

Abstract
Helicobacter pylori is a highly successful pathogen that colonizes the gastric mucosa of ∼50% of the world's population. Within this colonization niche, the bacteria encounter large fluctuations in nutrient availability. As such, it is critical that this organism regulate expression of key metabolic enzymes so that they are present when environmental conditions are optimal for growth. One such enzyme is the 2-oxoglutarate (α-ketoglutarate) oxidoreductase (OOR), which catalyzes the conversion of α-ketoglutarate to succinyl coenzyme A (succinyl-CoA) and CO2. Previous studies from our group suggested that the genes that encode the OOR are activated by iron-bound Fur (Fe-Fur); microarray analysis showed that expression of oorD, oorA, and oorC was altered in a fur mutant strain of H. pylori. The goal of the present work was to more thoroughly characterize expression of the oorDABC genes in H. pylori as well as to define the role of Fe-Fur in this process. Here we show that these four genes are cotranscribed as an operon and that expression of the operon is decreased in a fur mutant strain. Transcriptional start site mapping and promoter analysis revealed the presence of a canonical extended −10 element but a poorly conserved −35 element upstream of the +1. Additionally, we identified a conserved Fur binding sequence ∼130 bp upstream of the transcriptional start site. Transcriptional analysis using promoter fusions revealed that this binding sequence was required for Fe-Fur-mediated activation. Finally, fluorescence anisotropy assays indicate that Fe-Fur specifically bound this Fur box with a relatively high affinity (dissociation constant [Kd] = 200 nM). These findings provide novel insight into the genetic regulation of a key metabolic enzyme and add to our understanding of the diverse roles Fur plays in gene regulation in H. pylori.
INTRODUCTION
The Gram-negative pathogen Helicobacter pylori colonizes the gastric mucosa of more than one-half of the world's population (7, 22). While colonization usually results in asymptomatic carriage, a subset of infected individuals may progress to develop a range of disease states that include gastritis, gastric ulcers, and two types of gastric cancer (7, 22). Because of its unique colonization niche, H. pylori is continually faced with dramatic changes in environmental conditions such as pH and nutrient availability. As a result, this organism has evolved very efficient adaptive responses that enable it to persist within the gastric mucosa in spite of these drastic fluctuations. One crucial aspect of the adaptive process involves maintaining tightly controlled regulatory networks that differentially modulate gene expression in response to environmental stimuli.
In H. pylori, a key transcriptional regulator that maintains control of many of these adaptive regulatory networks is the ferric uptake regulator (Fur) (12, 23, 26). This regulator is found in many bacterial species and is traditionally associated with the regulation of iron uptake and storage systems. In H. pylori, Fur is critical for pH adaptation (6, 26), colonization (8, 26, 30), and mounting adaptive responses to oxidative (9, 11, 23, 26, 27) and nitrosative (35) stress conditions. Given the importance of Fur to these essential biological processes, perhaps it is not surprising that H. pylori Fur regulates a wide variety of genes that encode functionally diverse proteins that extend beyond just iron uptake and storage (23, 26). A somewhat unique aspect of Fur regulation in H. pylori is the ability to function beyond what is typically seen in other bacteria. Like Fur in other bacterial species, H. pylori Fur can function as an iron cofactored transcriptional repressor (Fe-Fur), where iron-dependent binding to specific DNA sequences (Fur boxes) prevents assembly of the transcriptional machinery at the core promoter elements (14, 17, 34). In addition, H. pylori Fur can also regulate gene expression in the iron-free or apo form of the protein (5, 9, 15, 16, 24). Previous studies have shown that apo-Fur can act both as a repressor (5, 9, 24) and as an activator (16). Furthermore, there is also evidence that Fe-Fur can activate transcription, although to date this regulatory function has been conclusively demonstrated to occur only at a single H. pylori promoter (1).
Previous transcriptional profiling experiments conducted by our group (26) identified another set of potentially Fe-Fur-activated genes; expression levels of oorD, oorA, and oorC were decreased in a fur mutant strain of H. pylori. At the chromosomal level, these genes appear to be part of an operon with oorB. The oorDABC genes encode a 2-oxoglutarate (α-ketoglutarate) oxidoreductase (OOR) (28), and OOR activity (reversible oxidative decarboxylation of 2-oxoglutarate to form succinyl coenzyme A [succinyl-CoA] and CO2) has been demonstrated in H. pylori using whole-cell lysates (28). Additional biochemical studies characterized the H. pylori OOR enzyme as an essential oxygen-labile heterotetrameric protein consisting of 4 polypeptide subunits of 10 kDa (OorD), 43 kDa (OorA), 33 kDa (OorB), and 24 kDa (OorC) (28). In addition to serving as a source of succinyl-CoA, it has also been proposed that the H. pylori OOR enzyme acts to provide NADPH with a respiratory donor electron (28). Thus, the OOR enzyme of H. pylori plays a multifaceted role in central metabolism and energy conservation.
While some of the basic biochemical characteristics of the H. pylori OOR enzyme have been described, there is currently little direct information about how expression of the genes that encode the essential enzyme are regulated. Given the importance of the OOR enzyme in the physiology of H. pylori and our previous preliminary evidence that suggested Fe-Fur activates expression of these genes (26), here we characterize transcriptional regulation of the oorDABC genes. We show that these genes are cotranscribed as a single operon from a single start site and that there is a conserved Fur box distally located upstream of the core promoter elements. Purified Fur bound specifically to this site, and this binding was essential for the Fur-mediated activation of oorDABC. These data add to the increasing number of genes regulated by H. pylori Fur via nonclassical (iron-bound repression) mechanisms and provide potential insight into how sensing changes in environmental cues such as iron availability can initialize changes in intracellular physiology.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All bacterial strains and plasmids used in this study are listed in Table 1, and oligonucleotides are listed in Table 2. H. pylori strains were maintained in brain heart infusion broth (Becton Dickinson) containing 10% fetal bovine serum (FBS) (Gibco) and 20% glycerol at −80°C. H. pylori strains were routinely cultured at 37°C under microaerophilic conditions (5% O2, 10% CO2, 85% N2) on horse blood agar (HBA) plates that contained 4% Columbia agar base (Neogen Corp.), 5% defibrinated horse blood (HemoStat Laboratories, Dixon, CA), 0.2% β-cyclodextrin (Sigma), 5 μg/ml amphotericin B (Amersco), 2.5 U/ml polymyxin B (Sigma), 5 μg/ml trimethoprim (Sigma), and 10 μg/ml vancomycin (Amersco). Liquid cultures of H. pylori were grown in brucella broth that contained 10% FBS and 10 μg/ml vancomycin. E. coli strains were maintained in LB with 40% glycerol at −80°C and routinely cultured at 37°C on 1.5% LB agar plates (MoBio) or in LB broth with shaking at 225 rpm. When applicable, bacterial growth medium was supplemented with 25 μg/ml kanamycin (Kan) (Gibco), 100 μg/ml ampicillin (Amp) (USB Corp.), or 8 μg/ml chloramphenicol (Cm) (EMD Chemicals, Inc.). The wild-type (WT) H. pylori strain G27 (4) or genetically modified G27 derivatives were used for all experiments in this study.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Reference |
|---|---|---|
| E. coli strains | ||
| DSM431 | BL21(DE3) Rosetta/pLys Δfur (pDSM430) Ampr Kanr Cmr | 9 |
| H. pylori strains | ||
| DSM1 | G27 WT | |
| DSM300 | G27 Δfur::cat Cmr | 26 |
| DSM925 | G27 Δfur (markerless) | 34 |
| DSM1079 | G27 WT (pDSM845) Kanr | This study |
| DSM1080 | G27 Δfur(pDSM845) Kanr | This study |
| DSM1081 | G27 WT (pDSM833) Kanr | This study |
| DSM1082 | G27 Δfur(pDSM833) Kanr | This study |
| Plasmids | ||
| pDSM430 | pET21A::fur | 9 |
| pDSM815 | pDSM199::cat | 34 |
| pDSM833 | pDSM815::PoorDABC (−FB)::cat | This study |
| pDSM845 | pDSM815::PoorDABC ::cat | This study |
Table 2.
Oligonucleotides used in this study
| Oligonucleotide | Sequence (5′–3′) |
|---|---|
| oorDA-up | GACAGGAAGGATTTCAAATTCGC |
| oorDA-dn | CGAGTGGGCATTCCGGTTGATGGG |
| oorAB-up | GAAAGCAAGCAAAAAGTCGGC |
| oorAB-dn | GCCCTACCATGCGTGGTGTGAACG |
| oorBC-up | GCGTCTCAAATGCTAAAATGG |
| oorBC-dn | GCTGATTTGAGCGACTGAAAGC |
| oorD-PE | GCGCGACAGCTTTAAAATCCCC |
| oorD_TSS-up | CGATTAGAGCCGTGGTTTTCC |
| oorD_TSS-dn | GAAATCCTTCCTGTCAGCCAC |
| oorD_Fb_up-F | ACCGATAACACCCCCTATAACACC |
| oorD_Fb_up-R | GAAAAAACGCCTCATTAACCTTAATTTGAAAGATTAATGTTTTTTAAATGCTCC |
| oorD_Fb_dn-F | GAGCATTTAAAAAACATTAATCTTTCAAATTAAGGTTAATGAGGCGTTTTTTC |
| oorD_Fb_dn-R | GTCTTCATTCACCCAAACGGC |
| oorD_EMSA-F | TTCAGCGCGACTTATAAGGAG |
| oorD_EMSA-R | AGCCCTAGCAATCAAAATTTGCC |
| oorD-FA | 5FluorT/TTCTAATAATCATTTTTAAAAT |
| oorD-FA_SCRAM | 5FluorT/TTCGTGTGTGTGTGTGTGAAAT |
| oorD_qRT-F | CCGTTTGGGTGAATGAAG |
| oorD_qRT-R | CACGCAACCGATACAACTC |
| G27_16S-F | ATGGATGCTAGTTGTTGGAGGGCT |
| G27_16S-R | TTAAACCACATGCTCCACCGCTTG |
| Cat_qRT-F | ATACCACGACGATTTCCGGCAGTT |
| Cat_qRT-R | ACTGGTGAAACTCACCAGGGATT |
RNA extraction, cDNA synthesis, and RT-PCR.
Exponential-phase liquid cultures of H. pylori (optical density at 600 nm [OD600], ∼0.5) were harvested onto a 0.45-μm filter by vacuum filtration and snap-frozen in liquid nitrogen. RNA was isolated using the TRIzol reagent (Invitrogen) as previously described (9). First-strand cDNA was synthesized using the Quantitect reverse transcription kit (Qiagen) according to the manufacturer's instructions. For each cDNA synthesis reaction, a corresponding control reaction with no reverse transcription enzyme was included. Quantitative real-time PCRs (qRT-PCRs) were performed using a Roto-gene Q instrument (Qiagen) in a total volume of 20 μl; each reaction mixture contained 1× Roto-Gene SYBR green RT-PCR master mix (Qiagen), 3 pmol each of the forward and reverse primers as indicated, and 1 μl of cDNA or NoRT control reaction as the template. Cycling conditions included an initial activation step of 5 min at 95°C, followed by 35 cycles of denaturing at 95°C for 10 s, annealing at 50°C for 20 s, and extension at 72°C for 30 s. SYBR green fluorescence was measured during each extension step. Relative gene expression was calculated as 2−ΔΔCT using 16S as the internal reference gene. A postrun melt curve analysis was performed to ensure specificity of amplification. Three biologically independent replicates of each experiment were performed.
oorDABC gene junction PCR.
To detect each of the oorDABC gene junctions, a qualitative reverse transcription-PCR was performed using GoTaq master mix (Promega), 3 pmol each of the forward and reverse primers as indicated, and 1 μl of cDNA (or NoRT control reaction mixture), which was synthesized using exponential-phase RNA from wild-type G27. Each reaction mixture was incubated for 5 min at 95°C, followed by 25 cycles of denaturation at 95°C for 30 s, annealing at 50°C for 30 s, and extension at 72°C for 1 min. PCR products were separated on 1% agarose gels and visualized by staining with ethidium bromide. Each PCR was performed at least twice using biologically independent cDNA templates.
Primer extension and TSS mapping.
Primer extension reactions were performed using 10 pmol of γ-32P-labeled oorD-RT primer, 5 μg of RNA harvested from exponential-phase wild-type or Δfur strain liquid cultures, and the AMV reverse transcription kit (Promega) according to the manufacturer's instructions. Dideoxy sequencing reactions were initiated using the γ-32P-labeled oorD-RT primer and the Sequenase PCR amplicon sequencing kit (USB) according to the manufacturer's instructions. Primer extension and sequencing reaction products were resolved by polyacrylamide gel electrophoresis. After electrophoresis, gels were exposed to phosphor screens and subsequently scanned using an FLA-multifunctional scanner (Fuji-film). Image analyses were performed using the Multi-gauge software package (Fuji-film). Transcriptional start site (TSS) mapping experiments were performed at least twice with comparable results.
Construction of PoorDABC::cat reporter plasmids and strains.
To evaluate the importance of the putative Fur box in the expression of the oorDABC promoter, we created plasmid-based transcriptional fusions to the chloramphenicol acetyltransferase reporter gene (cat). A 151-bp region of the oorDABC promoter containing the putative Fur box (TAATAATCATTTTTA) was PCR amplified using the primers PoorD-F and catEC-3′, digested with BamHI and PstI, and directionally cloned upstream of the promoterless cat reporter in pDSM815 (34) to create pDSM845 (pDSM815::PoorDABC::cat). This reporter plasmid is derived from pTM117 (10), which was developed and has been successfully used as a transcriptional reporter by our group (34). Similarly, to create the oorDABC promoter fusion without the Fur box, the oorD_5′(−) and oorD_3′(−) primers were used to amplify the portion of the oorDABC immediately downstream of the Fur box. This amplified fragment was then digested using BamHI and PstI and subsequently cloned into pDSM815 to create pDSM833 [pDSM815::PoorDABC(−FB)::cat]. Each promoter fusion plasmid was then transformed into the wild-type G27 strain DSM1 and the markerless G27 fur deletion strain DSM925 (34) to create strains DSM1079 (WT + pDSM845), DSM1080 (Δfur + pDSM845), DSM1081 (WT + pDSM833), and DSM1082 (Δfur + pDSM833). Transformants were selected on HBA plates that contained 25 μg/ml kanamycin, and plasmid integrity was verified by restriction digestion, PCR, and sequencing.
rFur purification.
Recombinant native H. pylori Fur (rFur) was purified as described previously (9). Briefly, an E. coli fur deletion strain (9) containing the H. pylori rFur expression plasmid pDSM430 (9) was grown to mid-exponential phase in LB broth, and fur expression was induced with 100 mM IPTG (isopropyl-d-thiogalactopyranoside) (Sigma) overnight at 30°C. Cells containing rFur were harvested by centrifugation and mechanically disrupted using a French pressure cell (Amicon). rFur was purified from pressed cell lysates by fast-protein liquid chromatography. Cell lysates were first passed through a HiTrap SP column for cation exchange-based purification using a salt gradient of 25 mM to 500 mM NaCl. Fractions containing rFur were collected and then passed through a Sephacryl-200 column for size exclusion purification. Peak fractions were collected and stored in 50 mM sodium phosphate, 500 mM NaCl, pH 8.0, at 4°C.
rFur FA.
Fluorescence anisotropy (FA) was used to measure the DNA recognition and binding properties of rFur and the putative oorDABC Fur box. Fluorescently labeled DNA that contained either the wild-type oorDABC Fur box (oorD-FA) or the scrambled Fur box sequence (oorD-FA_SCRAM) (Table 2) was incubated with increasing concentrations of Fur. Measurements were taken on an SS PC-1 spectrofluorimeter configured in the L format with an excitation wavelength of 495 nm and an emission wavelength of 519 nm. The band pass was 2 nm for the excitation and 1 nm for emission. In a typical experiment, a 15 nM solution of fluorescently labeled DNA in 30 mM Tris, 120 mM KCl, 16 mM dithiothreitol (DTT), 1 mM MnCl2, 20% glycerol, and 5 mM bovine serum albumin (BSA) at pH 8.0 was added to a 0.5-cm PL Spectrosil far-UV quartz window fluorescence cuvette (Starna Cells) (20). The anisotropy of the free DNA oligonucleotide was measured. rFur protein was then titrated into the cuvette from a stock solution containing 30 mM Tris, 120 mM KCl, 16 mM DTT, 1 mM MnCl2, and 20% glycerol at pH 8.0 in a stepwise fashion, and the resultant change in anisotropy was recorded. The protein was added until the anisotropy values reached a plateau, which indicated saturation. The data were analyzed by converting the anisotropy, r, to the bound fraction, Fbound (the fraction of Fur bound to DNA at a given DNA concentration), using the following equation (29):
where rfree is the anisotropy of the fluorescein-labeled oligonucleotide, rbound is the anisotropy of the DNA-protein complex at saturation, and Q is the quantum yield ratio of the bound form to free form and is calculated from the fluorescence intensity changes that occur (Q = Ibound/Ifree). Fbound was plotted against the protein concentration, treating Fur as a dimer, and fit using a one-site binding model:
where [P]is the protein (Fur) concentration and [D]is the DNA concentration. Each data point from the fluorescence anisotropy assay represents the average of 31 readings taken over a time course of 100 s. Each titration was carried out in triplicate.
Statistical analyses.
To assess the statistical significance of the differences in relative gene expression from the native oorDABC promoter identified by qRT-PCR, Student's t test was performed on log10-transformed fold change data. Differences in relative gene expression from the plasmid-based cat promoter fusions were determined using a paired Student's t test on normalized cat transcript levels.
RESULTS
The oorDABC genes are cotranscribed as an operon.
Previous transcriptional profiling studies using wild-type and fur mutant strains of H. pylori suggested that expression levels of oorD, oorA, and oorC were decreased in a fur mutant strain of H. pylori (26). At the chromosomal level, these genes appear to be part of an operon with oorB (Fig. 1A). Furthermore, the proteins produced from the oorDABC genes are thought to function together in a single multisubunit enzyme (28); genes encoding components that work together in this fashion are often expressed as a single transcriptional unit (40). Therefore, we hypothesized that the oorDABC genes are cotranscribed as an operon from a single promoter. To test this hypothesis, we designed PCR primers that would allow us to amplify the region that spans each of the gene junctions from genomic DNA (Fig. 1B) as well as a cDNA template; amplification from the cDNA template would occur only if the adjacent genes are cotranscribed. As shown in Fig. 1B, we were able to amplify the 295-bp oorDA, the 240-bp oorAB, and the 191-bp oorBC gene junctions. However, we were unable to amplify any of these junctions if no reverse transcriptase was added to the cDNA synthesis reaction. These data indicate that the oorDABC genes are in fact cotranscribed and constitute an operon.
Fig 1.

Genetic organization and transcription of the oorDABC operon. (A) The genes that encode the individual OOR subunits are organized in an operon. To determine whether these genes were cotranscribed, primers were designed for the oorDA, oorAB, and oorBC gene junctions. The approximate amplified regions are indicated by the small black bars (not drawn to scale). (B) PCR amplification of the oorDA, oorAB, and oorBC gene junctions using wild-type H. pylori genomic DNA produced amplicons of 295 bp, 240 bp, and 191 bp, respectively. (C) Qualitative reverse transcription-PCR (RT) of the oorDA, oorAB, and oorBC gene junctions produced amplicons of 295 bp, 240 bp, and 191 bp, indicating that these genes are cotranscribed as an operon. Control reactions for each reverse transcription-PCR (NoRT) did not produce amplification products, indicating that amplification was specific to the cDNA template. Reverse transcription-PCR and NoRT control reactions were performed in duplicate using biologically independent cDNA samples with similar results.
Expression of the oorDABC genes is activated by Fur under iron-replete conditions.
Given that our previous transcriptional profiling studies suggested that expression of the oorDABC genes was activated by Fur under iron-replete conditions (26), and because this type of regulation is different from the classical iron-dependent repression commonly associated with Fur regulation, we sought to further characterize regulation of the oorDABC promoter. Initially, we first sought to confirm our previous findings using qRT-PCR, since it is more sensitive than the methods used previously. Rather than measuring expression from each oor gene, we opted to use oorD expression as a readout for expression from the oorDABC promoter, since it is the first gene in the operon. Using qRT-PCR, we determined that under iron-replete conditions, expression of oorD was ∼3.5-fold less in the fur mutant strain than in the wild-type strain. This difference in expression was significant (P = 0.006, Student's t test). Thus, in agreement with our previous data (26), maximal transcription from the oorDABC promoter requires Fur.
Transcriptional start site mapping and identification of oorDABC promoter elements.
Given that the oorDABC genes are cotranscribed as an operon and that expression from this promoter is regulated by Fur, we next sought to define the promoter elements of these genes. To this end, we utilized primer extension to map the 5′ end of the oorDABC transcript and identify the transcriptional start site. As shown in Fig. 2A, the primer extension reaction produced a single cDNA product from samples obtained from both the wild-type and fur mutant strains. In agreement with our qRT-PCR data, the cDNA product was more abundant in the wild-type strain than in the fur mutant, suggesting that Fur activates expression from this promoter (Fig. 2A). In both the wild-type and fur mutant strains, the +1 was located at a guanine nucleotide located 54 bases upstream of the ATG start codon. Upstream of the transcription start site, we identified a well-conserved −10 element (TACAAT) that also contained a TGN motif immediately upstream, which is commonly seen in extended −10 regions (Fig. 2B). Extended −10 sequences are found in many H. pylori promoters (31, 36) and in this case may be important for efficient oorDABC expression, as this promoter does not contain a well-conserved −35 sequence. Instead, this region is replaced with a periodic AT-rich sequence that appears to typify many H. pylori promoters (31, 36). Further upstream of the core promoter elements, we identified a conserved putative Fur binding site (Fig. 2B). This AT-rich, nearly palindromic sequence (TAATAATCATTTTTTA) is centered ∼140 bases upstream of the transcription start site and differs from the core binding site for Fe-Fur by 1 nucleotide (34). Examination of this region in several sequenced H. pylori strains showed that this Fur box was highly conserved (data not shown). Of note, the stop codon of the upstream pabC gene lies within the 5′ end of the putative Fur box; since pabC has not been shown to be Fur regulated in previous studies (23, 26), the significance of the location of this Fur box remains unclear. While the binding site for Fur often overlaps the −10 and/or −35 regions for promoters that are repressed by Fur, this is not the case for promoters that are activated by this regulator; Fur typically binds upstream of the core promoter elements for these types of genes (1, 15). Thus, the positioning of the putative Fur binding site in the oorDABC promoter is consistent with the notion that Fe-Fur acts as an activator at this promoter.
Fig 2.

Mapping of the oorD TSS and promoter structure.(A) The oorD transcriptional start site (TSS) was mapped using RNA from wild-type and Δfur strains of H. pylori. First-strand cDNA was reverse transcribed from within the oorD coding region using the oorD-RT primer; only a single band was detected for both strains, indicating a single TSS. (B) The core promoter region contains a canonical −10 region (bold, underlined) as well as an extended −10 element but does not have a highly conserved −35 element. However, the region where a typical −35 sequence would lie is indicated for reference (bold, underlined). The putative Fur box (underlined, italics) is located ∼130 bp upstream of the TSS (+1), which is located 54 bp upstream of the ATG start codon (bold).
Fur specifically binds to the putative Fur box in the oorDABC promoter region.
Previous work from our lab demonstrated that Fe-Fur binds specifically to a 377-bp region upstream of the oorD coding region (26); however, that work did not determine where Fur binding occurred within this fragment. Given our identification of a putative Fur box located in the oorDABC promoter region that would have been contained in the larger fragment studied previously, we next sought to determine whether this sequence was in fact the target for Fur interaction. Given that fluorescence anisotropy (FA) has been successfully used to measure the DNA binding properties of metalloregulatory proteins, including Fur (20, 21, 41), we chose to use FA for these studies. To this end, we utilized a fluorescently labeled 23-bp fragment of the oorDABC promoter region that contained the putative Fur binding site (oorD-FA) as well as a derivation of the same fragment where the conserved Fur binding sequence was scrambled to a GT repeat sequence (oorD-FA_SCRAM). For each of these fluorescently labeled templates, we performed binding assays using a range of Fe-Fur concentrations; at concentrations of Fe-Fur where the protein bound the DNA template, the DNA:Fur complex displayed an increased anisotropy. As shown in Fig. 3, Fe-Fur bound to the template that contained the conserved Fur binding sequence, whereas Fur did not bind to the scrambled sequence to any significant extent. The solution-based anisotropy data obtained using Fe-Fur and the WT DNA template were used to fit an appropriate binding model and calculate a dissociation constant (Kd). The binding affinity of Fe-Fur with the labeled WT DNA template was 200 nM, which is consistent with Kd values for other Fe-Fur target promoters in H. pylori (our unpublished results).
Fig 3.
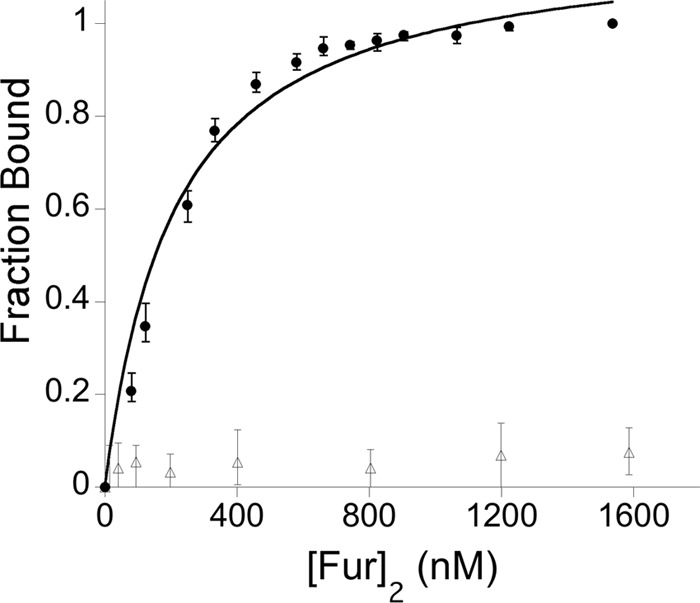
rFur binds specifically to the oorDABC Fur box. Fluorescently labeled PoorDABC DNA templates with the WT Fur box sequence (closed circles) or with a scrambled Fur box sequence (open triangles) were incubated with increasing concentrations of rFur. Fur binds specifically to the Fur box in the wild-type PoorDABC template but not to the scrambled sequence. After fitting to an appropriate binding model, the dissociation constant (Kd) was determined to be 200 nM. The y axis represents the fraction of labeled oligonucleotide duplex bound by Fur, and the x axis indicates the concentration of Fur dimer, [Fur]2.
The Fur box upstream of the oorDABC core promoter is essential for transcriptional activation.
While the binding of Fe-Fur to the Fur box in the oorDABC promoter region is consistent with Fur-dependent activation, we next wanted to ensure that Fur binding to this Fur box was in fact required for transcriptional activation at this promoter. To this end, we utilized plasmid-based promoter fusions to determine what, if any, effect loss of the Fur box had on transcription from the oorDABC promoter. The first of these fusion reporters contained the full-length wild-type promoter fused to a chloramphenicol acetyltransferase reporter gene (cat), while the other plasmid contained a slightly truncated promoter region that did not contain the putative Fur box. Each of these reporters was moved into both wild-type and fur mutant strains, and the contribution of the putative Fur box to activation of oorDABC was quantitated by measuring expression of cat by qRT-PCR in three biologically independent experiments (Table 3). First, we compared the levels of cat expression from the wild-type oorDABC promoter (PoorDABC) and Fur box deletion reporter [PoorDABC(−FB)] fusions in the wild-type and fur mutant strain backgrounds. Similar to the qRT-PCR results described above, and as expected if Fur was required for activation of expression, the level of cat expression from the wild-type oorDABC promoter (PoorDABC) was higher in the wild-type strain than in the fur mutant (mean fold activation of 3.7; P = 0.05, Student's t test) (Table 3). As expected, this difference in cat expression was not seen when the reporter plasmids did not contain the Fur box. In contrast, the PoorDABC(−FB) fusion was expressed at a slightly higher level in the fur mutant strain. When the expression of the fusions was compared only within the same strain background, the levels of cat expression were dramatically higher in the wild-type strain when the Fur box was present in the fusion (mean fold change of 27.2-fold higher). Since there was a high degree of variability in the degree of increase in reporter expression, this result was not statistically significant but showed a strong trend toward significance (P = 0.07). This increased level of expression required Fur, since a similar increase was not seen when these fusions were carried in the fur mutant strain; instead, the level of cat expression from the PoorDABC(−FB) fusion was dramatically higher in this strain background (Table 3). Collectively, the results from these experiments confirm our previous finding that oorDABC activation is Fur dependent and provide new evidence that activation requires the Fur box located in the −130 region of this promoter.
Table 3.
Activation of oorDABC in different strains of H. pyloria
| Strain | Fold activation |
|||
|---|---|---|---|---|
| Bio1 | Bio2 | Bio3 | Mean | |
| WT PoorDWT/Δfur PoorDWTstrain | 4.8 | 1.8 | 4.5 | 3.7* |
| WT PoorD(−FB)/Δfur PoorD(−FB)strain | 0.15 | 2 | 0.45 | 0.87 |
| WT PoorDWT/WT PoorD(−FB)strain | 6.7 | 50 | 25 | 27.2 |
| Δfur PoorDWT/Δfur PoorD(−FB)strain | 0.21 | 0.08 | 0.07 | 0.12* |
Activation was measured as cat reporter gene expression in WT or Δfur strains carrying oorD reporter plasmids with wild-type or mutated promoter sequences. *, statistically significant by Student's t test (P ≤ 0.05); Bio1, Bio2, Bio3, biologically independent experiments; WT, wild-type strain; PoorDWT, wild-type promoter; PoorD(−FB), Δfur box promoter; Δfur, fur deletion strain.
DISCUSSION
The ability to survive in the gastric mucosa requires that H. pylori be able to efficiently respond to changes in environmental conditions such as pH and fluctuation in nutrients. As with other bacterial pathogens, the major way that H. pylori adapts to changes within the host is by altering gene expression in response to external stimuli. However, unlike many other well-studied pathogens, H. pylori lacks the repertoire of regulatory proteins that usually facilitate these changes. Rather, H. pylori alters gene expression using only a few dedicated transcriptional regulators (4, 37). As a result, the regulatory proteins that are encoded by the H. pylori genome have evolved to function outside their canonical regulatory roles. One of these regulatory proteins that display extended function is Fur. Arguably the “master regulator” in H. pylori, Fur has been shown to repress and activate transcription in both the iron-cofactored and apo forms of the protein (1, 5, 9, 12, 14, 16, 17, 24). Of these types of regulation, Fe-dependent activation has been understudied; only nifS has previously been shown to be Fe-Fur activated in H. pylori (1). Therefore, to extend our knowledge of this type of regulation, here we characterize the role of Fur in the activation of oorDABC expression, which was previously suggested to be activated under iron-replete conditions (1).
The oorDABC genes constitute a single transcriptional unit that encodes a 2-oxoglutarate oxidoreductase (OOR) enzyme. This enzyme catalyzes the formation of succinyl-CoA, which is a major intermediate in carbon metabolism. In addition to generating succinyl-CoA, the oxidative decarboxylation of α-ketoglutarate (2-oxoglutarate) results in the transfer of electrons through the respiratory electron chain via NADPH (28). Thus, OOR function also plays a key role in both amino acid degradation/recycling and in maintaining the energy reserves within the bacterial cell. Since this catalytic step feeds into multiple physiologically important cellular processes, ensuring that this reaction occurs only when conditions are most favorable is critical for maintaining metabolic homeostasis within the cell. One way of controlling this reaction is to utilize environmental conditions as a signal to transcriptionally regulate the genes that encode the OOR enzyme complex. Given that it functions as a sensor of iron availability, Fur could be considered a surrogate for detection of nutritional availability in the environment: when iron levels are high, it is likely that other nutrients are available as well. Thus, by activating expression of the oorDABC genes when iron is abundant, H. pylori is able to prime a key component of its central metabolism and maximize efficiency while nutrients are readily available. In contrast, when nutrients are not readily available and iron is depleted, it is less advantageous to continue transcribing the oorDABC genes at high levels; thus, expression of these genes is reduced to a basal level. While Fur activation at this particular step in carbon metabolism has to yet be described in other organisms, Fur has been shown to activate expression of other metabolic genes in Neisseria meningitidis (15). Thus, the utilization of iron availability as a sensor of environmental growth conditions has apparently evolved in other bacterial pathogens.
The mechanism by which Fe-Fur represses transcription has been well characterized and has been shown to typically involve binding of Fur in the −10 and/or −35 region of the regulated promoter. This binding physically prevents the RNA polymerase (RNAP) from interacting with the promoter and initiating transcription (2, 14, 16, 18, 27). In contrast, the process of Fe-Fur activation occurs significantly upstream of the RNAP binding site. In this regard, Fe-Fur activation mimics activation by another H. pylori metalloregulator, NikR, which can also bind upstream of the core promoter elements to activate gene expression (25). For the oorDABC promoter, Fur binds a single distal Fur box (TAATAATCTTTTTA) centered ∼140 bp upstream of the +1; this binding sequence is highly conserved, nearly palindromic, and quite similar to the canonical Fe-Fur binding site defined in H. pylori and other organisms (3, 34). In addition, the highly conserved nature of this binding sequence among other sequenced H. pylori strains suggests that Fur-mediated regulation of oorDABC also occurs in these strains. To date, only one other H. pylori gene has been shown to be directly activated by Fe-Fur. This gene, nifS, encodes a protein involved in iron-sulfur cluster formation and is upregulated under oxidative stress conditions (1). The nifS promoter has been suggested to contain two predicted Fur binding sites; Alamuri et al. used DNase footprinting to show that Fur binds to these two regions that lie ∼175 bp and ∼225 bp upstream of the +1 (1). Interestingly, these reported Fur binding regions do not contain any similarity to the canonical Fe-Fur binding site recently identified for H. pylori (34). Fur activation at studied N. meningitidis promoters also occurs by binding to distal Fur boxes (binding sites were typically centered between the −80 and −100 regions) (15). Thus, promoters of Fe-Fur-activated genes tend to contain Fur boxes that lie distal to the core promoter elements, perhaps suggesting a similar activation mechanism at the molecular level.
The obvious question then becomes, “how does activation occur mechanistically?” While the answer to this question is not immediately obvious, several possibilities exist. First, Fur could have a direct physical role in recruiting the RNAP complex to the core promoter elements. However, this type of activation seems unlikely, given that Fur binds to the distal promoter regions, which would place the protein at such a distance that it would be unlikely to physically interact with RNAP. Second, Fur binding to the distal Fur box may induce a conformational change in the promoter DNA architecture that subsequently enhances either assembly of the RNAP complex or the process of transcription initiation. Finally, perhaps Fur binding upstream of the oorDABC promoter region modulates the function of another regulatory protein. Given that these genes are Fe-Fur activated in this scenario, binding of Fur would most likely block binding of a repressor protein. While we cannot currently rule out the first two mechanisms, previous studies suggest that Fur-dependent activation at the oorDABC promoter may in fact be the result of competition between two regulatory proteins.
Previous pH-dependent transcriptional profiling of H. pylori showed that expression of the OOR genes is activated at low pH (26, 38, 39). This pH-mediated activation is dependent upon the acid-responsive regulatory two-component system ArsRS (38). In this system, ArsS serves as the sensor kinase that senses low pH and initiates the phosphorelay that leads to phosphorylation of the response regulator ArsR. ArsR has been shown to regulate expression of numerous genes and appears to have the ability to interact with the promoters of some genes in the nonphosphorylated form while interacting with others only when phosphorylated (32, 33, 38). Previous electrophoretic mobility shift assays (EMSAs) showed that ArsR directly binds to the oorDABC promoter region and that this reaction did not require ArsR phosphorylation (38). Thus, in a normally growing cell at neutral pH, ArsR should exist primarily in the nonphosphorylated state and be able to bind the oorDABC promoter. Previous DNase I footprinting experiments using ArsR and ArsR target promoters highlighted an AT-rich region that is necessary for binding (19). Within this ArsR binding region, the authors identified two conserved DNA sequences (ATCATT and ATTAAA) that are important for binding (19). Mutation of the ATCATT sequence abrogated ArsR binding, whereas mutation of the ATTAAA sequence reduced binding affinity. As shown in Fig. 4B, the Fur binding sequence we identified in the oorDABC promoter region also contains the requisite ArsR core binding sequence (ATCATT), and this sequence directly overlaps the Fur box. Additionally, the secondary binding sequence (ATTAAA), with one nucleotide difference, is found slightly downstream from the Fur box. Thus, taken together these data support a proposed model wherein Fur and ArsR compete for binding at the oorDABC promoter; binding of Fur to the promoter would block binding of the ArsR protein, which likely acts as a repressor at this promoter. Thus, the Fur-mediated activation of oorDABC expression likely occurs via an antirepression mechanism (Fig. 4). This model could also explain the higher level of expression we observed when the PoorDABC(−FB) fusion was carried in the fur mutant strain; deletion of the Fur box also removes the sequence required for ArsR-mediated repression. Thus, the levels of oorDABC expressed are higher (Table 3). Since the Fur and ArsRS regulons share many genes in common and the proteins have similar binding sequences, it is possible that this type of regulatory interplay is a common phenomenon in H. pylori. Indeed, previous studies identified promoters that are subject to regulatory competition. For example, the exbB promoter region contains conserved binding sites for both Fur and the nickel-responsive regulator, NikR, and these two proteins bind to overlapping regions within this promoter (13). Thus, competition for binding sites among H. pylori regulators does occur, although the extent of competition between these proteins is for the most part underappreciated.
Fig 4.

Proposed model of Fur-dependent activation of oorDABC expression. (A) Under iron-replete conditions at normal pH, both Fur and ArsR compete for an overlapping binding site within the oorDABC promoter region. In the absence of a signal from the ArsR histidine kinase, ArsR may act as a repressor and bind to a region that overlaps the Fur box within the oorDABC promoter. When iron is abundant, Fur outcompetes ArsR for binding at this site, which derepresses oorDABC expression and results in increased oorDABC transcription. (B) Previously identified ArsR core binding sequences (bold text in boxes) overlap the Fur box (underlined) identified in this study. The overlap in these regions highlights the possibility of competition between Fur and ArsR.
In summary, we have shown Fur-mediated activation of the oorDABC genes, which encode the OOR enzyme in H. pylori (Fig. 1). We found that expression from this promoter occurs from a single start site (Fig. 2) and that Fur binds specifically to a conserved Fur box located ∼130 bp upstream of the core promoter region (Fig. 2). Furthermore, binding to this Fur box is required for activation of oorDABC gene expression (Table 3). These findings add to the growing complexity of Fur regulation in H. pylori. Although the precise mechanism of Fe-Fur activation is not yet clear, it likely occurs through an antirepression mechanism wherein Fur binding blocks binding of ArsR; we are currently investigating the possibility of direct competition between Fur and ArsR as a common regulatory phenomenon in this pathogen.
ACKNOWLEDGMENTS
Research in the laboratory of D. Scott Merrell is made possible by grant AI065529 from the NIAID.
The contents of this report are the sole responsibility of the authors and do not necessarily represent the official views of the DoD or the NIH.
Footnotes
Published ahead of print 21 September 2012
REFERENCES
- 1. Alamuri P, Mehta N, Burk A, Maier RJ. 2006. Regulation of the Helicobacter pylori Fe-S cluster synthesis protein NifS by iron, oxidative stress conditions, and fur. J. Bacteriol. 188:5325–5330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bagg A, Neilands JB. 1987. Ferric uptake regulation protein acts as a repressor, employing iron (II) as a cofactor to bind the operator of an iron transport operon in Escherichia coli. Biochemistry 26:5471–5477 [DOI] [PubMed] [Google Scholar]
- 3. Baichoo N, Helmann JD. 2002. Recognition of DNA by Fur: a reinterpretation of the Fur box consensus sequence. J. Bacteriol. 184:5826–5832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baltrus DA, et al. 2009. The complete genome sequence of Helicobacter pylori strain G27. J. Bacteriol. 191:447–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bereswill S, et al. 2000. Regulation of ferritin-mediated cytoplasmic iron storage by the ferric uptake regulator homolog (Fur) of Helicobacter pylori. J. Bacteriol. 182:5948–5953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bijlsma JJ, et al. 2002. The Helicobacter pylori homologue of the ferric uptake regulator is involved in acid resistance. Infect. Immun. 70:606–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blaser MJ. 1998. Helicobacter pylori and gastric diseases. BMJ 316:1507–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bury-Mone S, et al. 2004. Responsiveness to acidity via metal ion regulators mediates virulence in the gastric pathogen Helicobacter pylori. Mol. Microbiol. 53:623–638 [DOI] [PubMed] [Google Scholar]
- 9. Carpenter BM, et al. 2009. A single nucleotide change affects fur-dependent regulation of sodB in H. pylori. PLoS One 4:e5369 doi:10.1371/journal.pone.0005369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carpenter BM, et al. 2007. Expanding the Helicobacter pylori genetic toolbox: modification of an endogenous plasmid for use as a transcriptional reporter and complementation vector. Appl. Environ. Microbiol. 73:7506–7514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carpenter BM, Whitmire JM, Merrell DS. 2009. This is not your mother's repressor: the complex role of fur in pathogenesis. Infect. Immun. 77:2590–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Danielli A, et al. 2006. In vivo dissection of the Helicobacter pylori Fur regulatory circuit by genome-wide location analysis. J. Bacteriol. 188:4654–4662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Delany I, et al. 2005. In vitro analysis of protein-operator interactions of the NikR and fur metal-responsive regulators of coregulated genes in Helicobacter pylori. J. Bacteriol. 187:7703–7715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Delany I, Pacheco AB, Spohn G, Rappuoli R, Scarlato V. 2001. Iron-dependent transcription of the frpB gene of Helicobacter pylori is controlled by the Fur repressor protein. J. Bacteriol. 183:4932–4937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Delany I, Rappuoli R, Scarlato V. 2004. Fur functions as an activator and as a repressor of putative virulence genes in Neisseria meningitidis. Mol. Microbiol. 52:1081–1090 [DOI] [PubMed] [Google Scholar]
- 16. Delany I, Spohn G, Rappuoli R, Scarlato V. 2003. An anti-repression Fur operator upstream of the promoter is required for iron-mediated transcriptional autoregulation in Helicobacter pylori. Mol. Microbiol. 50:1329–1338 [DOI] [PubMed] [Google Scholar]
- 17. Delany I, Spohn G, Rappuoli R, Scarlato V. 2001. The Fur repressor controls transcription of iron-activated and -repressed genes in Helicobacter pylori. Mol. Microbiol. 42:1297–1309 [DOI] [PubMed] [Google Scholar]
- 18. de Lorenzo V, Wee S, Herrero M, Neilands JB. 1987. Operator sequences of the aerobactin operon of plasmid ColV-K30 binding the ferric uptake regulation (fur) repressor. J. Bacteriol. 169:2624–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dietz P, Gerlach G, Beier D. 2002. Identification of target genes regulated by the two-component system HP166-HP165 of Helicobacter pylori. J. Bacteriol. 184:350–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dosanjh NS, Hammerbacher NA, Michel SL. 2007. Characterization of the Helicobacter pylori NikR-P(ureA) DNA interaction: metal ion requirements and sequence specificity. Biochemistry 46:2520–2529 [DOI] [PubMed] [Google Scholar]
- 21. Dosanjh NS, Michel SL. 2006. Microbial nickel metalloregulation: NikRs for nickel ions. Curr. Opin. Chem. Biol. 10:123–130 [DOI] [PubMed] [Google Scholar]
- 22. Dunn BE, Cohen H, Blaser MJ. 1997. Helicobacter pylori. Clin. Microbiol. Rev. 10:720–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ernst FD, et al. 2005. Transcriptional profiling of Helicobacter pylori Fur- and iron-regulated gene expression. Microbiology 151:533–546 [DOI] [PubMed] [Google Scholar]
- 24. Ernst FD, et al. 2005. Iron-responsive regulation of the Helicobacter pylori iron-cofactored superoxide dismutase SodB is mediated by Fur. J. Bacteriol. 187:3687–3692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ernst FD, et al. 2005. The nickel-responsive regulator NikR controls activation and repression of gene transcription in Helicobacter pylori. Infect. Immun. 73:7252–7258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gancz H, Censini S, Merrell DS. 2006. Iron and pH homeostasis intersect at the level of Fur regulation in the gastric pathogen Helicobacter pylori. Infect. Immun. 74:602–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gilbreath JJ, Cody WL, Merrell DS, Hendrixson DR. 2011. Change is good: variations in common biological mechanisms in the epsilonproteobacterial genera Campylobacter and Helicobacter. Microbiol. Mol. Biol. Rev. 75:84–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hughes NJ, Clayton CL, Chalk PA, Kelly DJ. 1998. Helicobacter pylori porCDAB and oorDABC genes encode distinct pyruvate:flavodoxin and 2-oxoglutarate:acceptor oxidoreductases which mediate electron transport to NADP. J. Bacteriol. 180:1119–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lakowicz JR. 1999. Principles of fluorescence spectroscopy, 2nd ed Kluwer Academic/Plenum Publishers, Dordrecht, Netherlands [Google Scholar]
- 30. Miles S, et al. 2010. Detailed in vivo analysis of the role of Helicobacter pylori Fur in colonization and disease. Infect. Immun. 78:3073–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Petersen L, Larsen TS, Ussery DW, On SL, Krogh A. 2003. RpoD promoters in Campylobacter jejuni exhibit a strong periodic signal instead of a -35 box. J. Mol. Biol. 326:1361–1372 [DOI] [PubMed] [Google Scholar]
- 32. Pflock M, Dietz P, Schar J, Beier D. 2004. Genetic evidence for histidine kinase HP165 being an acid sensor of Helicobacter pylori. FEMS Microbiol. Lett. 234:51–61 [DOI] [PubMed] [Google Scholar]
- 33. Pflock M, et al. 2006. Characterization of the ArsRS regulon of Helicobacter pylori, involved in acid adaptation. J. Bacteriol. 188:3449–3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pich OQ, Carpenter BM, Gilbreath JJ, Merrell DS. 2012. Detailed analysis of Helicobacter pylori Fur-regulated promoters reveals a Fur box core sequence and novel Fur-regulated genes. Mol. Microbiol. 84:921–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qu W, et al. 2009. Helicobacter pylori proteins response to nitric oxide stress. J. Microbiol. 47:486–493 [DOI] [PubMed] [Google Scholar]
- 36. Sharma CM, et al. 2010. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464:250–255 [DOI] [PubMed] [Google Scholar]
- 37. Tomb JF, et al. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547 [DOI] [PubMed] [Google Scholar]
- 38. Wen Y, Feng J, Scott DR, Marcus EA, Sachs G. 2006. Involvement of the HP0165-HP0166 two-component system in expression of some acidic-pH-upregulated genes of Helicobacter pylori. J. Bacteriol. 188:1750–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wen Y, et al. 2003. Acid-adaptive genes of Helicobacter pylori. Infect. Immun. 71:5921–5939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yanofsky C, et al. 1981. The complete nucleotide sequence of the tryptophan operon of Escherichia coli. Nucleic Acids Res. 9:6647–6668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zheleznova EE, Crosa JH, Brennan RG. 2000. Characterization of the DNA- and metal-binding properties of Vibrio anguillarum fur reveals conservation of a structural Zn(2+) ion. J. Bacteriol. 182:6264–6267 [DOI] [PMC free article] [PubMed] [Google Scholar]