

Abstract
The C-terminal cell wall binding domains (CBDs) of phage endolysins direct the enzymes to their binding ligands on the bacterial cell wall with high affinity and specificity. The Listeria monocytogenes Ply118, Ply511, and PlyP40 endolysins feature related CBDs which recognize the directly cross-linked peptidoglycan backbone structure of Listeria. However, decoration with fluorescently labeled CBDs primarily occurs at the poles and septal regions of the rod-shaped cells. To elucidate the potential role of secondary cell wall-associated carbohydrates such as the abundant wall teichoic acid (WTA) on this phenomenon, we investigated CBD binding using L. monocytogenes serovar 1/2 and 4 cells deficient in WTA. Mutants were obtained by deletion of two redundant tagO homologues, whose products catalyze synthesis of the WTA linkage unit. While inactivation of either tagO1 (EGDe lmo0959) or tagO2 (EGDe lmo2519) alone did not affect WTA content, removal of both alleles following conditional complementation yielded WTA-deficient Listeria cells. Substitution of tagO from an isopropyl-β-d-thiogalactopyranoside-inducible single-copy integration vector restored the original phenotype. Although WTA-deficient cells are viable, they featured severe growth inhibition and an unusual coccoid morphology. In contrast to CBDs from other Listeria phage endolysins which directly utilize WTA as binding ligand, the data presented here show that WTAs are not required for attachment of CBD118, CBD511, and CBDP40. Instead, lack of the cell wall polymers enables unrestricted spatial access of CBDs to the cell wall surface, indicating that the abundant WTA can negatively regulate sidewall localization of the cell wall binding domains.
INTRODUCTION
Bacteriophage endolysins are cell wall-hydrolyzing enzymes produced during the late phase of gene expression in the lytic cycle of virus multiplication, mediating the release of progeny phages (3, 32). They are usually composed of two functional domains, an enzymatically active domain (EAD) at the N terminus and a cell wall binding domain (CBD) at the C-terminal part of the protein. Although endolysins have attracted attention as prospective tools and antimicrobial agents (3, 17, 21, 32), surprisingly little is known about the identity and structure of their cell wall-associated CBD binding ligands.
The cell envelope of a typical Gram-positive organism is composed of peptidoglycan, proteins, and secondary cell wall polymers, including teichoic acids (TAs) and other glycopolymers. Peptidoglycan together with the attached polymer chains plays a crucial role in bacterial physiology and assumes a variety of other functions (28, 35, 54). TAs represent the most abundant polymer associated with the cell walls of Gram-positive bacteria and include cell wall-anchored wall teichoic acids (WTAs) and membrane-anchored lipoteichoic acids (LTAs), differing in synthesis and chemical structure (37). WTAs are phosphate-containing, anionic carbohydrate polymers covalently attached to the peptidoglycan via a conserved linkage unit (37, 52). Although their structures may be highly variable between species or even strains (54), the repeating units of the WTA polymers often feature glycerol phosphate (e.g., in Bacillus subtilis 168) or ribitol phosphate (e.g., in Staphylococcus aureus and Listeria monocytogenes) backbones (56) and are often modified and substituted with different sugars or amino acids, such as d-alanine (27, 37). LTAs usually show less structural diversity (54). In L. monocytogenes, LTA polymers consist of hydrophilic poly(glycerol phosphate) chains decorated with d-alanine and galactosyl residues (23, 39, 50).
The CBD of Listeria phage endolysin PlyP35 specifically recognizes N-acetylglucosamine (GlcNAc) residues attached to the ribitol phosphate in WTA polymers of L. monocytogenes serovar 1/2 and 3 strains (11). In contrast, the CBDs of Listeria phage endolysins Ply118, Ply511, and PlyP40 (34, 43) investigated here are assumed to recognize a more common, serovar-independent motif in the peptidoglycan structure (43). Green fluorescent protein (GFP)-tagged CBD118, CBD511, and CBDP40 molecules mainly attach to poles and septal regions of Listeria cells (33, 43), while the nature of the binding ligands remains unclear. The specific spatial distribution of binding ligands on the Listeria cell surface could also suggest that CBD targeting might be regulated and/or affected by the presence of other components in the cell envelope, such as the extremely abundant WTA polymers, which were reported to constitute up to 70% of the Listeria cell wall dry weight (13, 14).
Although WTA polymers are not essential for viability in B. subtilis and S. aureus, they contribute to host-cell binding, immune evasion, and virulence properties (8, 53, 54, 56). B. subtilis and S. aureus cells devoid of WTAs have been obtained by inactivation of tagO, whose product initiates the first step in WTA synthesis, transfer of GlcNAc phosphate to its lipid carrier, undecaprenyl phosphate (45, 53). The situation in L. monocytogenes had not been investigated, until now. Therefore, the aim of this study was to define the ligands in the bacterial cell envelope responsible for binding of CBD118, CBD511, and CBDP40 and to explain the uneven spatial distribution of CBD molecules on Listeria cell walls. Toward this goal, we identified the key genes involved in WTA synthesis, which allowed us to construct WTA-deficient mutants in order to assess CBD binding and the role of WTA in this process.
MATERIALS AND METHODS
Bacteria, plasmids, and growth conditions.
The strains and plasmids used in this study are listed in Table 1. Escherichia coli strains were cultured aerobically at 37°C in Luria-Bertani (LB) medium with shaking and used for cloning, amplification of plasmids, and recombinant protein synthesis. L. monocytogenes strains were grown in brain heart infusion (BHI) medium or tryptose broth (TB) at 30°C with shaking. The following antibiotics were added to broth as selective agents when appropriate: ampicillin (100 μg/ml for E. coli), chloramphenicol (10 μg/ml for E. coli and L. monocytogenes), erythromycin (300 μg/ml for E. coli, 10 μg/ml for L. monocytogenes), and tetracycline (30 μg/ml for E. coli) (Sigma-Aldrich, Buchs SG, Switzerland). Media were supplemented with IPTG (isopropyl-β-d-thiogalactopyranoside; 240 μg/ml; Carl Roth GmbH, Karlsruhe, Germany) for IPTG-inducible gene expression, if applicable.
Table 1.
Bacterial strains and plasmidsa
| Strain or plasmid | Relevant characteristics | Reference or origin |
|---|---|---|
| Listeria monocytogenes strains | ||
| EGDe | Wild type; serovar 1/2a | J. Kreft |
| WSLC 1042 | Wild type; serovar 4b | ATCC 23074 |
| EGDeΔtagO1 | EGDeΔlmo0959; EGDe carrying a chromosomal deletion of lmo0959 | This study |
| EGDeΔtagO2 | EGDeΔlmo2519; EGDe carrying a chromosomal deletion of lmo2519 | This study |
| 1042ΔtagO1 | 1042ΔLMO1042_0979; WSLC 1042 carrying a chromosomal deletion of LMO1042_0979 | This study |
| 1042ΔtagO2 | 1042ΔLMO1042_2492; WSLC 1042 carrying a chromosomal deletion of LMO1042_2492 | This study |
| EGDeΔtagO1ΔtagO2::pLIV2(tagO1) | EGDe with a double deletion (Δlmo0959 and Δlmo2519) and lmo0959 expressed from an IPTG-inducible promoter; Camr IPTG (+/−)b | This study |
| 1042ΔtagO1ΔtagO2::pLIV2(tagO1) | WSLC 1042 with a double deletion (ΔLMO1042_0979 and ΔLMO1042_2492) and LMO1042_0979 expressed from an IPTG-inducible promoter; Camr IPTG (+/−) | This study |
| Escherichia coli strains | ||
| XL1-Blue MRF′ | Used for plasmid manipulations; Δ(mcrA)183 Δ(mcrCB-hsdSMR-mrr)173 endA1 supE44 thi-1 recA1 gyrA96 relA1 lac [F′ proAB lacIqZΔM15 Tn10 (Tetr)] | Stratagene |
| XL1-Blue MRF′ pHGFP_CBD118 | Used for protein expression of HGFP_CBD118; Ampr Tetr | 33 |
| XL1-Blue MRF′ pHGFP_CBD511 | Used for protein expression of HGFP_CBD511; Ampr Tetr | 43 |
| XL1-Blue MRF′ pHGFP_CBDP40 | Used for protein expression of HGFP_CBDP40; Ampr Tetr | 43 |
| XL1-Blue MRF′ pHGFP_CBDP35 | Used for protein expression of HGFP_CBDP35; Ampr Tetr | 43 |
| Plasmids | ||
| pKSV7 | Gram-negative bacterium/Gram-positive bacterium shuttle vector; thermosensitive; 6.9 kb; Camr | 44 |
| pAUL-A | Gram-negative bacterium/Gram-positive bacterium shuttle vector; thermosensitive; 9.2 kb; Eryr | 5 |
| pLIV2 | L. monocytogenes site-specific phage integration vector; IPTG-controlled expression; 6.1 kb; Camr | 24 |
| pKSV7(Δlmo0959) | pKSV7 with lmo0959 flanking regions; Camr | This study |
| pKSV7(Δlmo2519) | pKSV7 with lmo2519 flanking regions; Camr | This study |
| pKSV7(ΔLMO1042_0979) | pKSV7 with LMO1042_0979 flanking regions; Camr | This study |
| pKSV7(ΔLMO1042_2492) | pKSV7 with LMO1042_2492 flanking regions; Camr | This study |
| pAUL-A(Δlmo0959) | pAUL-A with lmo0959 flanking regions; Eryr | This study |
| pAUL-A(ΔLMO1042_0979) | pAUL-A with LMO1042_0979 flanking regions; Eryr | This study |
| pLIV2(Δlmo0959) | pLIV2 with lmo0959 under IPTG control; Camr | This study |
| pLIV2(Δlmo0979) | pLIV2 with LMO1042_0979 under IPTG control; Camr | This study |
Genes lmo0959 and LMO1042_0979 were designated tagO1; genes lmo2519 and LMO1042_2492 were named tagO2.
IPTG (+/−), IPTG was added when required to regulate the expression of tagO1 under the control of the inducible Pspac promoter.
General DNA techniques.
General molecular biology techniques were performed by using standard protocols (40). Restriction enzymes and T4 DNA ligase (Roche Diagnostics, Rotkreuz, Switzerland) were used according to the manufacturer's protocols. DNase, RNase, and proteinase K were purchased from Fermentas (Le Mont-sur-Lausanne, Switzerland). DNA fragments and PCR products used for cloning and construction of plasmids were created with Phusion high-fidelity DNA polymerase (Finnzymes, Espoo, Finland). PCR products and DNA restriction fragments were purified with a GenElute PCR cleanup kit (Sigma-Aldrich). Plasmids were purified using a GenElute plasmid miniprep kit (Sigma-Aldrich). Oligonucleotides (see Table S1 in the supplemental material) were designed using the sequence of L. monocytogenes serovar 1/2a strain EGDe (GenBank accession no. AL591824) and serovar 4b strain F2365 (GenBank accession no. AE017262) (20, 36). All PCR products, plasmids, and correct insertions into chromosomal DNA of L. monocytogenes strains were verified by DNA sequencing.
In silico analysis.
Identification and sequence alignment of putative tagO homologues and the corresponding amino acid sequences were performed for L. monocytogenes strains EGDe and F2365, Listeria innocua Clip 11262 (GenBank accession no. AL592022), Listeria seeligeri SLCC 3954 (serovar 1/2b; GenBank accession no. FN557490), and Listeria welshimeri SLCC 5334 (serovar 6b; GenBank accession no. AM263198). Sequences were compared to the respective proteins from B. subtilis 168 and S. aureus MN8 using the NCBI BLAST tool (www.ncbi.nlm.nih.gov) and the software CLC Main Workbench (Aarhus, Denmark).
Deletion of tagO alleles in L. monocytogenes.
Plasmids for allelic exchange and generation of L. monocytogenes in-frame deletion mutants EGDeΔtagO1 (EGDeΔlmo0959), EGDeΔtagO2 (EGDeΔlmo2519), 1042ΔtagO1 (1042ΔLMO1042_0979), and 1042ΔtagO2 (1042ΔLMO1042_2492) (Table 1) were constructed by splicing by overlap extension PCR (SOE-PCR) (25). Genes of L. monocytogenes serovar 4b strain WSLC 1042 were named according to the annotated genome sequences of L. monocytogenes serovar 4b strain F2365. The SOE-PCR primers listed in Table S1 in the supplemental material were designed to amplify upstream (primer pair A and B) and downstream (primer pair C and D) fragments of relevant genes by using EGDe or WSLC 1042 chromosomal DNA as the template. Fragments with in-frame deletions were then generated in an SOE-PCR using products AB and CD and the external primers A and D. The resulting products (AD) were digested with SacI and BamHI and ligated into the corresponding sites of the temperature-sensitive shuttle vector pKSV7 (44). The constructs were transformed into E. coli strain XL1-Blue MRF′, resulting in pKSV7(Δlmo2519) and pKSV7(Δlmo0959), used for strain EGDe, as well as pKSV7(ΔLMO1042_2492) and pKSV7(ΔLMO1042_0979), used for strain WSLC 1042.
Isolated plasmids were then electroporated into competent L. monocytogenes EGDe or WSLC 1042 using a previously published protocol (38). On the basis of the temperature-sensitive origin of replication of pKSV7, chromosomal integration was selected at 42°C (nonpermissive temperature) in the presence of antibiotics. After a series of passages and growth at the permissive temperature (30°C) in the absence of selective drugs, deletion of target genes was obtained via homologous recombination and allelic replacement (44). Colonies were screened for the loss of antibiotic resistance. Antibiotic-sensitive mutants EGDeΔtagO1 and EGDeΔtagO2, as well as 1042ΔtagO1 and 1042ΔtagO2, were confirmed by PCR and DNA sequencing to verify in-frame deletions.
Inducible expression of tagO.
L. monocytogenes tagO1 genes lmo0959 (strain EGDe) and LMO1042_0979 (strain WSLC 1042), including their native ribosome binding sites, were amplified from chromosomal DNA using the primer pairs listed in Table S1 in the supplemental material. PCR products were digested with BamHI and PstI and inserted into pLIV2 (24) immediately downstream of the IPTG-inducible Pspac promoter. pLIV2 is an integrative shuttle plasmid derived from pLIV1 (6) and the site-specific integration vector pPL2 (30). The constructs pLIV2(Δlmo0959) and pLIV2(Δlmo0979) were electroporated into E. coli strain XL1-Blue MRF′, and purified plasmids were used as described below.
Conditional ΔtagO1 ΔtagO2 double-deletion mutants.
Our initial attempts to delete both tagO alleles sequentially failed, since mutant bacteria were apparently not viable. In an alternative approach, we constructed conditional mutants using pLIV2(Δlmo0959) and pLIV2(Δlmo0979), to enable regulated expression of tagO under the control of the inducible promoter Pspac. Vector pLIV2(Δlmo0959) was introduced into EGDeΔtagO1, followed by selection for plasmid integration on BHI plates supplemented with chloramphenicol, yielding strain EGDeΔtagO1::pLIV2(Δlmo0959). Similarly, plasmid pLIV2(Δlmo0979) was used to generate strain 1042ΔtagO1::pLIV2(Δlmo0979). In the next step, conditional ΔtagO1 ΔtagO2 double-deletion mutants were constructed by allelic replacement using the temperature-sensitive shuttle vector pAUL-A (5) containing an erythromycin resistance marker. SOE-PCR products of Δlmo0959 and ΔLMO1042_0979 digested with SacI and BamHI were cloned into pAUL-A, yielding pAUL-A(Δlmo0959) and pAUL-A(ΔLMO1042_0979). After transformation into Listeria, insertion mutants were generated according to the procedure described above. Excision of chromosomally integrated plasmids was performed by repeated growth at 30°C in the absence of erythromycin but in the presence of chloramphenicol and 1 mM IPTG to maintain the pLIV2 recombinant plasmid in order to support cell growth and viability. Expression of genes under IPTG-inducible promoter control allowed the stepwise elimination of wild-type alleles. The resulting conditional tagO-null mutants were designated EGDeΔtagO1ΔtagO2::pLIV2(tagO1) and 1042ΔtagO1ΔtagO2::pLIV2(tagO1). All constructs were confirmed by PCR and sequence analysis.
Properties of tagO-deficient Listeria.
To determine the role of tagO for survival and viability of Listeria, growth of conditional tagO-null mutants was monitored in BHI medium without IPTG and compared to wild-type and tagO single-allele-deletion mutants. Conditional double-deletion mutants were inoculated into BHI supplemented with chloramphenicol (10 μg/ml) and IPTG (1 mM) and cultured overnight at 30°C with shaking. Parent strains EGDe and WSLC 1042 and the ΔtagO1 and ΔtagO2 mutants were grown in BHI without supplements. Then, cells were harvested, washed with BHI to remove residual IPTG and chloramphenicol, and resuspended in fresh BHI medium at a dilution of 1:100. The optical density at 600 nm (OD600) of each culture during growth at 30°C was then monitored.
Purification of Listeria cell walls.
Preparation of L. monocytogenes cell walls for the analysis of the cell wall phosphate content was performed as described previously (12, 15, 51, 55). Wild-type and mutant strains were cultivated in TB medium supplemented with antibiotics and IPTG, as required. The ΔtagO1 ΔtagO2 double-deletion mutants were precultured in TB medium containing IPTG, centrifuged, washed with IPTG-free medium, and resuspended into fresh IPTG-free medium at an OD600 of 0.01. Exponentially growing cells (OD600, 0.7) were heat killed at 100°C for 30 min, harvested by centrifugation (7,000 × g, 15 min, 4°C), and resuspended in SM buffer (50 mM Tris HCl, 100 mM NaCl, 8 mM MgSO4, pH 7.5). Cells were broken by two passages through a One-Shot cell disrupter (Constant cell disruption system; Northants, United Kingdom) at a pressure of 270 MPa. Larger debris and remaining intact cells were removed by centrifugation at 1,500 × g for 5 min. Cell walls were recovered by centrifugation of the supernatant (20,000 × g, 30 min, 4°C). The cell wall-containing pellet was washed three times with water and resuspended in SM buffer. DNase/RNase (25°C, 4 h) and proteinase K (25°C, 2 h) were added at a concentration of 100 μg/g cell wall wet weight. Then, the same volume of 8% (wt/vol) boiling SDS was added, followed by further incubation for 30 min at 100°C to remove noncovalently associated components such as membranes, proteins, and LTAs. SDS-insoluble material was collected by centrifugation (20,000 × g, 30 min, 20°C). Finally, the cell wall-containing pellet was washed five times in pure water by repeated centrifugation and resuspension, lyophilized, and stored at −20°C.
Determination of cell wall-associated phosphate.
To determine the relative amount of WTA in cell walls of wild-type and mutant strains, lyophilized purified cell walls (i.e., murein and covalently attached WTA) were assayed for total (cell wall-bound) phosphate as described elsewhere (12). Briefly, 0.1 mg purified peptidoglycan dissolved in 5 ml distilled water was treated with decomposition reagent (NANOCOLOR NanOx metal; Macherey-Nagel, Switzerland) for oxidative decomposition of cell walls according to the manufacturer's protocol. Subsequently, total phosphorus was determined using a photometric phosphate test (Spectroquant phosphate; Merck, Zug, Switzerland).
CBD binding and fluorescence microscopy.
GFP-tagged CBD proteins derived from phage endolysins Ply118, Ply511, PlyP40, and PlyP35 were produced in E. coli and purified by affinity chromatography as described earlier (33, 43). Protein concentration was determined using a NanoDrop ND-1000 analyzer, and purity was checked by SDS-PAGE analysis. Binding of GFP-CBD to bacteria was tested as described before (33). Cells were grown overnight in BHI medium (30°C with shaking) supplemented with antibiotics and IPTG as indicated. Then, cultures were diluted 10-fold into fresh medium and incubated for another 2 h. In case of the conditional double-deletion mutants, overnight cultures were sedimented by centrifugation, washed once with prewarmed BHI, resuspended in fresh medium, and used to inoculate growth medium without IPTG. Bacterial samples (2 to 5 ml) were collected in the late log phase (centrifugation at 7,000 × g for 1 min), washed with phosphate-buffered saline–Tween 20 (PBS-T) buffer (50 mM NaH2PO4, 120 mM NaCl, 0.1% Tween 20, pH 8.0), and resuspended in 1/10 volume PBS-T. For binding assays, 100 μl of the resuspended cells was mixed with 50 μl of an 800 nM solution of the GFP-CBD proteins, and the mixture was incubated for 5 min at room temperature. Cells and bound protein were pelleted at 16,000 × g for 1 min, resuspended and washed twice in 500 μl PBS-T, and finally, resuspended in 50 μl PBS-T and analyzed by confocal laser scanning fluorescence microscopy (Leica TCS SPE; Leica, Heerbrugg, Switzerland) using GFP-specific excitation and emission wavelengths.
To permeabilize and remove the outer membrane of E. coli cells for determination of CBD decoration of the A1γ peptidoglycan of Gram-negative bacteria, cells were treated as described previously (31). Briefly, exponentially growing E. coli cells were centrifuged (7,000 × g, 5 min, 10°C), resuspended in chloroform-saturated 50 mM Tris buffer (pH 7.7), and incubated for 45 min at room temperature. Finally, cells were washed to remove the solvent, adjusted to an OD600 of 1.0, and used for the CBD binding assays described above.
RESULTS
Two homologous tagO genes in L. monocytogenes.
The tagO products from B. subtilis and S. aureus have been shown to catalyze the first step in WTA biosynthesis, the transfer of GlcNAc to the bactoprenol carrier (45, 53). To provide a basis for the construction and analysis of WTA-deficient Listeria mutants, the first aim was to identify tagO homologues in the genomes of L. monocytogenes EGDe (serovar 1/2a) and F2365 (serovar 4b). Surprisingly, in silico analysis indicated two separate tagO orthologues with highly similar amino acid sequences (53% identity, 74% similarity) in the genomes of L. monocytogenes serovar 1/2 strain EGDe (genes lmo0959 and lmo2519; GenBank accession numbers NP_464484 and NP_466042, respectively) and in L. monocytogenes serovar 4b strain F2365 (genes LMOf2365_0979 and LMOf2365_2492; GenBank accession numbers YP_013580 and YP_015080, respectively) (Fig. 1). The genes are predicted to encode putative UDP-GlcNAc:undecaprenyl phosphate GlcNAc-1-phosphate transferases. Since they reveal significant homologies to the TagO proteins from B. subtilis and S. aureus (see Fig. S1 in the supplemental material; 42 to 49% amino acid identity and 66 to 71% similarity over the entire protein), we propose to use the tagO designation also for Listeria, although the specific transferase activities of these enzymes will have to be determined biochemically. Therefore, lmo0959 and LMOf2365_0979 are designated tagO1, while lmo2519 (previously annotated tagO [2]) and LMOf2365_2492 are named tagO2. In both cases, the apparently redundant tagO homologues are located at opposite positions on the bacterial genome and transcribed in different directions. At least lmo2519 appears to be part of an operon structure (49). We have identified tagO1 and tagO2 homologues in all other available Listeria sp. genomes (L. innocua Clip 11262, L. seeligeri SLCC 3954, and L. welshimeri SLCC 5334) and found them to be highly similar (Fig. 1).
Fig 1.

Genome regions harboring tagO homologues in the chromosomes of different Listeria strains. tagO1 (A) and tagO2 (B) genes from Listeria strains of different species and serovars (SV) are shown: L. monocytogenes (Lmo) EGDe (serovar 1/2a), L. monocytogenes strain F2365 (serovar 4b), L. innocua (Lin) Clip 11262 (serovar 6a), L. seeligeri (Lse) SLCC 3954 (serovar 1/2b), and L. welshimeri (Lwe) SLCC 5334 (serovar 6b). The tagO genes are shown as solid black arrows, and the individual gene designations are listed below. They are located at approximately opposite positions on the bacterial chromosomes.
Deletion of tagO in L. monocytogenes serovar 1/2a and 4b strains.
To determine the role of WTA polymers for recognition and binding of Listeria phage endolysins Ply118, Ply511, and PlyP40 (i.e., their CBDs), we constructed tagO deletion mutants in strains representing the two different WTA backbone types occurring in L. monocytogenes (18). Functional inactivation of tagO is expected to disrupt the first step of the WTA linkage unit biosynthetic pathway (45). We found that mutants featuring removal of a single allele (EGDeΔtagO1, EGDeΔtagO2, 1042ΔtagO1, and 1042ΔtagO2) showed absolutely no defects in growth or morphology. They were identical to the parent strains. This indicated that growth, division, and viability were not affected by removal of only one of the two apparently functionally redundant tagO alleles.
As a next step, we aimed to generate mutants defective in both genes, i.e., ΔtagO1 ΔtagO2 double deletions. However, all our attempts employing the classical two-step allelic replacement strategy were unsuccessful; i.e., the second exchange to inactivate the remaining tagO allele in EGDeΔtagO1::pKSV7(ΔtagO2) and EGDeΔtagO2::pKSV7(ΔtagO1) or in 1042ΔtagO1::pKSV7(ΔtagO2) and 1042ΔtagO2::pKSV7(ΔtagO1) failed. This finding suggested that at least one functional tagO locus is required and essential for normal cell growth and viability of Listeria. As a solution to the problem, we constructed double-deletion mutants which additionally feature conditional, regulated expression of tagO1 from an integrated single-copy plasmid, which yielded strains EGDeΔtagO1ΔtagO2::pLIV2(tagO1) and 1042ΔtagO1ΔtagO2::pLIV2(tagO1). Both mutants feature full in-frame chromosomal deletion of both tagO genes and a functional tagO1 allele under the control of an IPTG-inducible promoter. Both strains were viable and used for further studies.
Deletion of tagO results in loss of WTA.
To assess the impact of tagO deletion on WTA content in the Listeria peptidoglycan, the amount of phosphate (as a direct measure for WTA) present in purified cell walls of tagO single- and double-deletion mutants was determined and compared to that for the wild type. As shown in Fig. 2, deletion of only one of the two tagO alleles in strains EGDe and WSLC 1042 did not significantly affect the level of cell wall-associated phosphate in comparison to that for the wild type, confirming the functional redundancy of tagO1 and tagO2 in WTA biosynthesis. On the other hand, total phosphate associated with the cell walls of EGDeΔtagO1ΔtagO2::pLIV2(tagO1) and 1042ΔtagO1ΔtagO2::pLIV2(tagO1) double-deletion mutants grown in the absence of IPTG was drastically reduced, to approximately 30 to 35% of the wild-type level. In the presence of IPTG, total cell wall phosphate in the conditional double-deletion mutants was almost completely restored to about 85 to 90% of that of the parental strain (33).
Fig 2.

Cell wall teichoic acid depletion in L. monocytogenes tagO deletion mutants. Cell walls of serovar 1/2a (EGDe) and serovar 4b (WSLC 1042) wild-type strains, corresponding single-deletion mutants ΔtagO1 and ΔtagO2, and conditional double-deletion mutants ΔtagO1ΔtagO2::pLIV2(tagO1) (abbreviated ΔtagO1ΔtagO2) grown in the presence or absence of IPTG (strains are listed in Table 1) were purified and analyzed for total cell wall-associated phosphorus as an indicator for the presence or absence of covalently linked cell wall teichoic acid. The diagram shows the effect of TagO deficiency on WTA content. Total phosphate is shown as relative values, whereupon the content in wild-type cell walls was defined as 100%. All measurements were carried out as independent triplicates.
The L. monocytogenes serovar 1/2 EGDe mutant cells were also tested for decoration by fluorescent CBDP35, which specifically recognizes and binds to terminal GlcNAc residues attached to the poly(ribitol phosphate) backbone of Listeria WTA (11). Single tagO1 or tagO2 deficiency did not affect binding of CBDP35 (Table 2). In contrast, inactivation of both alleles in EGDeΔtagO1ΔtagO2::pLIV2(tagO1) resulted in loss of decoration by CBDP35 (Table 2). These findings correlate well with those of the chemical analysis described above and provide additional evidence for the drastically reduced WTA level in these cells.
Table 2.
Binding of GFP-tagged Listeria phage endolysin CBDs to Listeria cells
| L. monocytogenes strain | Serovar | IPTG growth conditiona | Presence of WTAb | Binding of CBDc |
|||
|---|---|---|---|---|---|---|---|
| PlyP35 | Ply511 | Ply118 | PlyP40 | ||||
| EGDe | 1/2a | + | ++ | ++ | ++ | ++ | |
| EGDeΔtagO1 | + | ++ | ++ | ++ | ++ | ||
| EGDeΔtagO2 | + | ++ | ++ | ++ | ++ | ||
| EGDeΔtagO1ΔtagO2::pLIV2(tagO1) | + | + | ++ | ++ | ++ | ++ | |
| EGDeΔtagO1ΔtagO2::pLIV2(tagO1) | − | − | − | ++ | ++ | ++ | |
| WSLC 1042 | 4b | + | − | + | − | ++ | |
| 1042ΔtagO1 | + | − | + | − | ++ | ||
| 1042ΔtagO2 | + | − | + | − | ++ | ||
| 1042ΔtagO1ΔtagO2::pLIV2(tagO1) | + | + | − | + | − | ++ | |
| 1042ΔtagO1ΔtagO2::pLIV2(tagO1) | − | − | − | ++ | ++ | ++ | |
When required, IPTG was added to a final concentration of 1 mM.
+, normal WTA content; −, reduced or no WTA content (determined by the amount of phosphate).
++, strong binding; +, weak binding; −, no binding (estimated by fluorescence microscopy, relative to wild-type EGDe or WSLC 1042).
WTA deficiency leads to impaired growth and coccoid cell morphology.
Single deletion of the tagO locus in EGDeΔtagO1, EGDeΔtagO2, 1042ΔtagO1, and 1042ΔtagO2 did not affect cell morphology or growth properties compared to those of the wild type (Fig. 3). In contrast, inactivation of both alleles in EGDeΔtagO1ΔtagO2::pLIV2(tagO1) and 1042ΔtagO1ΔtagO2::pLIV2(tagO1) was characterized by dramatic alterations in cell morphology (i.e., coccoid cells) and formation of aggregates when tagO expression was shut down following removal of IPTG. Additionally, WTA deficiency resulted in a substantially reduced growth rate, indicating a major role of WTA for normal cell growth and development. Transfer to IPTG-containing medium restored the normal phenotype (Fig. 2). These findings clearly indicate that the presence of at least one of the two redundant tagO homologues in Listeria is required for maintaining cell shape and growth response.
Fig 3.

Effect of tagO disruption on growth and cell morphology of L. monocytogenes. Growth response and changes in cell morphology were analyzed for parental, single ΔtagO1 and ΔtagO2 deletions, and conditional ΔtagO1 ΔtagO2 double-deletion mutants. All cells were cultured in BHI broth without antibiotics, and conditional ΔtagO1 ΔtagO2 double-deletion mutants were grown in the presence or absence of IPTG. Growth was monitored (as OD600) over the indicated time period. (A) L. monocytogenes serovar 1/2a wild-type strain EGDe and mutants EGDeΔtagO1, EGDeΔtagO2, and EGDeΔtagO1ΔtagO2::pLIV2(tagO1) with and without IPTG. (B) L. monocytogenes serovar 4b wild-type strain WSLC 1042 and mutants 1042ΔtagO1, 1042ΔtagO2, and 1042ΔtagO1ΔtagO2::pLIV2(tagO1) with and without IPTG. (C and D) Phase-contrast microscopy of cell samples taken from log-phase cultures of strain EGDe and derivatives (C) and strain WSLC 1042 and derivatives (D). In the double-deletion mutants, the drastic effect of complete tagO functional disruption on cell shape and morphology is clearly visible. Bars, 2 μm.
CBD118, CBD511, and CBDP40 do not require WTA for binding.
To determine the potential role of the WTA polymers for binding of CBD118, CBD511, and CBDP40, tagO null mutants were tested for decoration by GFP-tagged CBDs (Table 2; Fig. 4). CBD511 and CBDP40 decorate the entire cell surface of wild-type EGDe and WSLC 1042, with a clear preference (i.e., more intensive decoration) for septal and polar regions (43). The same binding pattern was observed for mutant strains EGDeΔtagO1, EGDeΔtagO2, 1042ΔtagO1, and 1042ΔtagO2. Interestingly, binding of CBD511 and CBDP40 to the WTA-deficient double tagO null mutant was no longer restricted to poles and septa but occurred at a high density and was evenly distributed over the entire lateral (sidewall) cell surface.
Fig 4.

Targeting of CBD118, CBD511, and CBDP40 to L. monocytogenes cells and wall teichoic acid-deficient mutants. Wild-type L. monocytogenes EGDe (serovar 1/2a) and WSLC 1042 (serovar 4b) and corresponding conditional tagO double-deletion mutants EGDeΔtagO1ΔtagO2::pLIV2(tagO1) and 1042ΔtagO1ΔtagO2::pLIV2(tagO1) (both indicated as ΔtagO1ΔtagO2) were exposed to different GFP-tagged Listeria phage CBD proteins (indicated on the left) (for details, see Materials and Methods). Conditional double-deletion mutants were grown in the presence or absence of IPTG. CBD binding was visualized by confocal laser scanning microscopy. The upper rows of each assay series show phase-contrast images of wild-type and mutant strains after CBD labeling, and the lower rows display the corresponding fluorescence images of the same field. Note the targeting of CBD118 to WTA-deficient L. monocytogenes WSLC 1042 cells. Bar, 2 μm.
Similar results were obtained for CBD118, which labeled the target cells at polar and septal regions (33). There was no difference in decoration of EGDe and the single-locus deletion mutants EGDeΔtagO1 and EGDeΔtagO2 (Table 2; Fig. 4). In contrast, CBD118 was found to decorate the entire cell surface of the WTA-deficient double-deletion mutant EGDeΔtagO1ΔtagO2::pLIV2(tagO1). Binding to these cells appeared to be significantly stronger than that for the parental bacteria, as judged from fluorescence microscopy imaging. Moreover, WTA-deficient double-deletion mutants of serovar 4b strain WSLC 1042 were also found to be labeled by CBD118. This was a very surprising result, given the fact that CBD118 normally recognizes Listeria serovar 1/2 and 3 cells and does not bind to strains featuring serovar 4, 5, or 6 (33). Conditional double-deletion mutants grown in the presence of IPTG fully restored the wild-type CBD binding properties (Fig. 4).
The peptidoglycan of Listeria belongs to the meso-diaminopimelic acid (m-DAP) cross-linked A1γ chemotype, which is also featured by many Gram-negative bacteria, such as E. coli (42). In this context, the ability of CBD118, CBD511, and CBDP40 to decorate the peptidoglycan of E. coli cells stripped of their outer membrane (Fig. 5) further supports our conclusion that the different CBDs share the ability to recognize and bind to the A1γ peptidoglycan backbone.
Fig 5.
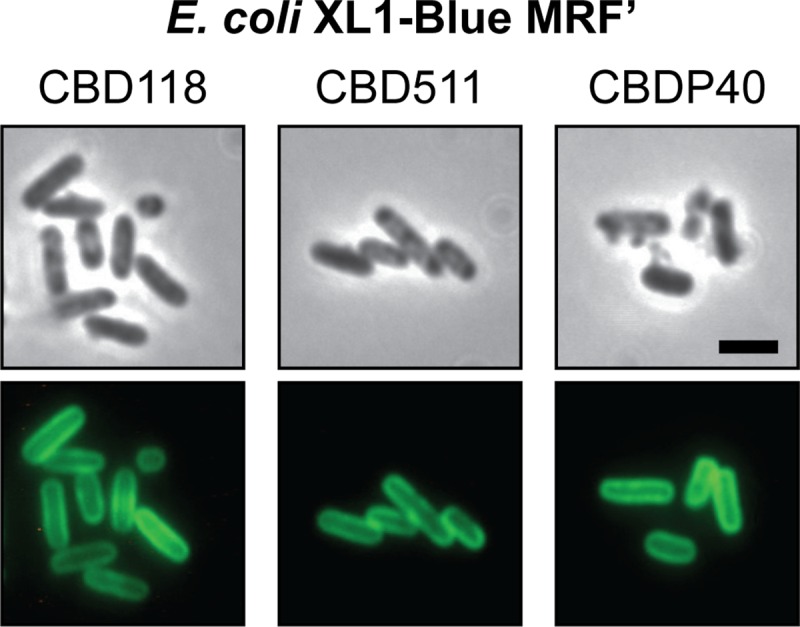
Binding of CBDs to E. coli peptidoglycan. Chloroform-treated E. coli XL1-Blue MRF′ cells were exposed to GFP-CBD118 (left), GFP-CBD511 (middle), and GFP-CBDP40 (right), to demonstrate the basic peptidoglycan-binding properties of the proteins. Samples were monitored by phase-contrast (upper row) and fluorescence (lower row) microscopy. Bar, 2 μm.
Taken together, our findings demonstrate that CBD118, CBD511, and CBDP40 directly interact with the peptidoglycan backbone structure and that the WTA polymers present on Listeria cells are not required for this interaction. Moreover, their presence seems to negatively regulate access of the endolysin ligands for binding of the CBDs.
DISCUSSION
The mostly C-terminally located cell wall binding domains of endolysins from bacteriophages infecting Gram-positive bacterial host cells recognize specific peptidoglycan-associated ligands on the surface of the bacteria with high affinity and specificity. However, surprisingly few endolysin ligands have so far been identified. The Cpl-1 enzyme from pneumococcal phage Cp-1 and other pneumococcal endolysins use choline-binding modules for anchoring to choline-containing teichoic acids of the pneumococcal cell wall (19, 21, 22). Further, the C terminus of endolysin Lyb5, encoded by Lactobacillus fermentum bacteriophage ΦPYB5, has been shown to bind to the peptidoglycan, mediated by LysM repeat regions (26). Regarding enzymes from Listeria phages, we recently reported that CBDP35 specifically recognizes terminal GlcNAc residues in the Listeria WTA molecules (11). In the work presented here, we show that the CBDs of Listeria phage endolysins Ply118, Ply511, and PlyP40 specifically interact with the peptidoglycan backbone structure of Listeria cells. In contrast to CBDP35, WTA polymers seem to be dispensable and are not required for recognition and binding. Instead, they apparently play a role in orchestrating the specific spatial localization of the endolysins to cell division sites and poles.
Use of GFP-tagged CBD fusion proteins to decorate cell walls of Listeria strains of different serovar types revealed individual binding partners of the different phage endolysins, which correlate very well with serovar- and strain-specific cell surface structures (33, 43). The binding patterns and properties of CBD118, CBD511, and CBDP40 differ fundamentally from those of other Listeria phage endolysin CBDs, such as CBD500, CBDPSA, and CBDP35 (29, 33, 43). The latter group of CBDs was found to recognize and bind to the entire cell surface, while CBD118, CBD511, and CBDP40 primarily associate with poles and septae (33, 43). Spatially distinct cell wall targeting has also been reported for other peptidoglycan hydrolases, such as Lactobacillus phage endolysin Lyb5 (26), the AcmA autolysin of Lactococcus lactis (46), and the Atl autolysin of S. aureus (41). The uneven distribution of these enzymes over the cell wall is also proposed to be due to the presence of secondary cell wall polymers such as teichoic acids that prevent targeting to the entire cell surface (41, 47).
Cell walls of L. monocytogenes contain two different polyanionic polymers: WTA, which is covalently linked to the peptidoglycan via a linkage unit, and LTA, which is anchored to the cytoplasmic membrane (35, 37). WTA polymers can account for up to 60 to 70% of the total dry mass of isolated Listeria cell walls (13, 14). During late log phase of bacterial growth, WTA polymers are evenly distributed over the entire cell surface. This is nicely illustrated by using fluorescent CBDP35 (11, 43), which targets the terminal GlcNAc substituted to position C-4 of the poly(ribitol phosphate) WTA backbone (11). In light of the findings described here, we postulate that the asymmetric distribution of CBD118, CBD511, and CBDP40 to the sites of cell division may also be based on WTA polymers. However, the polymers do not serve as binding ligands but seem to prevent an even distribution of the cell wall binding proteins to the peptidoglycan sacculus. The hypothesis was then tested and confirmed using WTA-deficient Listeria cells.
TagO catalyzes the initial step in synthesis of the WTA linkage unit, i.e., the formation of undecaprenyl-diphospho-N-acetylglucosamine (45, 48, 53). In order to gain better insight into WTA biosynthesis in L. monocytogenes, we made use of the fact that the WTA linkage units in Gram-positive bacteria feature conserved structures (1, 53). Based on sequence homologies with B. subtilis and S. aureus TagO, we identified two different Listeria genes designated tagO1 and tagO2. As suggested earlier (56), the designation “tag” (teichoic acid glycerol) was used instead of “tar” (for teichoic acid ribitol), because of the highly conserved mechanisms in biosynthesis of the polymers. Our in silico and experimental analysis of L. monocytogenes TagO1 and TagO2 functions demonstrated their functional redundancy.
It was shown that tagO is not essential for cell viability in B. subtilis and S. aureus (8, 9), although mutants revealed growth and division defects. Along this line, deletion of lmo2537 in L. monocytogenes, predicted to be involved in WTA linkage unit biosynthesis, resulted in a coccoid-like cell morphology (10) featuring a reduction in cell wall phosphate (48%) similar to that which we found for L. monocytogenes tagO double-deletion mutants. The low residual WTA level on these cells may be explained by leaky repression of tagO1 transcription from the IPTG-dependent Pspac promoter on pLIV2 in the absence of the inducer. It might also originate from other phosphate-containing components (7) covalently attached to the bacterial cell wall, whereas LTA has been completely removed. The initial failure to generate and/or isolate L. monocytogenes tagO double-null mutants without conditional complementation was likely due to the severe growth deficiencies, which render isolation of the desired mutants from a pool of normally growing cells impossible. However, the possibility that simultaneous deletion of both tagO copies may be lethal for Listeria cannot be excluded.
Our findings demonstrate that WTA polymers do not directly serve as binding ligands for CBD118, CBD511, and CBDP40. Instead, in the absence of WTA, both the spatial distribution of these CBDs and the quality of binding differed fundamentally from those for wild-type strains; i.e., the CBDs were targeted to the entire cell surface with high intensity. This finding indicated that these CBDs recognize and bind to the peptidoglycan backbone structure itself and that their cell wall binding pattern is apparently dependent on an exclusion-based regulation mechanism by the abundant WTA molecules. This situation appears to be somewhat similar to what was reported for the division-associated autolysins Atl of Staphylococcus aureus (41) and AcmA in L. lactis (46). In the latter case, the even distribution of the autolysin is hindered by LTA. With respect to Listeria (16, 39), however, LTAs can safely be excluded as potential ligands for CBD binding. This is not only because their removal does not affect CBD targeting to isolated and purified cell walls (33) but also because they are structurally identical among the different species and serovars of Listeria, which does not correlate with the variable Listeria phage CBD binding patterns (33, 43).
Considering that the peptidoglycan backbone of L. monocytogenes is of the same chemotype featured by E. coli and many other Gram-negative bacteria (42), our data suggest that CBD118, CBD511, and CBDP40 recognize and bind type A1γ murein (directly cross-linked m-DAP). This would again correlate well with the broad, serovar-independent binding of CBD511 and CBDP40 (43). The fact that the different CBDs show strong binding to E. coli murein also excludes an enhancing effect of the morphological change of the coccoid-like Listeria on decoration by the CBDs. In conclusion, binding does not appear to be affected by cell shape but, rather, appears to be affected by the presence or absence of the secondary polymers.
However, several differences between the three CBD polypeptides exist. Ply118 and Ply511 share considerable homology in their CBDs (43% identity and 80% similarity) (34), which implies that they may be targeted to the same ligands in the peptidoglycan structure. However, these two CBDs do display somewhat different recognition properties. CBD118 can decorate Bacillus megaterium (33), which also features A1γ peptidoglycan (42), while CBD511 does not. CBDP40 targets several non-Listeria strains, including some members from the genera Bacillus, Enterococcus, Staphylococcus, and Bifidobacterium (43). Although identical or at least similar peptidoglycan structures may represent plausible reasons for the cross-reactions, CBD118, CBD511, and CBDP40 still feature slightly different properties. These might be based on their similar but not identical amino acid sequences and on the existence of diverse and different peptidoglycan-binding motifs in the enzymes. CBDP40 has cell wall recognition motifs similar to bacterial SH3 and LysM motifs, whereas no such sequences could be identified in CBD118 and CBD511 (43). The peptidoglycan-binding properties of LysM domains appear to be dependent on other cell wall constituents, such as proteins, cell wall polymers, and even modifications of the peptidoglycan itself (4).
CBD118 offers the narrowest cell wall binding range, which correlates well with the host range of the parent A118 phage. This temperate phage infects Listeria serovar 1/2 strains only and does not recognize cells from serovars 4, 5, and 6 (33). Intriguingly, we show here that WTA-depleted serovar 4b cells (strain WSLC 1042) accept GFP-tagged CBD118 on the entire surface. This finding strongly suggests that the inability of CBD118 to target native serovar 4b cell walls is due to a mechanism of exclusion by the cell wall polymers.
In conclusion, the extremely abundant WTA polymers present on the Listeria cell surface assume crucial roles in the targeting and spatial distribution of phage endolysins CBD118, CBD511, and CBDP40 to septal and polar regions of the peptidoglycan. With increasing consideration of endolysins as potential antimicrobial agents and their cell wall binding domains as tools for immobilization and rapid detection of pathogens, molecular analysis of the endolysin and cell wall interactions becomes more and more important.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Yves Briers (Catholic University of Leuven, Leuven, Belgium), Sibylle Schmitter (ETH Zurich, Zurich, Switzerland), and Vladimir Lazarevic (Université de Lausanne, Lausanne, Switzerland) for helpful discussions. Karin Hotz (ETH Zurich, Zurich, Switzerland) is acknowledged for critical reading of the manuscript.
Footnotes
Published ahead of print 21 September 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Araki Y, Ito E. 1989. Linkage units in cell walls of Gram-positive bacteria. Crit. Rev. Microbiol. 17:121–135 [DOI] [PubMed] [Google Scholar]
- 2. Bierne H, Cossart P. 2007. Listeria monocytogenes surface proteins: from genome predictions to function. Microbiol. Mol. Biol. Rev. 71:377–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Borysowski J, Weber-Dabrowska B, Gorski A. 2006. Bacteriophage endolysins as a novel class of antibacterial agents. Exp. Biol. Med. (Maywood) 231:366–377 [DOI] [PubMed] [Google Scholar]
- 4. Buist G, Steen A, Kok J, Kuipers OP. 2008. LysM, a widely distributed protein motif for binding to (peptido)glycans. Mol. Microbiol. 68:838–847 [DOI] [PubMed] [Google Scholar]
- 5. Chakraborty T, et al. 1992. Coordinate regulation of virulence genes in Listeria monocytogenes requires the product of the prfA gene. J. Bacteriol. 174:568–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dancz CE, Haraga A, Portnoy DA, Higgins DE. 2002. Inducible control of virulence gene expression in Listeria monocytogenes: temporal requirement of listeriolysin O during intracellular infection. J. Bacteriol. 184:5935–5945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. D'Elia MA, Henderson JA, Beveridge TJ, Heinrichs DE, Brown ED. 2009. The N-acetylmannosamine transferase catalyzes the first committed step of teichoic acid assembly in Bacillus subtilis and Staphylococcus aureus. J. Bacteriol. 191:4030–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. D'Elia MA, Millar KE, Beveridge TJ, Brown ED. 2006. Wall teichoic acid polymers are dispensable for cell viability in Bacillus subtilis. J. Bacteriol. 188:8313–8316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. D'Elia MA, et al. 2006. Lesions in teichoic acid biosynthesis in Staphylococcus aureus lead to a lethal gain of function in the otherwise dispensable pathway. J. Bacteriol. 188:4183–4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dubail I, et al. 2006. Identification of an essential gene of Listeria monocytogenes involved in teichoic acid biogenesis. J. Bacteriol. 188:6580–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eugster MR, Haug MC, Huwiler SG, Loessner MJ. 2011. The cell wall binding domain of Listeria bacteriophage endolysin PlyP35 recognizes terminal GlcNAc residues in cell wall teichoic acid. Mol. Microbiol. 81:1419–1432 [DOI] [PubMed] [Google Scholar]
- 12. Eugster MR, Loessner MJ. 2011. Rapid analysis of Listeria monocytogenes cell wall teichoic acid carbohydrates by ESI-MS/MS. PLoS One 6:e21500 doi:10.1371/journal.pone.0021500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fiedler F. 1988. Biochemistry of the cell surface of Listeria strains: a locating general view. Infection 16(Suppl 2): 92–97 [DOI] [PubMed] [Google Scholar]
- 14. Fiedler F, Ruhland GJ. 1987. Structure of Listeria monocytogenes cell walls. Bull. Inst. Pasteur 85:287–300 [Google Scholar]
- 15. Fiedler F, Seger J, Schrettenbrunner A, Seeliger HPR. 1984. The biochemistry of murein and cell-wall teichoic-acids in the genus Listeria. Syst. Appl. Microbiol. 5:360–376 [Google Scholar]
- 16. Fischer W, Mannsfeld T, Hagen G. 1990. On the basic structure of poly(glycerophosphate) lipoteichoic acids. Biochem. Cell Biol. 68:33–43 [DOI] [PubMed] [Google Scholar]
- 17. Fischetti VA. 2005. Bacteriophage lytic enzymes: novel anti-infectives. Trends Microbiol. 13:491–496 [DOI] [PubMed] [Google Scholar]
- 18. Fujii H, et al. 1985. Structural study on teichoic acids of Listeria monocytogenes types 4a and 4d. J. Biochem. 97:883–891 [DOI] [PubMed] [Google Scholar]
- 19. Garcia E, et al. 1988. Molecular evolution of lytic enzymes of Streptococcus pneumoniae and its bacteriophages. Proc. Natl. Acad. Sci. U. S. A. 85:914–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Glaser P, et al. 2001. Comparative genomics of Listeria species. Science 294:849–852 [DOI] [PubMed] [Google Scholar]
- 21. Hermoso JA, Garcia JL, Garcia P. 2007. Taking aim on bacterial pathogens: from phage therapy to enzybiotics. Curr. Opin. Microbiol. 10:461–472 [DOI] [PubMed] [Google Scholar]
- 22. Hermoso JA, et al. 2003. Structural basis for selective recognition of pneumococcal cell wall by modular endolysin from phage Cp-1. Structure 11:1239–1249 [DOI] [PubMed] [Google Scholar]
- 23. Hether NW, Jackson LL. 1983. Lipoteichoic acid from Listeria monocytogenes. J. Bacteriol. 156:809–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Higgins DE, Buchrieser C, Freitag NE. 2006. Genetic tools for use with Listeria monocytogenes, p 620–633 In Fischetti VA, Novick RP, Ferretti JJ, Portnoy DA, Rood JI. (ed), Gram-positive pathogens. American Society for Microbiology, Washington, DC [Google Scholar]
- 25. Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68 [DOI] [PubMed] [Google Scholar]
- 26. Hu S, Kong J, Kong W, Guo T, Ji M. 2010. Characterization of a novel LysM domain from Lactobacillus fermentum bacteriophage endolysin and its use as an anchor to display heterologous proteins on the surfaces of lactic acid bacteria. Appl. Environ. Microbiol. 76:2410–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kohler T, Weidenmaier C, Peschel A. 2009. Wall teichoic acid protects Staphylococcus aureus against antimicrobial fatty acids from human skin. J. Bacteriol. 191:4482–4484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kohler T, et al. 2010. Teichoic acids, lipoteichoic acids and related cell wall glycopolymers of Gram-positive bacteria, p 75–91 In Microbial glycobiology. Academic Press, San Diego, CA [Google Scholar]
- 29. Korndörfer IP, et al. 2006. The crystal structure of the bacteriophage PSA endolysin reveals a unique fold responsible for specific recognition of Listeria cell walls. J. Mol. Biol. 364:678–689 [DOI] [PubMed] [Google Scholar]
- 30. Lauer P, Chow MY, Loessner MJ, Portnoy DA, Calendar R. 2002. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J. Bacteriol. 184:4177–4186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lavigne R, Briers Y, Hertveldt K, Robben J, Volckaert G. 2004. Identification and characterization of a highly thermostable bacteriophage lysozyme. Cell. Mol. Life Sci. 61:2753–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Loessner MJ. 2005. Bacteriophage endolysins—current state of research and applications. Curr. Opin. Microbiol. 8:480–487 [DOI] [PubMed] [Google Scholar]
- 33. Loessner MJ, Kramer K, Ebel F, Scherer S. 2002. C-terminal domains of Listeria monocytogenes bacteriophage murein hydrolases determine specific recognition and high-affinity binding to bacterial cell wall carbohydrates. Mol. Microbiol. 44:335–349 [DOI] [PubMed] [Google Scholar]
- 34. Loessner MJ, Wendlinger G, Scherer S. 1995. Heterogeneous endolysins in Listeria monocytogenes bacteriophages: a new class of enzymes and evidence for conserved holin genes within the siphoviral lysis cassettes. Mol. Microbiol. 16:1231–1241 [DOI] [PubMed] [Google Scholar]
- 35. Navarre WW, Schneewind O. 1999. Surface proteins of Gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 63:174–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nelson KE, et al. 2004. Whole genome comparisons of serotype 4b and 1/2a strains of the food-borne pathogen Listeria monocytogenes reveal new insights into the core genome components of this species. Nucleic Acids Res. 32:2386–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Neuhaus FC, Baddiley J. 2003. A continuum of anionic charge: structures and functions of d-alanyl-teichoic acids in Gram-positive bacteria. Microbiol. Mol. Biol. Rev. 67:686–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Park SF, Stewart GS. 1990. High-efficiency transformation of Listeria monocytogenes by electroporation of penicillin-treated cells. Gene 94:129–132 [DOI] [PubMed] [Google Scholar]
- 39. Ruhland GJ, Fiedler F. 1987. Occurrence and biochemistry of lipoteichoic acids in the genus Listeria. Syst. Appl. Microbiol. 9:40–46 [Google Scholar]
- 40. Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 41. Schlag M, et al. 2010. Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol. Microbiol. 75:864–873 [DOI] [PubMed] [Google Scholar]
- 42. Schleifer KH, Kandler O. 1972. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 36:407–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmelcher M, et al. 2010. Rapid multiplex detection and differentiation of Listeria cells using fluorescent phage endolysin cell wall binding domains. Appl. Environ. Microbiol. 76:5745–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smith K, Youngman P. 1992. Use of a new integrational vector to investigate compartment-specific expression of the Bacillus subtilis spoIIM gene. Biochimie 74:705–711 [DOI] [PubMed] [Google Scholar]
- 45. Soldo B, Lazarevic V, Karamata D. 2002. tagO is involved in the synthesis of all anionic cell-wall polymers in Bacillus subtilis 168. Microbiology 148:2079–2087 [DOI] [PubMed] [Google Scholar]
- 46. Steen A, et al. 2003. Cell wall attachment of a widely distributed peptidoglycan binding domain is hindered by cell wall constituents. J. Biol. Chem. 278:23874–23881 [DOI] [PubMed] [Google Scholar]
- 47. Steen A, et al. 2005. Autolysis of Lactococcus lactis is increased upon d-alanine depletion of peptidoglycan and lipoteichoic acids. J. Bacteriol. 187:114–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Swoboda JG, Campbell J, Meredith TC, Walker S. 2010. Wall teichoic acid function, biosynthesis, and inhibition. Chembiochem 11:35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Toledo-Arana A, et al. 2009. The Listeria transcriptional landscape from saprophytism to virulence. Nature 459:950–956 [DOI] [PubMed] [Google Scholar]
- 50. Uchikawa K, Sekikawa I, Azuma I. 1986. Structural studies on lipoteichoic acids from four Listeria strains. J. Bacteriol. 168:115–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Valyasevi R, Sandine WE, Geller BL. 1990. The bacteriophage kh receptor of Lactococcus lactis subsp. cremoris KH is the rhamnose of the extracellular wall polysaccharide. Appl. Environ. Microbiol. 56:1882–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ward JB. 1981. Teichoic and teichuronic acids: biosynthesis, assembly, and location. Microbiol. Rev. 45:211–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weidenmaier C, et al. 2004. Role of teichoic acids in Staphylococcus aureus nasal colonization, a major risk factor in nosocomial infections. Nat. Med. 10:243–245 [DOI] [PubMed] [Google Scholar]
- 54. Weidenmaier C, Peschel A. 2008. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat. Rev. Microbiol. 6:276–287 [DOI] [PubMed] [Google Scholar]
- 55. Wendlinger G, Loessner MJ, Scherer S. 1996. Bacteriophage receptors on Listeria monocytogenes cells are the N-acetylglucosamine and rhamnose substituents of teichoic acids or the peptidoglycan itself. Microbiology 142:985–992 [DOI] [PubMed] [Google Scholar]
- 56. Xia G, Peschel A. 2008. Toward the pathway of S. aureus WTA biosynthesis. Chem. Biol. 15:95–96 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.