor Nck and the actin nucleation promoter neural Wiskott-Aldrich syndrome protein (N-WASP). EPEC translocates effector molecules into host cells via type III secretion, and the interaction between the translocated intimin receptor (Tir) and the bacterial membrane protein intimin stimulates Nck and N-WASP recruitment, leading to the formation of actin pedestals beneath adherent bacteria. Vaccinia virus also recruits Nck and N-WASP to generate actin tails that promote cell-to-cell spread of the virus. In addition to Nck and N-WASP, WASP-interacting protein (WIP) localizes to vaccinia virus tails, and inhibition of actin tail formation upon ectopic expression of WIP mutants led to the suggestion that WIP is required for this process. Similar studies of WIP mutants, however, did not affect the ability of EPEC to form actin pedestals, arguing against an essential role for WIP in EPEC-induced actin assembly. In this study, we demonstrate that Nck and N-WASP are normally recruited by vaccinia virus and EPEC in the absence of WIP, and neither WIP nor the WIP family members CR16 and WIRE/WICH are essential for pathogen induced actin assembly. In addition, although Nck binds EPEC Tir directly, N-WASP is required for its localization during pedestal formation. Overall, these data highlight similar pathogenic strategies shared by EPEC and vaccinia virus by demonstrating a requirement for both Nck and N-WASP, but not WIP or WIP family members in pathogen-induced actin assembly.
INTRODUCTION
Manipulation of the host actin cytoskeleton is central to the pathogenesis of a variety of intra- and extracellular pathogens. Enteropathogenic Escherichia coli (EPEC), and vaccinia virus are important human pathogens that stimulate actin assembly to promote colonization and/or spread. Both pathogens mediate actin assembly by recruiting the host adaptor protein Nck, neural Wiskott-Aldrich syndrome protein (N-WASP), and the WASP-interacting protein (WIP) (55). When activated by host (e.g., Nck and Cdc42) or pathogen-encoded factors (see below), WASP family proteins, including WASP, N-WASP, and three WAVE/Scar molecules (49), efficiently stimulate the Arp2/3 complex to initiate actin assembly (8, 14, 22, 41, 42). This activity is central to diverse cellular processes, including formation of filopodia and lamellipodia (55), endocytosis (7, 26, 37), cell movement (24, 44, 51), and surface receptor signaling (6, 28).
During colonization of the intestinal epithelium, EPEC strains induce hallmark attaching-and-effacing (AE) lesions characterized by localized rearrangement of the actin cytoskeleton, the formation of actin-rich pedestals beneath adherent bacteria, and the destruction of adjacent microvilli (10, 12, 20, 21). To generate actin pedestals, EPEC utilizes a type III secretion system to deliver the translocated intimin receptor (Tir) to the apical plasma membrane of the host cell, where its extracellular loop binds the bacterial outer membrane protein intimin and stabilizes bacterial attachment (25). The C-terminal domain of Tir, localized in the host cell cytoplasm, initiates host signaling cascades that exploit the actin polymerizing ability of N-WASP to form actin-rich pedestals beneath adherent bacteria (21, 27, 29). EPEC Tir contains a critical C-terminal tyrosine residue (Y474) that is phosphorylated by host kinases upon translocation into the target cell (9). The phosphorylation of Y474 in EPEC Tir provides a binding site for the host adaptor protein Nck (9, 12, 21), and both Tir phosphorylation and Nck binding are essential for N-WASP recruitment and actin pedestal formation (9, 16, 17, 21, 29, 30). The related pathogen enterohemorrhagic E. coli (EHEC) also recruits N-WASP, but in an Nck-independent fashion (52, 53).
Vaccinia virus utilizes a similar signaling cascade to induce actin tails and enhance intercellular movement (46, 55). The vaccinia virus-encoded A36R coat protein also contains a C-terminal tyrosine (Y112), which, when phosphorylated by host kinases (38), provides an Nck binding site required for the recruitment of Nck and N-WASP (18). Immunolocalization studies have revealed the presence of Grb2, Nck, WIP, and N-WASP at the viral coat protein, and more recent fluorescence recovery after photobleaching (FRAP) experiments revealed that all four proteins undergo dynamic and continuous turnover during actin tail formation by vaccinia virus (54). It has been suggested that WIP mediates the recruitment of N-WASP to vaccinia virus, based on the observation that overexpression of the WASP-binding domain (WBD) of WIP acts as a dominant-negative inhibitor of vaccinia virus-induced actin tail formation and prevented N-WASP recruitment to the viral particle (36). Notably, FRAP studies also revealed that the baseline rates of exchange of Nck and WIP were nearly identical and significantly more rapid than the rate of N-WASP exchange, which is consistent with a model in which Nck and WIP may be recruited to A36R as a complex and together subsequently recruit N-WASP (54).
In contrast to vaccinia virus, Lommel et al. demonstrated that overexpression of the WIP WBD had no effect on the ability of EPEC to form pedestals on N-WASP-sufficient cells; the recruitment of N-WASP to EPEC Tir was also unaffected by expression of N-WASP lacking the WH1 domain, which is unable to interact with WIP (32). Based on the presence of functionally analogous phosphotyrosine motifs—Y112 and Y474 of vaccinia virus A36R and EPEC Tir, respectively—the apparent requirement for WIP in recruiting N-WASP to vaccinia virus-bound but not EPEC-bound Nck was unexpected.
The generation of mice and cell lines lacking N-WASP (47) and its interacting partners Nck (50), Cdc42 (13), and WIP (2) have enabled us to better characterize the intracellular host signaling cascades targeted by pathogens that manipulate the host actin polymerization machinery. We sought to characterize here more fully the molecular signaling events required for pathogen-induced actin assembly by EPEC and vaccinia virus. Utilizing targeted cell lines and immunolocalization studies, we demonstrate that vaccinia virus and EPEC both utilize and require Nck and N-WASP, but not WIP or WIP family members for actin assembly, and that Nck and N-WASP appear to be recruited by these pathogens in a mutually dependent manner.
MATERIALS AND METHODS
Gene targeting and cell preparations.
The generation of N-WASP−/− and Cdc42−/− fibroblast-like cells (FLC) from embryonic stem cells has been described (13, 47). WIP−/− FLC were isolated from lung tissue isolated from WIP knockout (KO) mice (2). Briefly, lung pieces from WT or WIP KO mice were washed with phosphate-buffered saline (PBS), minced and cultured in Iscove medium supplemented with 10% fetal bovine serum and penicillin-streptomycin (50 U/ml) for several days. After the removal of unattached debris, the adherent cells were treated with trypsin and maintained in culture.
Bacterial and viral strains.
EPEC JPN15/pMAR7 strain and EHEC TUV 93-0 were cultured in lysogeny broth for at least 8 h to late log phase the day prior to infection and then diluted 1:10 and grown overnight at 37°C without agitation. For EPEC, the media were supplemented with ampicillin (100 μg/ml). Shigella flexneri 2a strain 2457T was cultured overnight prior to infection. Vaccinia virus was either provided by Ramnik Xavier (Massachusetts General Hospital, Boston, MA) or produced in one of our laboratories (R.S.G., Children's Hospital, Boston, MA).
Retroviruses and vectors.
Retrovirus expressing rat N-WASP (19) was generated by cloning the coding sequence into a replication-defective retrovirus chicken matrix metalloproteinase plasmid (pCMMP) (31). N-WASP/GFP bicistronic viruses were constructed by cloning green fluorescent protein (GFP; containing an internal ribosome entry site) downstream of N-WASP, and recombinant virus was produced by transient transfection of the 293-GPG packaging cell line as previously described (39). Wild-type (WT) WIP was cloned in phase with GFP at the C terminus of WIP in the vector pcDNA3 as previously described (15). pELGFP-WIP was provided by Michael Way (36), and pEBB myc-tagged Nck SH3-1,2,3 mutant was provided by Bruce J. Mayer (50).
Infection assays.
Infection assays were described previously (13, 47). Briefly, FLC were inoculated and grown overnight on coverslips in Dulbecco modified Eagle medium supplemented with 10% fetal calf serum and overlaid with the bacterial or viral suspension. In bacterial experiments, to promote adhesion to the cells, the samples were centrifuged for 10 min at 2,000 × g at 23°C. The cells were incubated for 1 h at 37°C at a multiplicity of infection (MOI) of 200 for EPEC and S. flexneri and at an MOI of 400 for EHEC, followed by incubation in medium supplemented with gentamicin (50 μg/ml) for 3 h. The efficiency of Shigella and EPEC/EHEC infections were more than 10 and 90%, respectively. For vaccinia virus invasion, cells were infected at an MOI of 20 for 8 h without gentamicin, which resulted in an infection efficiency of 50 to 90%. After infection, coverslips were washed three to five times with PBS, fixed for 10 min with 3.7% fresh paraformaldehyde, and stained.
CR16 Western blotting.
Protein extracts from lung fibroblasts, as well as WT testes tissue (positive control), were separated by sodium dodecyl sulfate-gel electrophoresis and transferred onto nitrocellulose membrane as previously described (8). Immunoblotting was performed with rabbit polyclonal anti-CR16 (1:500) (generated in the R.S.G. laboratory), followed by horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (1:3,000; Cell Signaling, Danvers, MA), and visualized using enhanced chemiluminescence (GE Healthcare Life Sciences, Piscataway, NJ).
RNAi.
For WIRE knockdown, an ON-TARGETplus SMARTpool against mouse WIRE (Thermo Fisher Scientific/Dharmacon, Lafayette, CO) was transfected into WIP−/− and WT control fibroblasts using Lipofectamine 2000 according to the manufacturer's protocol. Control short interfering RNAs (siRNAs) for SMARTpool RNA interference (RNAi) included the ON-TARGETplus nontargeting pool (Thermo Fisher Scientific/Dharmacon). WIRE protein levels were determined 48 and 96 h after siRNA transfection by Western blotting and detection with rabbit polyclonal anti-WIRE antibody (Sigma, St. Louis, MO). Greater than 85% knockdown was confirmed by comparison of the optical density of WIRE bands relative to tubulin loading controls in ImageJ software.
Transfections.
For WIP and DN-Nck expression, DNA was transfected into 105 FLC 16 h prior to infection using Lipofectamine Plus (Invitrogen, Carlsbad, CA) or FuGENE6 (Roche Applied Science, Indianapolis, IN). The efficiency of transfection for each of these constructs ranged between 20 and 40%, with an overall transfection and infection efficiency of 2 to 15%.
Immunofluorescence microscopy.
The fixed cells were permeabilized with 0.5% Triton X-100 in PBS for 20 min and washed five times with 1% bovine serum albumin in PBS. Nck, N-WASP, and WIP were localized by incubating with anti-Nck rabbit polyclonal antibody (Upstate Biotechnology, Waltham, MA) at a 1:200 dilution, anti-N-WASP rabbit polyclonal antibody (provided by Rajat Rohatgi), or anti-WIP rabbit polyclonal antibody (provided by Raif Geha), respectively, for 1 h at room temperature. Subsequently, the cells were incubated with fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG antibody (Sigma) at a 1:200 dilution for 1 h. Myc-tagged DN-Nck was detected by incubation with anti-myc mouse monoclonal antibody (Oncogene, Cambridge, MA) at a 1:200 dilution for 1 h at room temperature. The cells were then incubated with FITC-conjugated anti-mouse IgG or TRITC (tetramethyl rhodamine isothiocyanate)-conjugated anti-mouse IgG antibody (Sigma, St. Louis, MO) at 1:200 dilution, followed by 1 μg of rhodamine-labeled phalloidin (Molecular Probes, Eugene, OR)/ml for 20 min at room temperature for F-actin staining. To identify bacterially infected cells, permeabilized cells were incubated with DAPI (4′,6′-diamidino-2-phenylindole; Molecular Probes, Eugene, OR) at 300 nM for 3 min at room temperature. Standard epifluorescence microscopy was performed using an AX70 upright microscope (Olympus, Tokyo, Japan). To determine the efficiency of pedestal formation for EPEC, the actin cytoskeleton of 100 bacterially infected cells were examined in five independent experiments by immunofluorescence. Cells containing at least 10 bacteria by DAPI staining were considered infected. To determine the relative tail lengths in vaccinia virus-infected or Shigella-infected WT and WIP−/− FLC, the lengths of 50 phalloidin-stained tails were measured using Photoshop 5.0 software (Adobe, San Jose, CA) by random sampling in three independent experiments.
RESULTS
Cdc42 is not required for EPEC-induced actin pedestal formation.
The Rho family GTPase Cdc42 interacts with the GTPase-binding domain (GBD) of N-WASP, and it was reported that WASP proteins lacking the GBD fail to localize to EPEC-induced actin pedestals, raising the possibility that Cdc42 plays a role in actin pedestal formation (27). However, inactivation of Cdc42 with Clostridium difficile toxin B or overexpression of N-WASP GBD did not prevent EPEC pedestal formation (5, 8), and an N-WASP mutant that is insensitive to Cdc42 activation can nonetheless mediate actin assembly by EPEC (32). To determine whether Cdc42 is required for pedestal formation, WT and Cdc42-deficient cells (13) were infected with EPEC. EHEC, which translocates the effector EspFU/TccP that directly activates N-WASP (14, 43), was included as a control. EPEC, like EHEC, efficiently formed pedestals in the absence of Cdc42 (Fig. 1), definitively ruling out an essential role for Cdc42 in EPEC pedestal formation.
Fig 1.

Actin pedestal formation by EPEC and EHEC does not require Cdc42. Pedestal formation by EPEC and EHEC is independent of Cdc42. EPEC and EHEC infection result in pedestal formation (arrows) in both WT (left panel) and Cdc42−/− FLCs (right panel) stained with phalloidin (F-actin, red) and DAPI (DNA, blue). Scale bar, 5 μm.
WIP is recruited to sites of actin assembly by diverse microbial pathogens but is not required for N-WASP recruitment or actin assembly.
Several pathogenic microbes usurp N-WASP to promote colonization and spread (48). WIP interacts with N-WASP and may play an important role in N-WASP-mediated actin assembly (34). WIP localizes to sites of actin assembly induced by vaccinia virus (47, 54), and Lommel et al. showed that WIP localizes along the entire length of the EPEC pedestal (32). WIP also localized to sites of actin assembly promoted by Shigella, a pathogen that does not utilize Nck (47). We first tested whether WIP was also found in actin pedestals induced by EHEC, another pathogen that generates assembly in the absence of Nck. In WT cells, WIP was recruited to the tips of pedestals induced by EHEC (Fig. 2A, upper panels, and see Fig. S2 in the supplemental material), indicating that pedestal localization of WIP does not require Nck. As expected, in agreement with previous studies (32), EPEC also recruited WIP to pedestals (Fig. 2A, upper panels, and see Fig. S2 in the supplemental material).
Fig 2.

WIP localizes to EPEC- and EHEC-induced pedestals but is not required for actin assembly by EPEC, EHEC, Shigella, or vaccinia virus. WT (top row) and WIP−/− (bottom row) fibroblasts were infected with EPEC, EHEC, Shigella, and vaccinia virus. Bacterial and viral DNA was identified by DAPI staining, and the sites of actin assembly were determined by staining with rhodamine-phalloidin. (A) WIP (arrows) localizes to actin pedestals induced by both EPEC and EHEC (upper panels); both EPEC and EHEC form actin-pedestals on WIP−/− cells (lower panels). (B) Shigella and vaccinia virus form actin tails efficiently on WT and WIP−/− FLC. Scale bar, 5 μm.
Given the localization of WIP at both EPEC and EHEC pedestals and the fact that it has been suggested that WIP may mediate the recruitment of N-WASP to pathogen-bound Nck (36, 45), we wanted to determine whether WIP was required for actin assembly induced by means of Nck-independent (EHEC and Shigella) or Nck-dependent (EPEC and vaccinia virus) mechanisms. After first confirming that WIP−/− FLC express normal levels of N-WASP and Nck (see Fig. S1 in the supplemental material), we found that EHEC efficiently formed pedestals on WIP−/− FLC similar to rates on WT FLC (Fig. 2A and Table 1). Furthermore, consistent with prior results showing no effect of WIP WASP-binding domain overexpression on Shigella tail formation (36), the percentages of Shigella-infected cells that had actin tails, and the relative tail lengths, were similar between WIP−/− FLC and WT controls (Fig. 2B and Table 1). These results indicate that although WIP is localized to sites of actin assembly induced by Shigella and EHEC, its presence neither depends on Nck, nor is it required for actin tail and pedestal formation, respectively.
Table 1.
The absence of WIP does not affect actin pedestal or actin tail formation by EPEC, EHEC, vaccinia virus, or Shigella
| Pathogen and parameter | Data (mean ± SD) for each host cell genotype |
P | |
|---|---|---|---|
| WT | WIP−/− | ||
| EPEC | |||
| Actin pedestals (%) | 99.6 ± 0.57 | 99.7 ± 0.58 | 0.65 |
| EHEC | |||
| Actin pedestals (%) | 90.3 ± 2.51 | 90.1 ± 4.7 | 0.81 |
| Shigella sp. | |||
| Infection rate (%) | 13.4 ± 4.3 | 9.1 ± 1.2 | 0.08 |
| Actin tails (%) | 69.3 ± 4.5 | 66.0 ± 6.0 | 0.32 |
| Avg tail length (μm) | 11.3 ± 5.0 | 10.7 ± 4.5 | 0.57 |
| Vaccinia virus | |||
| Infection rate (%) | 81.2 ± 9.7 | 80.1 ± 9.4 | 0.77 |
| Actin tails (%) | 81.7 ± 13.0 | 84.0 ± 11.6 | 0.18 |
| Avg tail length (μm) | 4.8 ± 1.3 | 4.9 ± 1.6 | 0.62 |
In contrast to Shigella and EHEC, vaccinia virus and EPEC utilize Nck to recruit host N-WASP, and previous reports suggested that WIP, by mediating the interaction between Nck and N-WASP, plays an important role in actin-tail formation by vaccinia virus (36) but, somewhat unexpectedly, not in actin pedestal formation by EPEC (32). To clarify this apparent dichotomy in the requirement for WIP in actin assembly, we assessed actin pedestal or tail formation by EPEC and vaccinia virus in WIP-deficient cells. Consistent with previous WIP mutant overexpression studies (32), EPEC efficiently formed pedestals on WIP−/− FLC similar to rates on WT FLC (Fig. 2A and Table 1). Moreover, we found that the percentage of vaccinia virus-infected cells that had actin tails was similar between WIP−/− FLC and WT controls (Fig. 2B and Table). There was also no difference in the relative tail length in vaccinia virus-infected WT or WIP−/− cells (Fig. 2B and Table 1). Together, these results demonstrate that WIP is not essential for EPEC-induced pedestal formation or for the actin-based motility of vaccinia virus.
Given that WIP was not required for EPEC- and vaccinia virus-based actin assembly, we hypothesized that WIP would not be required for either Nck or N-WASP recruitment. We first showed that N-WASP, but not Nck, was also recruited to EHEC actin pedestals on WIP-deficient cells, an observation consistent with the ability of EHEC to form pedestals in an Nck-independent manner (Fig. 3, bottom row). Importantly, N-WASP and Nck were also normally recruited by vaccinia virus and EPEC in WIP−/− cells (Fig. 3, top and middle rows).
Fig 3.

WIP is not essential for the recruitment of Nck or N-WASP during infection with vaccinia virus, EPEC or EHEC. F-actin (phalloidin staining, red) and DNA (DAPI, blue) were visualized in WT and WIP−/− FLC infected with vaccinia virus (top row), EPEC (middle row), or EHEC (lower row). Actin tails and pedestals are formed efficiently in WIP−/− FLC, and N-WASP (green) (arrows, left column) or Nck (green) (arrows, right column) are recruited to EPEC and vaccinia virus in the absence of WIP. *, Lack of Nck localization to EHEC. Scale bar, 5 μm.
WIP is recruited to EHEC, but not EPEC or vaccinia virus, upon ectopic expression of a dominant-negative Nck derivative.
Since WIP can bind to both Nck and N-WASP, WIP recruitment may be dependent on a direct interaction with either Nck and/or N-WASP. In order to test the role of Nck in WIP recruitment to both EPEC and vaccinia virus, we utilized a dominant-negative inhibitor of endogenous Nck (DN-Nck) that contains mutations in each of the SH3 domains (50). We first confirmed that the construct was effective by demonstrating that DN-Nck specifically blocked EPEC-induced but not EHEC-induced pedestal formation (21) (see Fig. S3 in the supplemental material) and blocked the recruitment of N-WASP to EPEC but not EHEC (Fig. 4, left panels). WIP recruitment to EHEC pedestals did not appear to depend on Nck because ectopic expression of the DN-Nck had no effect on pedestal localization by WIP (Fig. 4, bottom right). In contrast, DN-Nck expression blocked WIP recruitment to EPEC (Fig. 4, upper right panel). Similarly, and consistent with previously published work (13, 27), DN-Nck expression blocked both N-WASP and WIP recruitment to vaccinia virus (data not shown).
Fig 4.
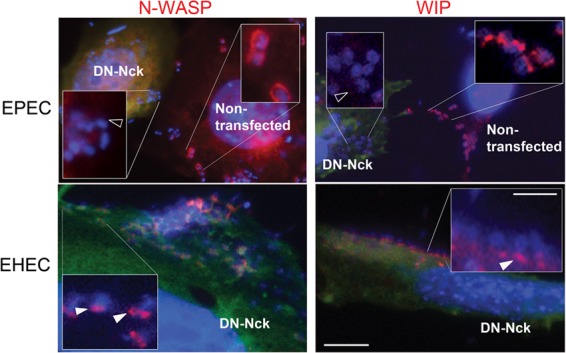
Neither N-WASP nor WIP is recruited to EPEC in the absence of Nck. FLC transfected with a dominant-negative inhibitor of Nck (green) were infected with either EPEC (upper row) or EHEC (lower row) and were analyzed by DAPI staining of DNA and immunostained for N-WASP (left panel, red) or WIP (right panel, red). N-WASP localizes to EHEC (closed arrowhead), but not EPEC in DN-Nck transfected cells (open arrowhead). WIP localizes to EHEC (closed arrowhead), but not EPEC (open arrowhead) in DN-Nck transfected cells. Green anti-myc staining is omitted from the insets to improve phalloidin and DAPI visualization. Scale bar, 10 μm; inset scale bar, 2.5 μm.
Localization of both WIP and Nck to EPEC and vaccinia virus is N-WASP dependent.
Given that the second SH3 domain within Nck interacts with WIP (4), the observation that WIP localization to sites of attachment by vaccinia virus or EPEC was blocked by the expression of an Nck derivative with defective SH3 domains is consistent with the simple model that Nck normally directly recruits WIP to sites of actin assembly induced by these pathogens. Alternatively, given that WIP and N-WASP are capable of direct interaction (15, 23, 34), it is also possible that the expression of a dominant-negative Nck derivative inhibited WIP recruitment by blocking the localization of N-WASP. Indeed, the ability of EHEC to recruit WIP to sites of pedestal formation (Fig. 4), in spite of the Nck-independent nature of EHEC pedestal formation, is consistent with N-WASP-dependent WIP recruitment. In addition, we previously demonstrated that WIP is recruited to sites of both vaccinia virus and Shigella actin tail formation in an N-WASP-dependent manner (47).
To determine whether N-WASP is required for WIP or Nck localization to sites of EPEC or EHEC pedestal formation, we infected N-WASP-deficient cells with these bacteria. As a control, in parallel we also infected these cells with vaccinia virus. All three pathogens were incapable of promoting actin assembly in these cells, an observation consistent with previous findings (21, 47). In addition, we confirmed that WIP and Nck recruitment to vaccinia virus actin tails required N-WASP (Fig. 5, lower row) (47). Finally, neither Nck nor WIP was localized at sites of adherent EHEC or EPEC in the absence of N-WASP (Fig. 5, top and middle rows). Since EHEC does not require Nck for either N-WASP recruitment or pedestal formation, these data are consistent with the suggestion that WIP is recruited to EHEC-induced actin pedestals indirectly through its interaction with N-WASP. Importantly, together with the above finding that ectopic expression of a DN-Nck derivative blocks N-WASP recruitment to sites of EPEC attachment, these data also suggest that, as is the case for vaccinia virus, N-WASP and Nck are dependent on each other, but not WIP, for localization to sites of EPEC-mediated actin assembly.
Fig 5.

N-WASP is required for the recruitment of both Nck and WIP. N-WASP−/− FLC were infected with either EPEC (upper row) or EHEC (middle row) or vaccinia virus (lower row) and were analyzed by DAPI staining of DNA (blue), phalloidin staining for actin (red), and immunostaining with Nck (left panels, green) or transfection with WIP-GFP (right panels, green). Neither Nck nor WIP localized to EPEC (arrows), EHEC (arrows) or vaccinia virus (arrows) in the absence of N-WASP. Scale bar, 5 μm.
WIP family members CR16 and WICH/WIRE do not contribute to actin rearrangements induced by EPEC or vaccinia virus in the absence of WIP.
In addition to WIP, the WIP family of proteins includes two additional WIP homologues that are capable of binding N-WASP and may be able to functionally substitute for WIP: CR16 and WIRE/WICH (WIP related/WIP CR16 homologous) (3, 35). In order to determine whether the apparent dispensability of WIP for actin pedestal and tail formation was a result of functional redundancy of CR16 or WIRE for WIP, we first confirmed by Western blotting that only WIRE, and not CR16, was expressed in fibroblasts (see Fig. 4A in the supplemental material). We next generated WIRE knockdown and WIP−/−/WIRE knockdown lines by siRNA-mediated depletion of WIRE in WT and WIP−/− cells. We confirmed by Western blot and densitometry that there was >85% knockdown of WIRE (see Fig. S4B in the supplemental material). We found no significant differences, compared to WT cells or WIP−/− cells, in the rates of infection, the frequency of actin tails or actin pedestals, or the morphology of these actin structures during infection with EPEC or vaccinia virus in WIRE-depleted cell lines (Fig. 6; see also Table S1 in the supplemental material). These data confirm that neither WIP, nor CR16, nor WIRE/WICH is required for EPEC- and vaccinia virus-induced actin rearrangement.
Fig 6.
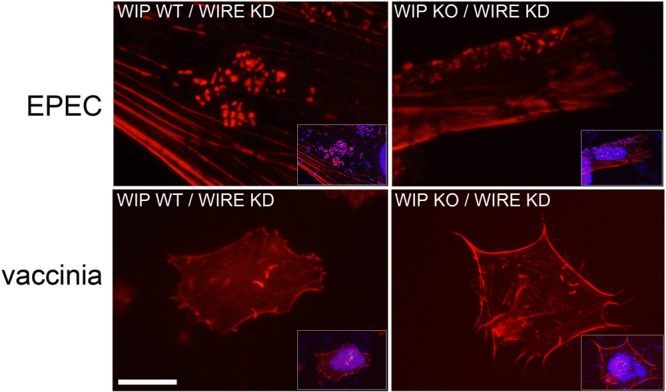
The WIP family member WIRE/WICH does not substitute for WIP in WIP−/− cells during infection with EPEC or vaccinia virus. EPEC (upper row) and vaccinia virus (lower row) were equally able to induce actin pedestals and actin tails, respectively, in WIP WT (left panels) or KO cells (right panels) depleted of the WIRE/WICH by siRNA. Red is actin staining with Alexa Fluor 594-conjugated phalloidin; insets show DAPI staining of nuclei (omitted from the figure for clarity). Scale bar, 5 μm.
DISCUSSION
In order to characterize the role of N-WASP and its interacting partners (WIP, Cdc42, and Nck) in regulating the actin cytoskeleton, we generated cell lines with germ line deletion or dominant-negative inhibition of these proteins (1, 2, 13, 47). These cell lines have enabled us to characterize more fully the host molecular signaling events required for pathogen-induced actin-assembly by EPEC, EHEC, and vaccinia virus and demonstrate conclusively that Cdc42 is not required for EPEC- or EHEC-induced pedestal formation.
We have demonstrated that WIP is not required for actin assembly induced by either EPEC or vaccinia virus. Although both N-WASP and WIP are recruited to sites of actin assembly by EPEC (Fig. 2A and 4) and vaccinia virus (Fig. 3), only N-WASP, and not WIP, was required for actin pedestal formation and actin-based motility (Fig. 3) (32, 47). For vaccinia virus, Nck has been implicated as the link between virally expressed A36R and WIP, which in turn binds N-WASP (36, 45). Like vaccinia virus, EPEC also utilizes Nck as a link between a bacterially expressed protein (Tir) and downstream host signaling molecules (9, 21). We have demonstrated that EPEC and vaccinia virus do not require WIP for Nck localization or for the recruitment of N-WASP in initiating actin polymerization. A direct interaction between Nck and N-WASP, which has been confirmed in vitro (40, 42), is consistent with our findings. However, reports have suggested that the region of N-WASP previously shown to bind Nck, the proline-rich region domain, is not required for either the localization of N-WASP to pedestals or for the formation of pedestals by EPEC (27, 33). Given the more recent demonstration that EPEC generates actin pedestals by (at least) three pathways, only one of which is Nck-dependent (11), the lack of discernible phenotypes associated with N-WASP deletion derivatives might be due to redundancy. For example, an Nck-independent pathway involving the N-WASP WH1 domain might facilitate N-WASP localization and pedestal formation in the absence of the Nck-binding N-WASP proline-rich domain.
Our results suggest that the recruitment of Nck, WIP, and N-WASP to EPEC or vaccinia virus does not proceed in a simple linear fashion. Despite the fact that Nck can bind to phosphorylated Tir (9, 21), we found that Nck does not localize to EPEC or vaccinia virus in the absence of N-WASP (Fig. 5). These findings contrast with previous reports in which GFP-tagged Nck was observed beneath extracellular vaccinia virus particles in N-WASP−/− cells (54). That we did not see endogenous Nck staining suggests that N-WASP may be required to stabilize the nascent interaction between Tir and Nck or may facilitate the translocation of Nck to the cytoplasmic tail of Tir, but that GFP-Nck, ectopically expressed at relatively high levels, might still localize to vaccinia virus in the absence of N-WASP. We propose that EPEC Tir and vaccinia virus A36R require a complex that contains N-WASP and Nck for recruitment to the pathogen (Fig. 7). A role for N-WASP upstream of Nck is also consistent with FRAP studies demonstrating that the rate of N-WASP exchange is the major determinant of both Nck and WIP turnover at the vaccinia virus coat protein (54). It has been proposed that the observed differences in the rates of Nck and N-WASP exchange may be due to additional stabilizing interactions between the WASP-homology 2 (WH2) domain of N-WASP and free barbed ends of growing actin filaments (54), and this model is compatible with our data demonstrating that the presence of both Nck and N-WASP at the viral particle is required for WIP recruitment to vaccinia virus. Although WIP can be found as part of this Nck/N-WASP complex at the site of actin assembly, its presence is not required for actin assembly induced by EPEC or vaccinia virus.
Fig 7.
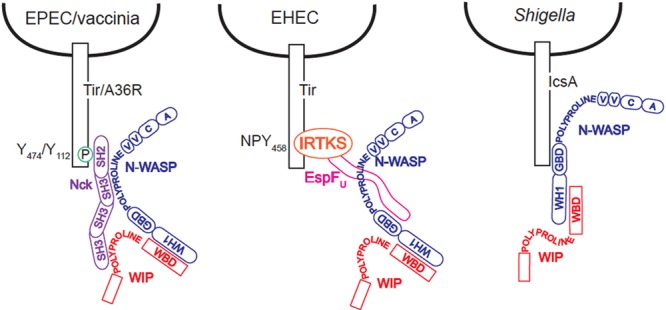
Model for strategies of Nck/N-WASP recruitment and activation shared by EPEC and vaccinia virus and contrasted with EHEC and Shigella.
We also determined that it is unlikely that other WIP family members—CR16 and WIRE/WICH—serve a functionally redundant role in stabilizing the Nck/N-WASP complex. CR16 is not expressed in the fibroblasts used in these experiments, and depletion of WIRE/WICH did not affect the rates of infection or the ability of EPEC or vaccinia virus to induce actin rearrangements in WIP−/− cells. Our findings are also consistent with prior studies involving the overexpression of either the WBD of WIP or WIPΔWBD that indicated that an N-WASP/WIP interaction was dispensable for EPEC pedestal formation (32). The observation that overexpression of the WBD of WIP blocked vaccinia virus actin tail formation, suggesting an essential role for WIP in this process (36), may have resulted from reduced actin polymerization activity through nonspecific sequestration of N-WASP. Given that we show here that N-WASP is required for Nck recruitment by vaccinia virus, the prior observation that overexpression of the WBD of WIP also blocked the recruitment of Nck to vaccinia virus (36) may also have been a consequence of its effect on N-WASP.
Overall, our studies highlight similar pathogenic strategies shared by EPEC and vaccinia virus in the Nck-dependent recruitment of an N-WASP/WIP complex to homologous phosphotyrosine motifs in EPEC Tir and vaccinia virus A36R. We demonstrate a dual requirement for both Nck and N-WASP, but not WIP or WIP family members, in actin assembly induced by EPEC and vaccinia virus.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health grants R01AI46454 (to J.M.L.), P01HL059561 (to R.S.G. and S.B.S.), R01AI052354 (to S.B.S.), and F32DK088442 (to J.J.G.), as well as a Career Development Award from the Crohn's and Colitis Foundation of America (to J.J.G.) and the Harvard Digestive Disease Center (5P30DK034854 to S.B.S. and J.J.G.).
Footnotes
Published ahead of print 10 September 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Anton I, et al. 2003. WIP participates in actin reorganization and ruffle formation induced by PDGF. J. Cell Sci. 116:2443–2451 [DOI] [PubMed] [Google Scholar]
- 2. Anton IM, et al. 2002. WIP deficiency reveals a differential role for WIP and the actin cytoskeleton in T and B cell activation. Immunity 16:193–204 [DOI] [PubMed] [Google Scholar]
- 3. Anton IM, Jones GE. 2006. WIP: a multifunctional protein involved in actin cytoskeleton regulation. Eur. J. Cell Biol. 85(3–4):295–304 [DOI] [PubMed] [Google Scholar]
- 4. Anton IM, Lu W, Mayer BJ, Ramesh N, Geha RS. 1998. The Wiskott-Aldrich syndrome protein-interacting protein (WIP) binds to the adaptor protein Nck. J. Biol. Chem. 273:20992–20995 [DOI] [PubMed] [Google Scholar]
- 5. Ben-Ami G, et al. 1998. Agents that inhibit Rho, Rac, and Cdc42 do not block formation of actin pedestals in HeLa cells infected with enteropathogenic Escherichia coli. Infect. Immun. 66:1755–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Benesch S, et al. 2005. N-WASP deficiency impairs EGF internalization and actin assembly at clathrin-coated pits. J. Cell Sci. 118(Pt 14):3103–3115 [DOI] [PubMed] [Google Scholar]
- 7. Bu W, Chou AM, Lim KB, Sudhaharan T, Ahmed S. 2009. The Toca-1-N-WASP complex links filopodial formation to endocytosis. J. Biol. Chem. 284:11622–11636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Campellone KG, et al. 2008. Repetitive N-WASP-binding elements of the enterohemorrhagic Escherichia coli effector EspF(U) synergistically activate actin assembly. PLoS Pathog. 4:e1000191 doi:10.1371/journal.ppat.1000191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Campellone KG, Giese A, Tipper DJ, Leong JM. 2002. A tyrosine-phosphorylated 12-amino-acid sequence of enteropathogenic Escherichia coli Tir binds the host adaptor protein Nck and is required for Nck localization to actin pedestals. Mol. Microbiol. 43:1227–1241 [DOI] [PubMed] [Google Scholar]
- 10. Campellone KG, Leong JM. 2003. Tails of two Tirs: actin pedestal formation by enteropathogenic Escherichia coli and enterohemorrhagic E. coli O157:H7. Curr. Opin. Microbiol. 6:82–90 [DOI] [PubMed] [Google Scholar]
- 11. Campellone KG, Leong JM. 2005. Nck-independent actin assembly is mediated by two phosphorylated tyrosines within enteropathogenic Escherichia coli Tir. Mol. Microbiol. 56:416–432 [DOI] [PubMed] [Google Scholar]
- 12. Campellone KG, et al. 2004. Clustering of Nck by a 12-residue Tir phosphopeptide is sufficient to trigger localized actin assembly. J. Cell Biol. 164:407–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen F, et al. 2000. Cdc42 is required for PIP(2)-induced actin polymerization and early development but not for cell viability. Curr. Biol. 10:758–765 [DOI] [PubMed] [Google Scholar]
- 14. Cheng HC, Skehan BM, Campellone KG, Leong JM, Rosen MK. 2008. Structural mechanism of WASP activation by the enterohaemorrhagic Escherichia coli effector EspF(U). Nature 454:1009–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de la Fuente MA, et al. 2007. WIP is a chaperone for Wiskott-Aldrich syndrome protein (WASP). Proc. Natl. Acad. Sci. U. S. A. 104:926–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. DeVinney R, Puente JL, Gauthier A, Goosney D, Finlay BB. 2001. Enterohaemorrhagic and enteropathogenic Escherichia coli use a different Tir-based mechanism for pedestal formation. Mol. Microbiol. 41:1445–1458 [DOI] [PubMed] [Google Scholar]
- 17. Finlay BB, Rosenshine I, Donnenberg MS, Kaper JB. 1992. Cytoskeletal composition of attaching and effacing lesions associated with enteropathogenic Escherichia coli adherence to HeLa cells. Infect. Immun. 60:2541–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frischknecht F, et al. 1999. Actin-based motility of vaccinia virus mimics receptor tyrosine kinase signalling. Nature 401:926–929 [DOI] [PubMed] [Google Scholar]
- 19. Fukuoka M, Miki H, Takenawa T. 1997. Identification of N-WASP homologs in human and rat brain. Gene 196:43–48 [DOI] [PubMed] [Google Scholar]
- 20. Garmendia J, et al. 2004. TccP is an enterohaemorrhagic Escherichia coli O157:H7 type III effector protein that couples Tir to the actin-cytoskeleton. Cell Microbiol. 6:1167–1183 [DOI] [PubMed] [Google Scholar]
- 21. Gruenheid S, et al. 2001. Enteropathogenic Escherichia coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat. Cell Biol. 3:856–859 [DOI] [PubMed] [Google Scholar]
- 22. Higgs HN, Pollard TD. 2000. Activation by Cdc42 and PIP(2) of Wiskott-Aldrich syndrome protein (WASp) stimulates actin nucleation by Arp2/3 complex. J. Cell Biol. 150:1311–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ho HY, et al. 2004. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell 118:203–216 [DOI] [PubMed] [Google Scholar]
- 24. Isaac BM, et al. 2010. N-WASP has the ability to compensate for the loss of WASP in macrophage podosome formation and chemotaxis. Exp. Cell Res. 316:3406–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jerse AE, Yu J, Tall BD, Kaper JB. 1990. A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc. Natl. Acad. Sci. U. S. A. 87:7839–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaksonen M, Sun Y, Drubin DG. 2003. A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell 115:475–487 [DOI] [PubMed] [Google Scholar]
- 27. Kalman D, et al. 1999. Enteropathogenic Escherichia coli acts through WASP and Arp2/3 complex to form actin pedestals. Nat. Cell Biol. 1:389–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kempiak SJ, Yip SC, Backer JM, Segall JE. 2003. Local signaling by the EGF receptor. J. Cell Biol. 162:781–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kenny B. 1999. Phosphorylation of tyrosine 474 of the enteropathogenic Escherichia coli (EPEC) Tir receptor molecule is essential for actin nucleating activity and is preceded by additional host modifications. Mol. Microbiol. 31:1229–1241 [DOI] [PubMed] [Google Scholar]
- 30. Kenny B, et al. 1997. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91:511–520 [DOI] [PubMed] [Google Scholar]
- 31. Klein C, Bueler H, Mulligan RC. 2000. Comparative analysis of genetically modified dendritic cells and tumor cells as therapeutic cancer vaccines. J. Exp. Med. 191:1699–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lommel S, Benesch S, Rohde M, Wehland J, Rottner K. 2004. Enterohaemorrhagic and enteropathogenic Escherichia coli use different mechanisms for actin pedestal formation that converge on N-WASP. Cell Microbiol. 6:243–254 [DOI] [PubMed] [Google Scholar]
- 33. Lommel S, et al. 2001. Actin pedestal formation by enteropathogenic Escherichia coli and intracellular motility of Shigella flexneri are abolished in N-WASP-defective cells. EMBO Rep. 2:850–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martinez-Quiles N, et al. 2001. WIP regulates N-WASP-mediated actin polymerization and filopodium formation. Nat. Cell Biol. 3:484–491 [DOI] [PubMed] [Google Scholar]
- 35. Meng L, Rajmohan R, Yu S, Thanabalu T. 2007. Actin binding and proline-rich motifs of CR16 play redundant role in growth of vrp1Δ cells. Biochem. Biophys. Res. Commun. 357:289–294 [DOI] [PubMed] [Google Scholar]
- 36. Moreau V, et al. 2000. A complex of N-WASP and WIP integrates signalling cascades that lead to actin polymerization. Nat. Cell Biol. 2:441–448 [DOI] [PubMed] [Google Scholar]
- 37. Naqvi SN, Zahn R, Mitchell DA, Stevenson BJ, Munn AL. 1998. The WASp homologue Las17p functions with the WIP homologue End5p/verprolin and is essential for endocytosis in yeast. Curr. Biol. 8:959–962 [DOI] [PubMed] [Google Scholar]
- 38. Newsome TP, Weisswange I, Frischknecht F, Way M. 2006. Abl collaborates with Src family kinases to stimulate actin-based motility of vaccinia virus. Cell Microbiol. 8:233–241 [DOI] [PubMed] [Google Scholar]
- 39. Ory DS, Neugeboren BA, Mulligan RC. 1996. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc. Natl. Acad. Sci. U. S. A. 93:11400–11406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rivero-Lezcano OM, Marcilla A, Sameshima JH, Robbins KC. 1995. Wiskott-Aldrich syndrome protein physically associates with Nck through Src homology 3 domains. Mol. Cell. Biol. 15:5725–5731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rohatgi R, et al. 1999. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97:221–231 [DOI] [PubMed] [Google Scholar]
- 42. Rohatgi R, Nollau P, Ho HY, Kirschner MW, Mayer BJ. 2001. Nck and phosphatidylinositol 4,5-bisphosphate synergistically activate actin polymerization through the N-WASP-Arp2/3 pathway. J. Biol. Chem. 276:26448–26452 [DOI] [PubMed] [Google Scholar]
- 43. Sallee NA, et al. 2008. The pathogen protein EspF(U) hijacks actin polymerization using mimicry and multivalency. Nature 454:1005–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sanchez AM, et al. 2010. Estrogen receptor-alpha promotes breast cancer cell motility and invasion via focal adhesion kinase and N-WASP. Mol. Endocrinol. 24:2114–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Scaplehorn N, et al. 2002. Grb2 and Nck act cooperatively to promote actin-based motility of vaccinia virus. Curr. Biol. 12:740–745 [DOI] [PubMed] [Google Scholar]
- 46. Smith GL, Murphy BJ, Law M. 2003. Vaccinia virus motility. Annu. Rev. Microbiol. 57:323–342 [DOI] [PubMed] [Google Scholar]
- 47. Snapper SB, et al. 2001. N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nat. Cell Biol. 3:897–904 [DOI] [PubMed] [Google Scholar]
- 48. Stevens JM, Galyov EE, Stevens MP. 2006. Actin-dependent movement of bacterial pathogens. Nat. Rev. Microbiol. 4:91–101 [DOI] [PubMed] [Google Scholar]
- 49. Takenawa T, Suetsugu S. 2007. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell. Biol. 8:37–48 [DOI] [PubMed] [Google Scholar]
- 50. Tanaka M, Gupta R, Mayer BJ. 1995. Differential inhibition of signaling pathways by dominant-negative SH2/SH3 adapter proteins. Mol. Cell. Biol. 15:6829–6837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tsuboi S. 2006. A complex of Wiskott-Aldrich syndrome protein with mammalian verprolins plays an important role in monocyte chemotaxis. J. Immunol. 176:6576–6585 [DOI] [PubMed] [Google Scholar]
- 52. Vingadassalom D, et al. 2009. Insulin receptor tyrosine kinase substrate links the Escherichia coli O157:H7 actin assembly effectors Tir and EspF(U) during pedestal formation. Proc. Natl. Acad. Sci. U. S. A. 106:6754–6759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weiss SM, et al. 2009. IRSp53 links the enterohemorrhagic Escherichia coli effectors Tir and EspFU for actin pedestal formation. Cell Host Microbe 5:244–258 [DOI] [PubMed] [Google Scholar]
- 54. Weisswange I, Newsome TP, Schleich S, Way M. 2009. The rate of N-WASP exchange limits the extent of ARP2/3-complex-dependent actin-based motility. Nature 458:87–91 [DOI] [PubMed] [Google Scholar]
- 55. Welch MD, Mullins RD. 2002. Cellular control of actin nucleation. Annu. Rev. Cell Dev. Biol. 18:247–288 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.