

Abstract
FMS-like tyrosine kinase-3 ligand (Flt3L) is a dendritic cell (DC) growth and differentiation factor with potential in antitumor therapies and antibacterial immunization strategies. However, the effect of systemic Flt3L treatment on lung-protective immunity against bacterial infection is incompletely defined. Here, we examined the impact of deficient (in Flt3L knockout [KO] mice), normal (in wild-type [WT] mice), or increased Flt3L availability (in WT mice pretreated with Flt3L for 3, 5, or 7 days) on lung DC subset profiles and lung-protective immunity against the major lung-tropic pathogen, Streptococcus pneumoniae. Although in Flt3L-deficient mice the numbers of DCs positive for CD11b (CD11bpos DCs) and for CD103 (CD103pos DCs) were diminished, lung permeability, a marker of injury, was unaltered in response to S. pneumoniae. In contrast, WT mice pretreated with Flt3L particularly responded with increased numbers of CD11bpos DCs and with less pronounced numbers of CD103pos DCs and impaired bacterial clearance and with increased lung permeability following S. pneumoniae challenge. Notably, infection of Flt3L-pretreated mice with S. pneumoniae lacking the pore-forming toxin, pneumolysin (PLY), resulted in substantially less lung CD11bpos DCs activation and reduced lung permeability. Collectively, this study establishes that Flt3L treatment enhances the accumulation of proinflammatory activated lung CD11bpos DCs which contribute to acute lung injury in response to PLY released by S. pneumoniae.
INTRODUCTION
FMS-like tyrosine kinase-3 ligand (Flt3L) is a hematopoietic growth factor promoting the proliferation, differentiation, and survival of progenitor cells in the bone marrow (33, 43). Flt3L treatment of mice has been shown to promote immune reconstitution after myeloablative chemotherapy and bone marrow transplantation (7). Systemic administration of human Flt3L to mice results in the strong mobilization and expansion of myeloid dendritic cell (DC) subsets in lymphoid and nonlymphoid tissues, including the lung (36, 37). In the past few years, Flt3L has been used in immunization strategies aimed at preventing airborne bacterial infections. For example, Flt3L administration, when used in murine vaccination studies, enhances mucosal immunity against Streptococcus pneumoniae and improves protective immunity against Mycobacterium tuberculosis (24, 48). Moreover, in preclinical cancer models, Flt3L administration expands DC populations and supports the generation of a specific antitumor response in mice, resulting in tumor regression (2, 34). In addition, several clinical studies conducted in patients with multiple types of cancer, including breast, lung, or metastatic colon cancer, demonstrated that Flt3L markedly increases DC mobilization, providing opportunities for Flt3L use in antitumor immunotherapies (11, 13, 40). Given the expanding therapeutic potential of Flt3L, it is imperative to understand the effects of Flt3L-induced DC mobilization on the host response to infection or injury in all tissues including the lung.
Resident lung DCs are strategically located between the alveolar epithelium and lung vascular endothelium, where they form a dense network of sentinel cells specialized to sample inhaled pathogens and apathogenic particles (23). In the murine lung, two primary subsets of the classical migratory myeloid DC populations exist: (i) DCs positive for CD11b (CD11bpos DCs), which are those with cell surface antigen expression of high levels of CD11b and expression of major histocompatibility complex class II (MHC-II) and the absence of CD103 expression (CD11bhigh MHCIIpos CD103neg cells), and (ii) CD103pos DCs, defined by cell surface antigen expression of low levels of CD11b and expression of MHC-II and CD103 (CD11blow MHCIIpos CD103pos cells) (18, 46). Current knowledge reveals that lung CD103pos DCs are critically dependent on Flt3L for their differentiation from bone marrow-derived pre-DCs. In contrast, additional cytokines including granulocyte-macrophage colony-stimulating factor (GM-CSF) support the growth and differentiation of lung CD11bpos DCs, which are more heterogeneous and can include an additional population of nonresident CD11bpos DCs derived from circulating blood monocytes in response to lung inflammation or infection (29, 41). An increasing amount of data reveals that CD11bpos DCs and CD103pos DCs differ in function. For example, CD11bpos DCs respond to lipopolysaccharide (LPS)-induced lung inflammation with potent chemokine production (3), whereas CD103pos DCs are involved in viral antigen transport to draining lymph nodes and induce CD8 T cell proliferation during influenza virus infection (22). Also, CD103pos DCs, but not CD11bpos DCs, were found to play a major role in uptake of apoptotic cells in the lungs of mice, thereby contributing to the reestablishment of lung homeostasis (8).
However, despite these advances, our understanding of the functional differences between lung CD11bpos DCs and CD103pos DCs remains limited. Although some studies suggest that DCs may enhance immunity against microbes or tumors, our prior data suggest that augmenting lung DC numbers may impair bacterial clearance and promote lung injury. Specifically, we previously showed that systemic pretreatment of mice with Flt3L severely aggravated the lung inflammatory response to bacterial infection, which was characterized by decreased bacterial clearance and increased lung barrier dysfunction in mice (49, 53). Moreover, other groups have reported controversial data showing that Flt3L treatment of mice either impaired protective immunity against intracellular pathogens (1, 49, 53) or improved resistance against Pseudomonas aeruginosa in a mouse model of burn wound infection (4). However, neither our previous study nor reports from other groups addressed the effect of Flt3L treatment of mice on the accumulation and activation profiles of lung CD11bpos DC versus CD103pos DC subsets and their possible roles as proinflammatory effectors during bacterial infections.
In the current study, we examined the impact of deficient, normal, and increased Flt3L availability on numbers and activation phenotypes of lung CD11bpos DC and CD103pos DC subsets, and we further explored the contribution of the pneumococcal virulence factor, pneumolysin (PLY), to lung DC subset activation, lung permeability, and bacterial clearance.
MATERIALS AND METHODS
Mice.
Wild-type (WT) C57BL/6N mice were purchased from Charles River (Sulzfeld, Germany). Flt3L knockout (KO) mice (C57BL/6-flt3Ltm1Imx) (39) and respective C57BL/6N control mice were obtained from Taconic (Germantown, NY). Inducible nitric oxide (iNOS) KO mice (B6.129P2-NOS2tm1Lau/J) (28) and corresponding C57BL/6J WT controls were purchased from Jackson Laboratories (Sacramento, CA). Mice were kept under conventional conditions with free access to food and water and were used in all experiments at 8 to 12 weeks of age, according to the guidelines of the Institutional Animal Care and Use Committee of Hannover Medical School. Animal experiments were approved by our local government authorities.
Culture and quantification of S. pneumoniae bacteria.
We used a PLY-producing clinical isolate of capsular group 19 S. pneumoniae strain EF3030 (6). In selected experiments, an otherwise isogenic PLY-deficient derivate of serotype 19 S. pneumoniae strain EF3030 (EF3030ΔPLY) (19, 51) was employed. Bacteria were grown in Todd-Hewitt broth (Oxoid, Basingstoke, United Kingdom) supplemented with 20% fetal calf serum (FCS) (PAA Laboratories, Pasching, Austria) to mid-log phase. Aliquots were snap-frozen in liquid nitrogen and stored at −80°C. For quantification, serial dilutions of S. pneumoniae were plated on sheep blood agar plates (BD Biosciences, Heidelberg, Germany) and incubated at 37°C in 5% CO2 for 18 h, followed by determination of the numbers of CFU, as described previously (20, 53).
Application of Flt3L or iNOS inhibitor 1400W and infection of mice with S. pneumoniae.
Mice were either pretreated with subcutaneous (s.c.) injections of Flt3L (10 μg/mouse) or vehicle (phosphate-buffered saline [PBS]) (PAA Laboratories) supplemented with 0.1% human serum albumin (Behring, Marburg, Germany) for 3, 5, or 7 consecutive days. For infection experiments, Flt3L KO mice or Flt3L-pretreated WT mice or respective control WT mice were anesthetized with xylazine (5 mg/kg of body weight) (Bayer, Leverkusen, Germany) and ketamine (75 mg/kg) (Albrecht, Aulendorf, Germany) and then orotracheally (o.t.) intubated with a 29-gauge Abbocath catheter (Abbott, Wiesbaden, Germany), which was inserted into the trachea under visual control with transillumination of the neck region (17, 21). Mice received o.t. instillations of 107 CFU of S. pneumoniae in a volume of 50 μl of PBS. Mock-infected mice received applications of PBS only. Subsequently, mice were brought back to their cages with free access to food and water. In selected experiments, Flt3L-pretreated mice received intraperitoneal (i.p.) injections of iNOS inhibitor 1400W (Cayman Chemical, Ann Arbor, MI) (5 mg/kg of body weight, dissolved in dimethyl sulfoxide [DMSO; Roth, Karlsruhe, Germany]) or vehicle (PBS-DMSO), starting at 1 h prior to infection, with subsequent applications every 12 h until the mice were euthanized at the time points designated in the figures (12).
Determination of bacterial loads in bronchoalveolar lavage (BAL) fluid and lung tissue.
At various time points postinfection, S. pneumoniae-infected Flt3L KO mice, Flt3L-pretreated WT mice, and WT control mice were euthanized with an overdose of isoflurane (Baxter, Unterschleissheim, Germany), and bacterial loads in both BAL fluid and lung tissue homogenates collected from the same mice were determined as outlined previously in detail (17, 47, 53). Briefly, the trachea was cannulated with a shortened 20-gauge needle, and 300-μl aliquots of ice-cold sterile PBS supplemented with EDTA (Versen; Biochrom, Berlin, Germany) was instilled and aspirated until a first BAL fluid volume of 1.5 ml was obtained. Subsequently, an additional BAL fluid volume of 4.5 ml was collected by rinsing the lung with 500-μl aliquots of PBS-EDTA. After collection of BAL fluids, individual lung lobes were removed, dissected, and homogenized in 2 ml of Hanks' balanced salt solution (HBSS) without supplements (PAA Laboratories) using a tissue homogenizer (IKA T18; IKA, Staufen, Germany) and then filtered through 100-μm-pore-size cell strainers (BD Biosciences). Bacterial loads in BAL fluids and lung tissue homogenates were determined by plating 100 μl of the respective aliquots in 10-fold serial dilutions on sheep blood agar plates (BD Biosciences), followed by incubation of the plates at 37°C in 5% CO2 for 18 h and subsequent determination of total CFU counts.
Determination of neutrophils in BAL fluids.
For quantification of neutrophils in BAL fluids, BAL fluid samples (whole-lung washes) were centrifuged (400 × g at 4°C for 9 min), and resulting cell pellets were resuspended in RPMI 1640 medium (PAA Laboratories)–10% FCS. Alveolar-recruited neutrophils were quantified in Pappenheim-stained cytocentrifuge preparations according to their staining patterns and morphological criteria, including cell size and nuclear shape, and absolute leukocyte numbers were calculated by multiplication of the respective values by total BAL fluid cell counts.
Determination of lung permeability.
Analysis of lung permeability in various experimental groups of mice (infected or uninfected Flt3L KO mice, Flt3L-pretreated WT mice, or the respective control WT mice) was done as described recently in detail (20, 53). The lung permeability index is defined as the ratio of fluorescence signals of undiluted BAL fluid samples relative to fluorescence signals of 1/100 diluted serum samples.
Purification of CD11c-positive cells from lung tissue and immuno-phenotypic analysis of lung DC subsets.
Lungs from mice of the various experimental groups outlined above were subjected to BAL followed by perfusion of the lungs via the right ventricle with HBSS until lung lobes were visually free of blood. Lung lobes were removed in a manner to avoid contaminations with lymphatic tissue or conducting airways; the lobes were cut into small pieces and incubated in digestion solution consisting of RPMI medium supplemented with collagenase A (5 mg/ml) and DNase I (1 mg/ml) (both from Roche, IN) for 90 min at 37°C. The digested tissue was then filtered through a 40-μm-pore-size cell strainer (BD Biosciences), and digestion was stopped by adding RPMI medium–10% FCS (44, 45, 53). Next, CD11c-positive leukocytes, including CD11bpos DC and CD103pos lung DC subsets, were purified from digested tissue by using a CD11c magnetic bead cell sorting (MACS) purification kit (Miltenyi Biotec, Bergisch Gladbach, Germany) following the instructions of the manufacturer. Briefly, cells were centrifuged at 300 × g for 10 min at 4°C, and the pellet was resuspended in MACS buffer. After centrifugation, cells were incubated with 10 μl of anti-CD11c antibody-conjugated magnetic beads/107 cells (Miltenyi Biotec) for 15 min at 4°C. Following incubation, cells were washed and then passed over a MACS magnetic separation (MS) column to separate the CD11c-positive cells (∼90% purity). Purified lung CD11c-positive cells were further subjected to flow cytometric immuno-phenotypic analysis of lung DC subsets. After cells were blocked with Octagam (Octapharm, Langenfeld, Germany), they were stained for 20 min at 4°C with appropriately diluted fluorochrome-conjugated monoclonal antibodies with specificities for the following cell surface molecules: phycoerythrin (PE)-Cy7-conjugated anti-CD11b, PE-Cy5.5-conjugated anti-CD11c, PE-conjugated anti-MHC class II (all obtained from BD Biosciences), and allophycocyanin (APC)-conjugated anti-CD103 antibodies (eBioscience, San Diego, CA). Subsequently, cells were washed twice in PBS–0.1% bovine serum albumin–0.02% Na-azide and centrifuged at 300 × g for 3 min at 4°C. Immuno-phenotypic analysis of lung CD11c-positive cells was performed on a FACSCanto flow cytometer (BD Biosciences). The identification of CD11bpos and CD103pos DCs was performed according to their forward scatter (FSC) area/side scatter (SSC) area characteristics and their low-green autofluorescence properties, in conjunction with their CD11cpos CD11bhigh MHCIIpos CD103neg (CD11bpos DCs) or CD11cpos CD11blow MHCIIpos CD103pos (CD103pos DCs) immuno-phenotypic profiles. Data analysis and postacquisition compensation of spectral overlaps between the fluorescence channels were performed using FACSDiva software (BD Biosciences). For determination of total cell numbers, percentages of DC subsets were multiplied by the respective total cell counts of purified CD11c-positive cells (18, 45, 53).
Flow sorting of lung DC subsets.
High-speed cell sorting of lung CD11bpos DC and CD103pos DC subsets for subsequent real-time reverse transcription-PCR (RT-PCR) and Western blot analysis was performed with appropriately stained lung CD11c-positive cell preparations, as described above. Sorting was performed on a high-speed FACSAria II flow cytometer (BD Biosciences), which was prepared for aseptic sorting according to the manufacturer's instructions. CD11bpos DC and CD103pos DC subsets were gated as outlined above. After appropriate gating and compensation setting, cells were sorted with an 85-μm nozzle at a flow rate of ∼10,000 particles per second. The complete sorting process was performed at a constant temperature of 4°C. Resort analysis of sorted cells revealed sort purities of >98% (44, 45).
Total cellular RNA isolation, cDNA synthesis, and real-time RT-PCR.
Total cellular RNA was isolated from flow-sorted lung CD11bpos DCs and CD103pos DCs from Flt3L-pretreated, mock-infected, or S. pneumoniae-infected mice using an RNeasy Micro Kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions. One hundred nanograms of total cellular RNA was used for cDNA synthesis. Quantitative real-time RT-PCR was performed on an ABI 7300 Real-Time PCR System (PE Applied Biosystems, Warrington, United Kingdom) using SYBR green (Eurogentec, Seraing, Belgium) for semiquantitative assessment of gene expression profiles, as described recently (44, 45). PCR primers for quantification of β-actin (NM_007393.3), Toll-like receptor 2 (TLR2) (NM_011905.3), TLR4 (NM_021297), macrophage inflammatory protein 2 (MIP-2) (NM_009140), tumor necrosis factor alpha (TNF-α) (M13049), Fas ligand (FasL; NM_001205243.1), and iNOS (NM_010927.3) were designed with Primer Express software (PE Applied Biosystems), based on gene sequence data retrieved from GenBank, and utilized at a concentration of 0.4 μM. For normalization, β-actin was used as a housekeeping gene, and mean fold changes were calculated using the 2−ΔΔCT (where CT is threshold cycle) method (31). All samples were tested in duplicate. One no-template control was included per amplification run.
Western blot analysis.
Flow-sorted lung CD11bpos DCs were subjected to Western blot analysis of iNOS and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) protein expression. Briefly, CD11bpos DCs from lung tissue of vehicle- or Flt3L-pretreated mice infected with S. pneumoniae were transferred to ice-cold lysis buffer containing protease inhibitors (100 μl per 1 × 106 to 2 × 106 cells). Cell lysates were incubated on ice for 30 min, followed by centrifugation (25 min at 11,000 × g) and collection of the supernatants. Protein concentrations were determined using a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, Waltham, MA), according to the manufacturer's instructions. Equal protein amounts of cell lysates (20 μg) were loaded onto a 12.5% SDS polyacrylamide gel (Criterion; Bio-Rad, Hercules, CA) for electrophoresis. Using semidry-blotting technology, separated proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA), and nonspecific antibody binding was blocked by incubation of membranes in 5% milk powder for 1 h. After being washed, membranes were incubated with appropriately diluted rabbit anti-mouse iNOS antibody (Cell Signaling Technologies, Danvers, MA) or rabbit anti-mouse GAPDH antibody (Cell Signaling Technologies), following incubation with peroxidase-conjugated donkey anti-rabbit polyclonal IgG(H+L) (Jackson ImmunoResearch Laboratories, Suffolk, United Kingdom). Expression of immunogenic proteins was determined by adding Lumigen detection reagent (GE Healthcare, Buckinghamshire, United Kingdom) and enhanced chemiluminescence (ECL) signals were evaluated using a Vilber/Lourmat Chemi-Smart 5000 analyzer (Vilber/Lourmat, Eberhardzell, Germany) (45).
Quantification of nitric oxide in lung homogenate.
Nitric oxide (NO) release in lung tissue of Flt3L-pretreated and S. pneumoniae-infected mice and control mice was assessed by using a nitric oxide quantification kit (Active Motif, Rixensart, Belgium), following the instructions of the manufacturer. Briefly, lungs were perfused as described above and homogenized in 1 ml of PBS. Supernatants of lung homogenates were filtered through a 10-kDa micropore filter (Amicon Ultra; Millipore) to remove high-molecular-weight proteins. Subsequently, samples were placed in a 96-well microtiter plate, and nitrate reductase and cofactors were added to convert nitrate to nitrite. Nitrite concentrations were assayed using Griess reagent. Photometric measurements were performed using a spectrophotometer (Versa; Molecular Devices, Sunnyvale, CA) operating at 540-nm absorbance and 620-nm reference wavelengths.
Statistics.
The data are shown as means ± standard errors of the means (SEM). Comparisons between two or more groups were performed using a Mann-Whitney U Test or Kruskal-Wallis H test, respectively. Significant differences between the groups were analyzed using SPSS Statistics (IBM, Armonk, NY). Significant differences between groups were assumed with P values of <0.05.
RESULTS
Effect of Flt3L on the accumulation of CD11bpos and CD103pos DC subsets in the lungs of uninfected mice.
In initial experiments, we characterized CD11bpos DC and CD103pos DC subset profiles in the lungs of WT mice, Flt3L KO mice, and WT mice pretreated with Flt3L for 3, 5, and 7 consecutive days. As shown in Fig. 1A, flow cytometric immuno-phenotypic analysis of CD11c-positive cells isolated from lung parenchymal tissue of WT mice revealed two distinct subsets of low-green autofluorescent lung DC populations (population 4 [P4] and P5), defined by their differential CD11b and CD103 cell surface expression levels. Cells of P4 were found to be CD11cpos CD11bhigh MHCIIpos CD103neg, thus identifying these cells as CD11bpos DCs. Cells in P5 exhibited a CD11cpos MHCIIpos CD103high CD11blow antigen expression profile, thus representing CD103pos DCs, consistent with recent reports (18, 46). High-green autofluorescent cells shown in the dot plots in Fig. 1A to C represent lung macrophages (53). The CD11bpos DC subset was also detected in lung tissue of Flt3L KO mice and WT mice pretreated with Flt3L (Fig. 1B and C, P4). In contrast, CD103pos DCs were nearly undetectable in the lungs of Flt3L KO mice, consistent with recent reports (15), but were easily detectable in the lungs of Flt3L-treated WT mice (Fig. 1C, P5). Quantification of lung CD11bpos DCs and CD103pos DCs in the lungs of mice of the various experimental groups showed that under baseline conditions, Flt3L KO mice exhibited an ∼3-fold reduction in the numbers of lung CD11bpos DCs compared with levels in WT mice (Fig. 1D), whereas WT mice treated with Flt3L had significantly increased numbers of lung CD11bpos DCs as early as 3 days posttreatment, with a further increase by days 5 and 7 of Flt3L pretreatment, compared to untreated control mice (Fig. 1E, d 0). Flt3L KO mice displayed a marked reduction in CD103pos DCs relative to WT mice, whereas treatment of WT mice with Flt3L for 7 days did not increase numbers of lung CD103pos DCs (Fig. 1F and G).
Fig 1.

Determination of CD11bpos and CD103pos DC subsets in the lungs of uninfected Flt3L KO mice and Flt3L-treated WT mice. Lungs of WT mice, Flt3L KO mice, and Flt3L-treated WT mice (10 μg of Flt3L s.c./mouse) were subjected to BAL followed by MACS purification of CD11c-positive cells from lung tissue digests. (A to C) Representative flow cytometry dot plots showing the gating strategy for identification of lung CD11bpos and CD103pos DCs in WT mice (A), Flt3L KO mice (B), and WT mice treated with Flt3L for 5 consecutive days (C). Cells were gated according to their FSC versus SSC characteristics (P1) followed by hierarchical subgating according to their FSC versus low-green autofluorescence properties (P2), followed by subgating of the cells according to their CD11b versus CD11c (P3) and MHCII versus CD103 characteristics (P4, CD11bpos DCs; P5, CD103pos DCs). (D to G) Fluorescence-activated cell sorting-based quantification of total numbers of lung CD11bpos DCs (D and E) and lung CD103pos DCs (F and G) in mice of the designated experimental groups, as indicated. Values are shown as means ± SEM (n = 3 to 9 mice per time point). *, P < 0.05; **, P < 0.01 (compared with untreated WT mice). FITC, fluorescein isothiocyanate.
Effect of pneumococcal infection on lung DC subset accumulation in Flt3L KO mice and WT mice pretreated with Flt3L.
Based on these initial findings, we next investigated the effect of intratracheal infection with S. pneumoniae in mice in which numbers of lung DCs were either decreased (Flt3L KO mice), normal (WT mice), or increased (WT mice pretreated with Flt3L for 3, 5, or 7 days). Our results show that the numbers of lung CD11bpos DCs increased postinfection in both WT mice and Flt3L KO mice (Fig. 2A); note that numbers of CD11bpos DCs were about 3-fold lower in Flt3L KO mice than in WT mice. In contrast to CD11bpos DCs, the subset of CD103pos DCs did not expand postinfection in WT mice, and only a minor increase was observed in the lungs of infected Flt3L KO mice (Fig. 2B). In WT mice with increased numbers of lung DCs due to pretreatment with Flt3L for 3, 5, or 7 days, we observed a substantially additional accumulation of lung CD11bpos DCs and, albeit much less pronounced, of lung CD103pos DCs after infection with S. pneumoniae (relative to infected vehicle-treated mice) (Fig. 2C to H).
Fig 2.
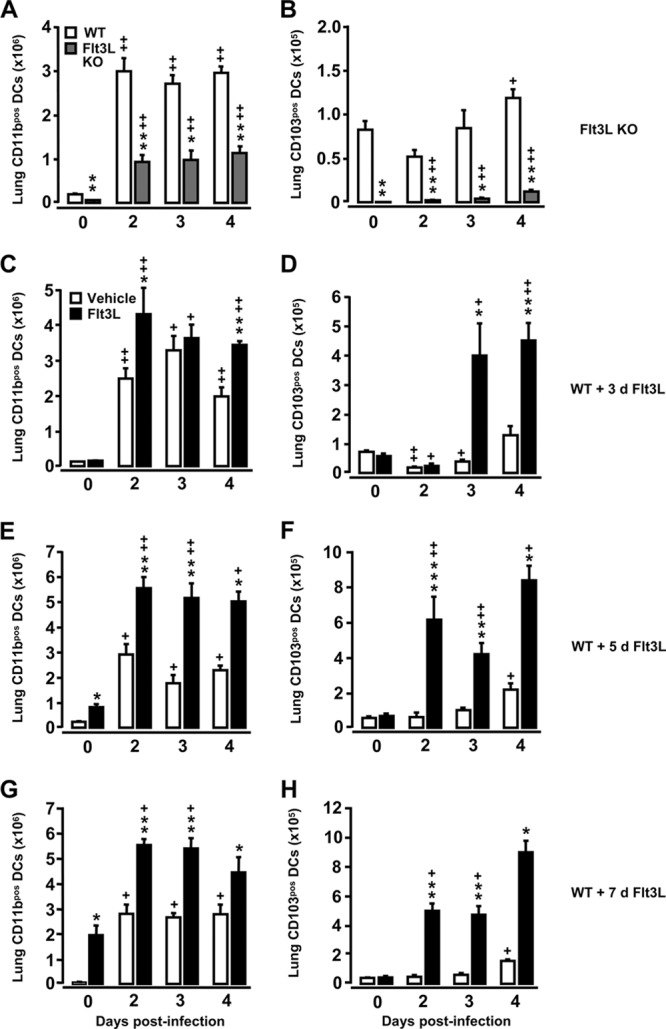
Recruitment kinetics of lung CD11bpos DCs and lung CD103pos DCs in Flt3L KO mice and Flt3L-pretreated WT mice challenged with S. pneumoniae. WT mice (white bars), Flt3L KO mice (A and B, gray bars), and WT mice pretreated with Flt3L for 3 days (C and D), 5 days (E and F), or 7 days (G and H, black bars) were left uninfected (day 0) or were infected intratracheally with S. pneumoniae. At the indicated time points, mice were euthanized, and CD11c-positive cells collected from lung parenchymal tissue digests were subjected to immuno-phenotypic characterization of DC subset profiles. Data are presented as means ± SEM (n = 3 to 11 mice per time point and treatment group). Significance compared to WT mice is shown as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Significance compared to uninfected mice is shown as follows: +, P < 0.05; ++, P < 0.01. d, day.
Effect of Flt3L deficiency or pretreatment of mice with Flt3L on lung protective immunity against S. pneumoniae.
We next examined what effect the observed differences in lung DC subset recruitment profiles observed in Flt3L KO mice versus Flt3L-pretreated WT mice would have on lung protective immunity against pneumococcal challenge. As shown in Fig. 3A, Flt3L KO mice demonstrated slightly but significantly increased bacterial loads in their lungs relative to control mice which were not due to reduced alveolar neutrophil recruitment in the Flt3L KO mice (Fig. 3B). Notably, clearance of S. pneumoniae from the lungs of WT mice pretreated with Flt3L for 5 or 7 days (but not 3 days) was significantly impaired (Fig. 3C, E, and G), even though the numbers of recruited alveolar neutrophils were equivalent to those of similarly infected WT mice that received vehicle pretreatment only (Fig. 3D, F, and H). Overall, in all treatment groups we observed mortality rates of <10% with no significant differences noted between groups, and within the observation period of 4 days, more than 90% of the mice were not able to completely clear pneumococci from their lungs (data not shown).
Fig 3.
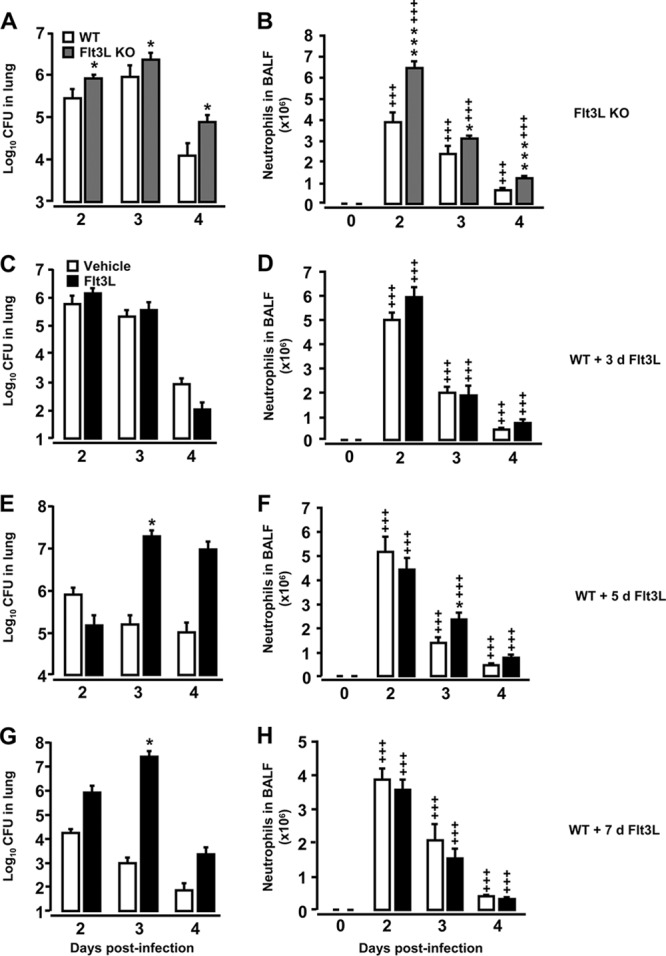
Bacterial loads in lung tissue of Flt3L KO mice and Flt3L-pretreated WT mice challenged with S. pneumoniae. Mice of the designated experimental group were infected with S. pneumoniae and then subjected to analysis of bacterial loads in their lungs. (A and B) Bacterial loads and respective BAL fluid neutrophil counts in untreated (0 h) or S. pneumoniae-infected WT mice and Flt3L KO mice, as indicated. (C to H) Bacterial loads and respective BAL fluid neutrophil counts in vehicle-treated, S. pneumoniae-infected WT mice or WT mice pretreated with Flt3L (10 μg/mouse; black bars) for 3 days (C and D), 5 days (E and F), or 7 days (G and H), as indicated. Note that the lung CFU data are pooled from CFU data determined in lung tissue homogenates and respective BAL fluid supernatants from the same mice. The data are presented as means ± SEM (n = 4 to 10 mice per time point and treatment group). *, P < 0.05; ***, P < 0.001 (both, compared with S. pneumoniae-infected control mice); +++, P < 0.001 relative to the respective uninfected mice (day zero time points).
Infection-induced lung leakage is increased in WT mice pretreated with Flt3L but not in Flt3L KO mice.
Next, we examined whether alterations in DC numbers prior to infection altered lung leakage, a marker of lung injury, following pulmonary challenge with S. pneumoniae. Results demonstrate that despite diminished numbers of lung DCs at baseline and postinfection, the degree of lung leakage in Flt3L KO mice was similar to that of WT mice both under baseline conditions and in response to pneumococcal infection (Fig. 4A). In contrast, in WT mice with enhanced numbers of lung DCs due to pretreatment with Flt3L for either 3, 5, or 7 days, infection with S. pneumoniae resulted in increased lung leakage relative to similarly infected, vehicle-treated WT mice (Fig. 4B to D). These data illustrate that even slightly increased lung DC counts (WT mice treated with Flt3L for just 3 days) were sufficient to cause lung barrier dysfunction in mice after bacterial challenge, whereas reduced lung DC counts, as observed in Flt3L KO mice, did not result in lung leakage after pneumococcal challenge.
Fig 4.

Lung permeability of Flt3L KO mice and Flt3L-pretreated WT mice infected with S. pneumoniae. WT mice (white bars), Flt3L KO mice (A, gray bars), or WT mice pretreated with Flt3L for 3 days (B), 5 days (C), or 7 days (D) with Flt3L (black bars) were either left uninfected (d 0) or were infected with S. pneumoniae for various time intervals, as indicated. One hour before sacrifice, mice received intravenous injection of FITC-labeled human albumin (1 mg/mouse), fluorescence measurement of lung permeability changes was performed as outlined in Materials and Methods. Lung permeability values are given in arbitrary units (AU). Data are shown as means ± SEM (n = 3 to 10 mice per time point and experimental group). Significant increases compared to vehicle-treated, S. pneumoniae-infected WT mice are shown as follows: *, P < 0.05; ** P < 0.01; *** P < 0.001. Significance with uninfected mice at day zero is shown as follows: +, P < 0.05; ++, P < 0.01.
Infection of Flt3L-pretreated mice with S. pneumoniae results in drastically increased iNOS gene expression in lung CD11bpos DCs.
To assess a possible role for lung CD11bpos DC versus CD103pos DC subsets as effector cells in acute lung injury developing in S. pneumoniae-infected Flt3L-pretreated mice, CD11bpos DCs and CD103pos DCs were flow sorted and then subjected to real-time RT-PCR analysis of selected genes with relevance to the pathobiology of pneumococcal lung infection. As shown in Fig. 5A, pneumococcal challenge of Flt3L-pretreated mice resulted in enhanced expression of TLR4 mRNA (∼3-fold) and, albeit less pronounced, of TLR2 mRNA (particularly in CD11bpos DCs but not CD103pos DCs) relative to Flt3L-pretreated, mock-infected mice. Gene expression of MIP-2, a chemokine known to play a role in neutrophil recruitment, was higher in CD11bpos DCs at day 1 of infection than in CD103pos DCs, whereas TNF-α mRNA levels were similar in the two lung DC subsets (Fig. 5B). Examination of the expression of the apoptosis-inducing TNF superfamily member FasL gene in CD11bpos DCs versus CD103pos DCs revealed that FasL mRNA was barely detectable in both DC subsets (Fig. 5C).
Fig 5.

Expression profiles in lung DC subsets and lung NO levels in Flt3L-pretreated mice infected with S. pneumoniae. (A to D) WT mice were pretreated with Flt3L for 5 consecutive days followed by mock infection (50 μl of PBS intratracheally) or S. pneumoniae. At days 1 and 3 postinfection, lung CD11bpos and CD103pos DCs were purified by high-speed cell sorting and subjected to real-time RT-PCR analysis as outlined in Materials and Methods. (A to D) Fold change in mRNA levels of TLR2 and TLR4 (A), MIP-2 and TNF-α (B), FasL (C), and iNOS (D) in lung CD11bpos DCs versus lung CD103pos DCs of S. pneumoniae-infected WT mice pretreated with Flt3L for 5 days relative to values of mock-infected WT mice pretreated with Flt3L for 5 days. (E) Representative Western blot analysis of iNOS protein expression in flow-sorted CD11bpos DCs purified from lungs of vehicle- or Flt3L-pretreated (5 days) WT mice infected with S. pneumoniae for 3 days. (F) WT mice were pretreated with vehicle or Flt3L for 5 days prior to mock infection (50 μl of PBS) or infection with S. pneumoniae for 3 days, followed by determination of nitrite/nitrate levels using Griess reagent. In panels A to D and F, data are presented as means ± SEM (n = 4 to 6 mice per time point and experimental group). *, P < 0.05 compared to CD103pos DCs (A to D). (E) Equal amounts of protein (20 μg) were loaded onto the gel, and GAPDH protein expression was analyzed as a control for similar protein loading of the gel. Bone marrow-derived DCs stimulated with LPS for 6 h were used as positive controls for iNOS (52). For one Western blot analysis, protein collected from sorted CD11bpos DCs of n = 8 mice was pooled. (F) Significant increases compared to vehicle-pretreated, S. pneumoniae (S. pn.)-infected mice (**, P < 0.01) and relative to Flt3L-pretreated, mock-infected mice (++, P < 0.01) are indicated.
Our analysis of iNOS gene expression demonstrated that iNOS mRNA levels were markedly increased in lung CD11bpos DCs but not CD103pos DCs of Flt3L-pretreated, S. pneumoniae-infected mice (compared to mock-infected mice), with ∼13,000-fold and ∼3,500-fold increased iNOS mRNA levels in CD11bpos DCs observed on days 1 and 3 after pneumococcal challenge (Fig. 5D). To confirm our mRNA data, we demonstrated that CD11bpos DCs of Flt3L-treated, S. pneumoniae-infected mice expressed increased iNOS protein levels (Fig. 5E) and that mice had evidence of increased NO release in their lungs, which was not observed in uninfected mice treated only with Flt3L or in S. pneumoniae-infected WT mice (Fig. 5F). Collectively, these data show that Flt3L-elicited CD11bpos DCs, but not CD103pos DCs, display a proinflammatory phenotype following pulmonary infection with S. pneumoniae and suggest that CD11bpos DCs may be important contributors to the increased lung injury we observed in Flt3L-pretreated mice infected with S. pneumoniae.
iNOS deficiency does not protect Flt3L-treated mice from S. pneumoniae-induced acute lung injury.
Various studies in mice and humans have suggested that iNOS-dependent NO release by lung inflammatory cells not only promotes bacterial clearance but may also contribute to acute lung injury (9, 27, 50, 54). Based on our observation that Flt3L pretreatment of mice induced increased iNOS protein expression in lung CD11bpos DCs and NO production in lung tissue after pneumococcal challenge, we next questioned whether iNOS deficiency would ameliorate acute lung injury in Flt3L-pretreated mice infected with S. pneumoniae. As shown in Fig. 6A, iNOS deficiency resulted in only a slight, but nonsignificantly reduced, lung leakage in Flt3L-pretreated, S. pneumoniae-infected mice. At the same time, numbers of BAL fluid neutrophils were not different between groups (data not shown), thereby arguing against a major impact of neutrophil-derived NO on the observed lung leakage in these mice. Similarly, iNOS deficiency also did not affect their overall lung bacterial clearance capacity (Fig. 6B), and, again, administration of the selective iNOS inhibitor, 1400W, to infected Flt3L-pretreated mice did not improve lung permeability (relative to similarly infected and Flt3L-pretreated WT mice), despite the normalization of NO levels (Fig. 6D and 6C to E). Collectively, these results largely exclude a major role for NO produced by lung CD11bpos DCs in the lung leakage we observed in Flt3L-treated mice challenged with S. pneumoniae.
Fig 6.
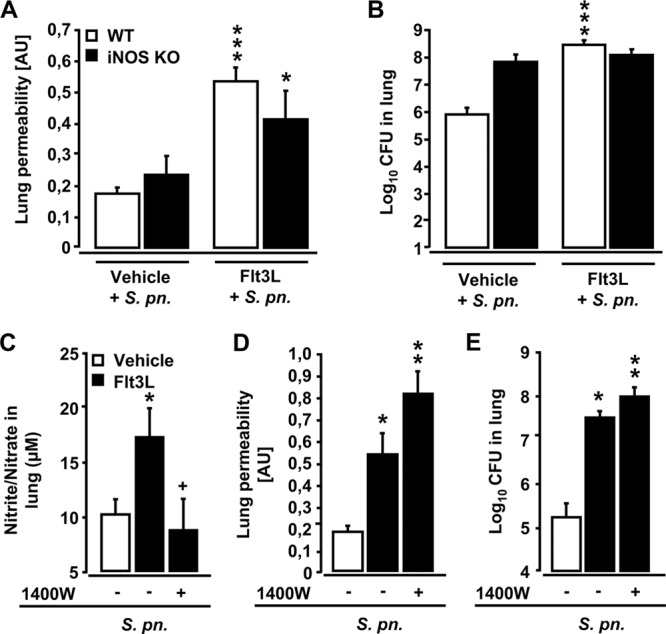
Effect of deletion or inhibition of iNOS on lung permeability and bacterial loads in Flt3L-pretreated and S. pneumoniae-infected mice. (A and B) Wild-type mice and iNOS KO mice were pretreated with Flt3L for 5 days and were infected with S. pneumoniae for 3 days. Mice were treated with FITC-labeled human albumin (1 mg/mouse, intravenously). After 1 h, lung permeability changes were determined (A), and values are shown in arbitrary units (AU). Bacterial loads in the lungs of mice of the respective experimental groups are shown in panel B. The CFU data are pooled from CFU data determined in lung tissue homogenates and respective BAL fluid supernatants from the same mice. (C to E) WT mice pretreated with vehicle or Flt3L for 5 days were infected with S. pneumoniae for 3 days in the absence (−) or presence (+) of the specific iNOS inhibitor 1400W (applied at 5 mg/kg, i.p.), as indicated. Panel C shows nitrite and nitrate levels in lung tissue homogenates of perfused lungs of mice of the indicated experimental groups. One hour before sacrifice, mice were injected intravenously with FITC-labeled human albumin (1 mg/mouse) for fluorometric analysis of lung permeability (D), and values are depicted in arbitrary units (AU). Bacterial loads in the lungs of mice were determined as indicated (E). The lung CFU data are pooled from CFU data determined in lung tissue homogenates and respective BAL fluid supernatants from the same mice. Values are depicted as means ± SEM (n = 4 to 14 mice per experimental group and time point). Asterisks indicate a significant increase (*, P < 0.05; ***, P < 0.001) relative to vehicle-treated, S. pneumoniae-infected mice in panels A and B. In panels C to E, significant differences compared to S. pneumoniae-infected vehicle-treated mice are indicated as follows: *, P < 0.05; **, P < 0.01. A significant difference relative to Flt3L-pretreated S. pneumoniae-infected mice receiving vehicle only is indicated in panel C (+, P < 0.05).
Impact of pneumococcal PLY on acute lung injury in Flt3L-pretreated mice infected with S. pneumoniae.
Lung CD11bpos DCs of Flt3L-pretreated mice displayed increased TLR4 gene expression following pneumococcal challenge, and TLR4 is a well-known receptor for the major pneumococcal virulence factor PLY (51). Therefore, we next examined the role of PLY on DC activation and the development of lung injury in Flt3L-pretreated mice by utilizing an isogenic PLY-deficient serotype 19 S. pneumoniae strain (EF3030ΔPLY). Real-time RT-PCR analysis revealed that flow-sorted lung CD11bpos DCs from Flt3L-treated mice infected with PLY-deficient S. pneumoniae demonstrated significantly reduced expression of TLR4 and iNOS mRNA relative to lung CD11bpos DCs of Flt3L-pretreated mice infected with WT S. pneumoniae (Fig. 7A). This decreased activation of lung CD11bpos DCs was accompanied by substantially reduced lung permeability and improved lung bacterial clearance in these mice relative to Flt3L-pretreated mice infected with PLY-producing S. pneumoniae (Fig. 7B and C). Of note, PLY expression by S. pneumoniae had no effect on total DC accumulation in the lungs of Flt3L-pretreated mice (data not shown). Collectively, these data demonstrate that pathogen-derived virulence factors such as PLY rather than host-derived factors such as iNOS had a major impact on the outcome of pneumococcal infection in Flt3L-pretreated mice.
Fig 7.

Gene expression profiles of lung CD11bpos DCs and lung permeability and bacterial loads in Flt3L-pretreated mice infected with PLY-deficient S. pneumoniae. (A) WT mice were pretreated with Flt3L for 5 days followed by mock infection (50 μl of PBS o.t.) or infection with PLY-competent or PLY-deficient S. pneumoniae. At days 1 and 3 postinfection, lung CD11bpos DCs were purified by high-speed cell sorting and analyzed by real-time RT-PCR as outlined in Materials and Methods. Fold changes of TLR4 and iNOS mRNA levels were calculated in lung CD11bpos DCs from Flt3L-pretreated mice infected with S. pneumoniae (S. pn. WT) and lung CD11bpos DCs from Flt3L-pretreated mice infected with PLY-deficient S. pneumoniae (S. pn. ΔPLY), relative to Flt3L-treated, mock-infected mice. Lung permeability changes (B) and lung bacterial loads (C) were determined in WT mice pretreated with vehicle or Flt3L for 5 days followed by infection with S. pneumoniae (white bars) or PLY-deficient S. pneumoniae (black bars) for 3 days. Lung permeability values are shown in arbitrary units (AU). CFU data are pooled from CFU data determined in lung tissue homogenates and respective BAL fluids from the same mice. Data are presented as means ± SEM (n = 3 to 10 mice per group and time point). In panel A, significant decrease (*, P < 0.05) relative to CD11bpos DCs from Flt3L-treated mice infected with S. pneumoniae at the respective time point is indicated. Significant differences relative to Flt3L-treated mice infected with S. pneumoniae (*, P < 0.05; **, P < 0.01) and significant increases relative to vehicle-treated mice infected with S. pneumoniae (++, P < 0.01) are indicated in panels B and C.
DISCUSSION
In the current study, we examined the impact of deficient (Flt3L KO), normal (WT), or increased Flt3L availability (WT mice pretreated with Flt3L) on lung DC subset profiles and lung-protective immunity against the major lung-tropic pathogen, S. pneumoniae. Flt3L-deficient mice exhibited strongly reduced numbers of lung CD11bpos DCs and an even greater reduction in CD103pos DCs but did not respond with increased lung permeability to infection with S. pneumoniae. In contrast, short-term treatment of WT mice with Flt3L for just 3 days was sufficient to perturb lung innate immunity against S. pneumoniae. Importantly, particularly lung CD11bpos DCs but not CD103pos DCs from Flt3L-treated, S. pneumoniae-infected mice responded to infection with proinflammatory gene activation, including iNOS. At the same time, elevated levels of NO were found in lung tissue of Flt3L-treated mice infected with S. pneumoniae, suggesting that lung CD11bpos DCs are an important source of NO in this model. However, neither deletion nor pharmacologic inhibition of NO ameliorated lung barrier dysfunction in Flt3L-treated mice after pneumococcal challenge. Rather, the pneumococcal virulence factor PLY was identified to contribute to increased lung leakage observed in Flt3L-pretreated, infected mice. Collectively, we believe that within the reported complex host-pathogen interaction, Flt3L treatment resulting in increased lung DC accumulation makes the host more vulnerable to pathogen-derived virulence factors such as PLY, which itself was observed in the current study to exert a major effect on acute lung injury in Flt3L-pretreated mice challenged with S. pneumoniae. These data are of clinical relevance in view of safety considerations for Flt3L-based immunization strategies or immunomodulatory therapies in patients undergoing myeloablative chemotherapies.
The developmental pathways of lung DC subsets from bone marrow progenitors are still incompletely defined. Under the control of Flt3L, DCs are presumed to differentiate from a macrophage and DC precursor (MDP) in the bone marrow, which gives rise to a common DC precursor (CDP) and to circulating blood monocytes (10, 30). The CDP further gives rise to pre-DCs, which migrate from the bone marrow through the blood into nonlymphoid tissue to generate CD103pos DCs, at least in the steady state, and this process appears to be dependent on Flt3L. Nonlymphoid tissue CD11bpos DCs are more heterogeneous (15). In the current study, we found that in Flt3L KO mice, lung CD103pos DCs were nearly absent both under baseline conditions and following infection with S. pneumoniae. These findings support an essential role for Flt3L in maintaining lung CD103pos DC pool sizes under baseline and inflammatory conditions. In the same Flt3L KO mice, numbers of CD11bpos DCs were reduced by approximately 50% in the lungs at baseline and following infection, thus confirming the dependence of this DC subset on both Flt3L and other developmental cytokines (15, 25).
Our experimental approach provided us the opportunity to study the numbers of CD11bpos DCs and CD103pos DCs in mice in which DC numbers were normal (WT mice), decreased (Flt3L KO mice), or increased in stepwise fashion by the administration of Flt3L for 3, 5, or 7 days. Our results show that repetitive treatment of WT mice with Flt3L led to a stepwise accumulation of the two myeloid lung DC subsets under baseline conditions, which was enhanced further after pneumococcal challenge. We observed that the diminished numbers of both CD11bpos DCs and CD103pos DCs in Flt3L KO mice were associated with diminished bacterial clearance following pneumococcal infection compared to WT mice with normal lung DC distribution profiles. These findings demonstrate that lung DCs are indispensable for innate immune responses to S. pneumoniae. Importantly, despite increased lung bacterial loads, Flt3L KO mice did not develop increased lung leakage relative to S. pneumoniae-infected WT mice, suggesting that a decrease in lung DC pool sizes favors lung resistance against S. pneumoniae-induced lung leakage.
Conversely, expansion of the lung DC pool following pretreatment of WT mice with Flt3L for just 3 or 5 days was sufficient to increase lung permeability in mice in response to S. pneumoniae. Numbers of alveolar recruited neutrophils were similar in Flt3L-pretreated and untreated, S. pneumoniae-infected WT mice, ruling out a major impact of corecruited neutrophils on the observed changes in lung leakage and injury. These findings refine our previous observations (53) by showing that even short-term treatment of mice with Flt3L for just 3 days is sufficient to perturb lung protective immunity against inhaled bacterial pathogens. These findings have important safety implications for therapeutic interventions involving the use of Flt3L as an immunomodulatory agent in the treatment of human diseases.
By employing high-speed cell sorting in conjunction with real-time PCR approaches, we are the first to report major differences in inflammatory activation profiles between CD11bpos DCs and CD103pos DCs isolated from the lungs of Flt3L-pretreated, S. pneumoniae-infected mice. We found that lung CD11bpos DCs, but not CD103pos DCs, exhibited a robust proinflammatory activation profile in response to pneumococcal challenge, including increased mRNA levels for the important pattern recognition receptors TLR2 and TLR4 as well as the neutrophil chemoattractant MIP-2. We further identified a major induction of iNOS mRNA and protein in lung CD11bpos DCs, but not CD103pos DCs, accompanied by substantially increased NO levels in lung tissue of Flt3L-pretreated mice infected with S. pneumoniae. Notably, increased iNOS activity and NO are known to be important inflammatory mediators in the pathogenesis of acute lung injury (9, 27, 50, 54), and iNOS-producing DCs have been shown to actively contribute to tissue damage and immune pathology during infection with various types of pathogens, such as parasites or influenza virus (16, 29). Our data suggest that the overall increased lung NO levels in Flt3L-treated, S. pneumoniae-infected mice are the result of increased accumulation and inflammatory activation of iNOS-producing CD11bpos DCs in lung tissue of these mice. However, neither iNOS deletion nor pharmacologic blockade in infected Flt3L-pretreated mice had a detrimental effect on bacterial clearance, nor did it reduce the amount of lung leakage observed in these mice, thus ruling out a major impact of Flt3L-induced NO to mediate lung barrier dysfunction in mice.
Despite similarly increased lung DC numbers, Flt3L-treated mice infected with PLY-deficient pneumococci exhibited significantly reduced lung permeability and bacterial loads in their lungs compared to Flt3L-treated mice infected with PLY-competent S. pneumoniae. These results provide compelling evidence that in DC-enriched mice, pathogen-associated molecular patterns such as PLY released by S. pneumoniae are central to the induction of lung permeability changes. At higher lytic concentrations, PLY directly causes tissue damage by means of its transmembrane pore-forming properties (14, 26, 38, 42). In this regard, a recent study identified protein kinase C-α and arginase 1 as important mediators to contribute to PLY-induced pulmonary endothelial dysfunction (32). Thus, a direct cytolytic effect of PLY represents one possible mechanism by which S. pneumoniae may have caused lung leakage in Flt3L-treated mice. However, besides its cytolytic activity, PLY is also known to act as a pathogen-associated molecular pattern (5, 35). Since TLR4 is a receptor for PLY and since PLY-deficient S. pneumoniae failed to induce TLR4 gene expression in lung CD11bpos DCs of Flt3L-treated mice, which at the same time lacked increased lung permeability, the PLY-dependent lung leakage observed in the current model may, at least in part, be mediated by engagement of TLR4 expressed by CD11bpos DCs. Future studies aiming to specifically ablate CD11bpos DCs while leaving other important lung CD11b-expressing leukocyte subsets unaffected are needed to further unravel the role of lung CD11bpos DCs in lung barrier dysfunction in Flt3L-treated mice.
Collectively, this study provides a comprehensive analysis of the effect of Flt3L deficiency or Flt3L pretreatment on the accumulation and inflammatory activation of lung CD11bpos and CD103pos DC subsets under baseline conditions and after infection with PLY-competent and -deficient S. pneumoniae. The data provided may be important for safety considerations involving hematopoietic growth factor applications as immunomodulatory strategies in humans.
ACKNOWLEDGMENTS
Flt3L was generously provided by Celldex Therapeutics (Needham, MA).
Ulrich A. Maus is a member of the German Center for Lung Research (DZL).
Footnotes
Published ahead of print 24 September 2012
REFERENCES
- 1. Alaniz RC, Sandall S, Thomas EK, Wilson CB. 2004. Increased dendritic cell numbers impair protective immunity to intracellular bacteria despite augmenting antigen-specific CD8+ T lymphocyte responses. J. Immunol. 172:3725–3735 [DOI] [PubMed] [Google Scholar]
- 2. Barnard Z, et al. 2012. Expression of FMS-like tyrosine kinase 3 ligand by oncolytic herpes simplex virus type I prolongs survival in mice bearing established syngeneic intracranial malignant glioma. Neurosurgery 71:741–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beaty SR, Rose CE, Jr, Sung SS. 2007. Diverse and potent chemokine production by lung CD11bhigh dendritic cells in homeostasis and in allergic lung inflammation. J. Immunol. 178:1882–1895 [DOI] [PubMed] [Google Scholar]
- 4. Bohannon J, et al. 2008. Prophylactic treatment with FMS-like tyrosine kinase-3 ligand after burn injury enhances global immune responses to infection. J. Immunol. 180:3038–3048 [DOI] [PubMed] [Google Scholar]
- 5. Braun JS, Novak R, Gao G, Murray PJ, Shenep JL. 1999. Pneumolysin, a protein toxin of Streptococcus pneumoniae, induces nitric oxide production from macrophages. Infect. Immun. 67:3750–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Briles DE, et al. 2003. Immunizations with pneumococcal surface protein A and pneumolysin are protective against pneumonia in a murine model of pulmonary infection with Streptococcus pneumoniae. J. Infect. Dis. 188:339–348 [DOI] [PubMed] [Google Scholar]
- 7. Buza-Vidas N, et al. 2007. Crucial role of FLT3 ligand in immune reconstitution after bone marrow transplantation and high-dose chemotherapy. Blood 110:424–432 [DOI] [PubMed] [Google Scholar]
- 8. Desch AN, et al. 2011. CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J. Exp. Med. 208:1789–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Farley KS, et al. 2006. Effects of macrophage inducible nitric oxide synthase in murine septic lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 290:L1164–1172 [DOI] [PubMed] [Google Scholar]
- 10. Fogg DK, et al. 2006. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 311:83–87 [DOI] [PubMed] [Google Scholar]
- 11. Fong L, et al. 2001. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc. Natl. Acad. Sci. U. S. A. 98:8809–8814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garvey EP, et al. 1997. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 272:4959–4963 [DOI] [PubMed] [Google Scholar]
- 13. Gasparetto C, et al. 2002. Mobilization of dendritic cells from patients with breast cancer into peripheral blood stem cell leukapheresis samples using Flt-3-Ligand and G-CSF or GM-CSF. Cytokine 18:8–19 [DOI] [PubMed] [Google Scholar]
- 14. Gilbert RJ, et al. 1999. Two structural transitions in membrane pore formation by pneumolysin, the pore-forming toxin of Streptococcus pneumoniae. Cell 97:647–655 [DOI] [PubMed] [Google Scholar]
- 15. Ginhoux F, et al. 2009. The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med. 206:3115–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guilliams M, et al. 2009. IL-10 dampens TNF/inducible nitric oxide synthase-producing dendritic cell-mediated pathogenicity during parasitic infection. J. Immunol. 182:1107–1118 [DOI] [PubMed] [Google Scholar]
- 17. Hahn I, et al. 2011. Cathepsin G and neutrophil elastase play critical and non-redundant roles in lung protective immunity against Streptococcus pneumoniae in mice. Infect. Immun. 79:4893–4901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hahn I, et al. 2011. Dendritic cell depletion and repopulation in the lung after irradiation and bone marrow transplantation in mice. Am. J. Respir. Cell Mol. Biol. 45:534–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harvey RM, Ogunniyi AD, Chen AY, Paton JC. 2011. Pneumolysin with low hemolytic activity confers an early growth advantage to Streptococcus pneumoniae in the blood. Infect. Immun. 79:4122–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Henken S, et al. 2010. Efficacy profiles of daptomycin for treatment of invasive and noninvasive pulmonary infections with Streptococcus pneumoniae. Antimicrob. Agents Chemother. 54:707–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Henken S, et al. 2010. Evaluation of biophotonic imaging to estimate bacterial burden in mice infected with highly virulent compared to less virulent Streptococcus pneumoniae serotypes. Antimicrob. Agents Chemother. 54:3155–3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ho AW, et al. 2011. Lung CD103+ dendritic cells efficiently transport influenza virus to the lymph node and load viral antigen onto MHC Class I for presentation to CD8 T Cells. J. Immunol. 187:6011–6021 [DOI] [PubMed] [Google Scholar]
- 23. Holt PG. 2005. Pulmonary dendritic cells in local immunity to inert and pathogenic antigens in the respiratory tract. Proc. Am. Thorac. Soc. 2:116–120 [DOI] [PubMed] [Google Scholar]
- 24. Kataoka K, et al. 2011. The nasal dendritic cell-targeting Flt3 ligand as a safe adjuvant elicits effective protection against fatal pneumococcal pneumonia. Infect. Immun. 79:2819–2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kingston D, et al. 2009. The concerted action of GM-CSF and Flt3-ligand on in vivo dendritic cell homeostasis. Blood 114:835–843 [DOI] [PubMed] [Google Scholar]
- 26. Kirkham LA, et al. 2006. Construction and immunological characterization of a novel nontoxic protective pneumolysin mutant for use in future pneumococcal vaccines. Infect. Immun. 74:586–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kristof AS, Goldberg P, Laubach V, Hussain SN. 1998. Role of inducible nitric oxide synthase in endotoxin-induced acute lung injury. Am. J. Respir. Crit. Care Med. 158:1883–1889 [DOI] [PubMed] [Google Scholar]
- 28. Laubach VE, Shesely EG, Smithies O, Sherman PA. 1995. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc. Natl. Acad. Sci. U. S. A. 92:10688–10692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. 2008. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J. Immunol. 180:2562–2572 [DOI] [PubMed] [Google Scholar]
- 30. Liu K, Nussenzweig MC. 2010. Origin and development of dendritic cells. Immunol. Rev. 234:45–54 [DOI] [PubMed] [Google Scholar]
- 31. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 32. Lucas R, et al. 10 May 2012. Protein kinase C-α and arginase I mediate pneumolysin-induced pulmonary endothelial hyperpermeability. Am. J. Respir. Cell Mol. Biol. doi:10.1165/rcmb.2011-0332OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lyman SD, et al. 1993. Molecular cloning of a ligand for the Flt3/Flk-2 tyrosine kinase receptor: a proliferative factor for primitive hematopoietic cells. Cell 75:1157–1167 [DOI] [PubMed] [Google Scholar]
- 34. Lynch DH, et al. 1997. Flt3 ligand induces tumor regression and antitumor immune responses in vivo. Nat. Med. 3:625–631 [DOI] [PubMed] [Google Scholar]
- 35. Malley R, et al. 2003. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl. Acad. Sci. U. S. A. 100:1966–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maraskovsky E, et al. 1996. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J. Exp. Med. 184:1953–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Masten BJ, Olson GK, Kusewitt DF, Lipscomb MF. 2004. Flt3 ligand preferentially increases the number of functionally active myeloid dendritic cells in the lungs of mice. J. Immunol. 172:4077–4083 [DOI] [PubMed] [Google Scholar]
- 38. Maus UA, et al. 2004. Pneumolysin-induced lung injury is independent of leukocyte trafficking into the alveolar space. J. Immunol. 173:1307–1312 [DOI] [PubMed] [Google Scholar]
- 39. McKenna HJ, et al. 2000. Mice lacking Flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood 95:3489–3497 [PubMed] [Google Scholar]
- 40. Morse MA, et al. 2000. Preoperative mobilization of circulating dendritic cells by Flt3 ligand administration to patients with metastatic colon cancer. J. Clin. Oncol. 18:3883–3893 [DOI] [PubMed] [Google Scholar]
- 41. Osterholzer JJ, et al. 2009. Accumulation of CD11b+ lung dendritic cells in response to fungal infection results from the CCR2-mediated recruitment and differentiation of Ly-6Chigh monocytes. J. Immunol. 183:8044–8053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rubins JB, et al. 1993. Toxicity of pneumolysin to pulmonary alveolar epithelial cells. Infect. Immun. 61:1352–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Singh P, et al. 2012. Blockade of prostaglandin E2 signaling through EP1 and EP3 receptors attenuates Flt3L-dependent dendritic cell development from hematopoietic progenitor cells. Blood 119:1671–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Srivastava M, et al. 2007. Mediator responses of alveolar macrophages and kinetics of mononuclear phagocyte subset recruitment during acute primary and secondary mycobacterial infections in the lungs of mice. Cell Microbiol. 9:738–752 [DOI] [PubMed] [Google Scholar]
- 45. Steinwede K, et al. 2011. Local delivery of GM-CSF protects mice from lethal pneumococcal pneumonia. J. Immunol. 187:5346–5356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sung SS, et al. 2006. A major lung CD103 (αE)-β7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J. Immunol. 176:2161–2172 [DOI] [PubMed] [Google Scholar]
- 47. Taut K, et al. 2008. Macrophage turnover kinetics in the lungs of mice infected with Streptococcus pneumoniae. Am. J. Respir. Cell Mol. Biol. 38:105–113 [DOI] [PubMed] [Google Scholar]
- 48. Triccas JA, et al. 2007. Effects of DNA- and Mycobacterium bovis BCG-based delivery of the Flt3 ligand on protective immunity to Mycobacterium tuberculosis. Infect. Immun. 75:5368–5375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. von Wulffen W, et al. 2007. Lung dendritic cells elicited by FMS-like tyrosine 3-kinase ligand amplify the lung inflammatory response to lipopolysaccharide. Am. J. Respir. Crit. Care Med. 176:892–901 [DOI] [PubMed] [Google Scholar]
- 50. Wang LF, et al. 2002. Role of inducible nitric oxide synthase in pulmonary microvascular protein leak in murine sepsis. Am. J. Respir. Crit. Care Med. 165:1634–1639 [DOI] [PubMed] [Google Scholar]
- 51. Weber M, et al. 2012. Hepatic induction of cholesterol biosynthesis reflects a remote adaptive response to pneumococcal pneumonia. FASEB J. 26:2424–2436 [DOI] [PubMed] [Google Scholar]
- 52. Wilkins-Rodriguez AA, Escalona-Montano AR, Aguirre-Garcia M, Becker I, Gutierrez-Kobeh L. 2010. Regulation of the expression of nitric oxide synthase by Leishmania mexicana amastigotes in murine dendritic cells. Exp. Parasitol. 126:426–434 [DOI] [PubMed] [Google Scholar]
- 53. Winter C, et al. 2007. FMS-like tyrosine kinase 3 ligand aggravates the lung inflammatory response to Streptococcus pneumoniae infection in mice: role of dendritic cells. J. Immunol. 179:3099–3108 [DOI] [PubMed] [Google Scholar]
- 54. Zhu S, Ware LB, Geiser T, Matthay MA, Matalon S. 2001. Increased levels of nitrate and surfactant protein a nitration in the pulmonary edema fluid of patients with acute lung injury. Am. J. Respir. Crit. Care Med. 163:166–172 [DOI] [PubMed] [Google Scholar]