

Abstract
The maintenance of adequate tissue O2 levels in skeletal muscle is vital for normal physiology and requires a well regulated and appropriately distributed convective O2 supply. Inherent in this fundamental physiological process is the requirement for a mechanism which both senses tissue O2 need and locally adjusts flow to appropriately meet that need. Over the past several years we and others have suggested that, in skeletal muscle, O2 carrying erythrocytes participate in the regulation of total blood flow and its distribution by releasing ATP. Importantly, the release of this vasoactive molecule must be both rapid and well controlled if it is to serve an important physiological role. Here we provide insights into three distinct regulated signalling pathways within the erythrocyte that are activated by exposure to reduced O2 tension or in response to binding of agonists to the prostacyclin or β-adrenergic receptors. Although much has been learned about the role of the erythrocyte in perfusion of skeletal muscle, much remains to be understood. However, what is clear is that the long established passive carrier of O2 also contributes to the regulation of the distribution of microvascular perfusion in skeletal muscle by virtue of its capacity to release ATP.

Mary L. Ellsworth, PhD (Albany Medical College) and Randy S. Sprague, MD (St Louis University) have collaborated on studies related to the role of ATP released from erythrocytes in the regulation of the distribution of microvascular perfusion. In association with colleagues and students from the US and Canada, they have described the effect of low oxygen tension on erythrocyte ATP release and the impact of that release on vascular function and oxygen supply, and have characterized the signal transduction pathways which regulate the ATP release in response to both physiological and pharmacological stimuli. Recent studies have focused on the implications of defects in these release pathways on oxygen transport in a number of pathological conditions including diabetes, prediabetes and pulmonary hypertension.
The concept that blood gains a ‘vital essence’ in the lungs which it then transports throughout the body has been considered doctrine since it was first described by Greek philosophers of the Ionian school in Asia Minor in the 6th century BC (Cournand, 1982). However, it was not until 1628 that a detailed description of the systemic circulation and the observation that blood is pumped by the heart were provided by the English physician William Harvey (1889). Thirty years later, the Dutch biologists Jan Swammerdam and Anton van Leeuwenhoek each reported that they had visualized erythrocytes, seeing them as flexible discs in a ‘milky’ medium (Bessis & Delpech, 1981). Despite acknowledgement that erythrocytes were present in the circulation, it was not until 1840 that the oxygen (O2) carrying protein they contain, haemoglobin, was discovered by F. L. Hünefeld (1840). The reversible oxygenation and competitive binding of O2 to haemoglobin was detailed a few years later by Felix Hoppe-Seyler (1866). For most of the next century, the erythrocyte was considered to be a flexible, haemoglobin containing cell whose sole purpose is to carry oxygen from the lungs to the tissues.
In a 1914 book entitled The Respiratory Function of the Blood, Joseph Barcroft stated that ‘the cell takes what it needs and leaves the rest’. Barcroft then suggested that it is only logical that the following sequence must occur. ‘Firstly the call for oxygen, secondly the mechanism by which the call elicits a response, the immediate response consisting of the carriage of oxygen to the tissue by the blood and its transference from the blood to the cell. Thirdly in the background you have the further mechanism by which the blood acquires its oxygen’ (Barcroft, 1914). Barcroft explored possible scenarios to explain how these three components are able to satisfy the tissues’‘call for oxygen’ but none could account for the complexity of the response.
As Christian Bohr's assistant, August Krogh became interested in problems connected with gas exchange in the living organism in the early part of the 20th century. His studies demonstrated an increased utilization of the O2 of the blood during muscular work, which he explained by an increase in the diffusion surface resulting from the opening of previously closed capillaries of skeletal muscle (Krogh, 1959). However he was unable to determine a mechanism by which perfusion distribution was altered to precisely meet the metabolic needs of that tissue. Since the time of Krogh, investigations have revealed that there is not a preponderance of closed capillaries in resting skeletal muscle which would be opened during exercise (Poole et al. 2011) and that, in addition, the capillary is not the sole site of oxygen transfer (Ellsworth et al. 2009). These findings make the determination of factors responsible for appropriate oxygen supply significantly more complex. In the intervening years, most studies have focused on mechanisms by which the resistance vessels increase and decrease their diameter via alterations in the activity of the autonomic nervous system and/or levels of vasoactive mediators. Although both mechanisms are likely to contribute to overall tissue perfusion, neither appears capable of the exquisite control necessary for the optimal matching of O2 supply with demand in skeletal muscle.
Release of ATP from erythrocytes in response to reduced O2 tension
A 1993 study of capillary O2 transport in hamster skeletal muscle (Stein & Ellsworth, 1993) set the stage for a series of studies exploring the possibility that the erythrocyte, by virtue of its capacity to release the vasoactive mediator ATP in response to a decrease in O2 saturation, could serve as the mechanism by which Barcroft's ‘call for oxygen’ might be answered. In an earlier paper, Bergfeld & Forrester (1992) showed that ATP was released from healthy human erythrocytes upon exposure to reduced O2 tension in the presence of hypercapnia. It has subsequently been established that isolated human, rat, rabbit and hamster erythrocytes release ATP when exposed to reduced O2 tension in the presence of normal levels of CO2 indicating that it is the decrease in O2 which is the driving force for ATP release (Ellsworth et al. 1995; Ellsworth, 2000; Dietrich et al. 2000). It has subsequently been suggested that ATP release is evoked by the conformational change in the membrane associated haemoglobin molecules which occurs as they desaturate in response to exposure to reduced O2 levels (Jagger et al. 2001) with the optimal response in the range of the animal's P50 (Sprague & Ellsworth, 2012).
ATP, once released from the erythrocyte, can bind to purinergic receptors located on the vascular endothelium, which initiates a conducted vasodilator response (McCullough et al. 1997; Collins et al. 1998) augmenting blood flow to the specific local tissue region in need (Sprague et al. 2010). Thus, the mobile erythrocyte, by releasing both O2 and ATP in regions of low O2 tension contributes to the optimization of O2 delivery to skeletal muscle enabling it to function appropriately.
The egress of ATP from erythrocytes upon exposure to low O2 tension occurs by a mechanism that is both rapid and well regulated (Dietrich et al. 2000; Wan et al. 2008). Critical for this release is the activation of a discrete signalling pathway within the erythrocyte likely initiated by the conformational change induced by the offloading of O2 from membrane-associated haemoglobin (Jagger et al. 2001; Wan et al. 2008; Sridharan et al. 2010b; Forsyth et al. 2011). Key components of this signalling pathway include the heterotrimeric G protein, Gi (Sprague et al. 2002; Olearczyk et al. 2004a), adenylyl cyclase (Sprague et al. 1998, 2001, 2008), protein kinase A (PKA) (Sprague et al. 2001), the cystic fibrosis transmembrane conductance regulator (CFTR) (Sprague et al. 1998) and pannexin 1, which serves as the final ATP conduit (Sridharan et al. 2010a). The release of ATP via this pathway requires an increase in intracellular cAMP. The level of this second messenger is controlled by the relative activities of adenylyl cyclase and phosphodiesterase 3. The details of this pathway are depicted in Fig. 1 and have been the subject of several recent reviews (Ellsworth et al. 2009; Sprague et al. 2011; Sprague & Ellsworth, 2012). ATP release in response to exposure to low O2 tension has been suggested to contribute to the appropriate distribution of perfusion in skeletal muscle. However, ATP release from erythrocytes also occurs when these cells are exposed to the endogenous vasoactive mediators, prostacyclin (PGI2) (Olearczyk et al. 2001; Sprague et al. 2008) and adrenaline (Olearczyk et al. 2001; Sprague et al. 2001), both of which can be released locally into the circulation.
Figure 1. Proposed signalling pathway for low O2-induced ATP release from erythrocytes.
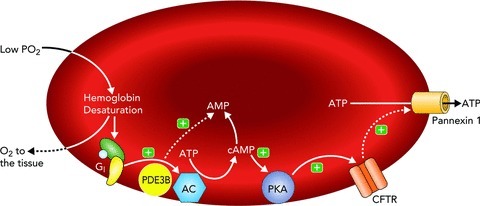
When exposed to low O2, membrane-associated haemoglobin releases O2. The associated conformational change in the haemoglobin molecule associated with its desaturation results in local membrane deformation and activation of Gi. This leads to activation of AC and increase in cAMP, the level of which is regulated by PDE3B activity. Increases in cAMP activate PKA and, subsequently, CFTR. The final conduit for ATP release in this pathway is pannexin 1. Abbreviations: Gi, heterotrimeric G protein; PDE3B, phosphodiesterase 3B; cAMP, cyclic adenosine monophosphate; AMP, adenosine monophosphate; AC, adenylyl cyclase; PKA, protein kinase A; CFTR, cystic fibrosis transmembrane conductance regulator; ATP, adenosine triphosphate; (+), activation; (−), inhibition.
Receptor mediated ATP release from erythrocytes: prostacyclin
In microvessels, increased endothelial shear stress (Ngai & Yao, 2010), as would occur with an increase in the velocity of blood flow, results in the synthesis and/or release of vasodilators, which include nitric oxide (NO) and several products of arachidonic acid metabolism, including PGI2 (Koller & Kaley, 1990; Osanai et al. 2000). Both NO and PGI2 stimulate vasodilatation via direct action on vascular smooth muscle. However, in addition, exposure of erythrocytes to PGI2 analogues, including iloprost and UT-15C, stimulates receptor-mediated increases in cAMP synthesis and ATP release from human erythrocytes in a concentration-dependent manner (Sprague et al. 2008). Thus, blood vessels exposed to increased shear stress vasodilate as the result of both direct effects of locally released NO and PGI2 on vascular smooth muscle and the initiation of a conducted vasodilatation secondary to PGI2-mediated release of ATP from erythrocytes.
The regulated release of ATP in response to PGI2 requires that erythrocytes possess the prostacyclin receptor (IPR) and that activation of the IPR initiates a signalling pathway. Over the past several years it has been established that the IPR signalling pathway is distinct from the pathway for ATP release activated by exposure of these cells to reduced O2 tension (Fig. 2). In non-erythrocytes, the IPR is coupled to the heterotrimeric G protein (Gs) and its activation results in increased production of cAMP via adenylyl cyclase. Erythrocytes of humans and rabbits possess both the IPR and the heterotrimeric G protein, Gs, and exposure of these cells to PGI2 analogues results in increases in cAMP and ATP release (Olearczyk et al. 2001, Sprague et al. 2008). That these responses were receptor mediated was confirmed by studies in which increases in cAMP and ATP release were prevented by pre-incubation of erythrocytes with the IPR receptor antagonist CAY10441 (Sprague et al. 2008). In subsequent studies, it was shown that increases in cAMP in this pathway are regulated by the activity of phosphodiesterase 3 (PDE3) (Hanson et al. 2008). This PDE has two distinct isoforms, one cytosolic (PDE3A) and the other membrane bound (PDE3B), both of which are present in human erythrocytes (Hanson et al. 2008, Adderley et al. 2010a, 2011b). PDE3A has been associated with IPR activation in platelets and pulmonary vessels and thus is the PDE3 isoform likely to be associated with this pathway in human erythrocytes.
Figure 2. Proposed signalling pathways for ATP release from erythrocytes via activation of the Gs-coupled IPR or βAR.
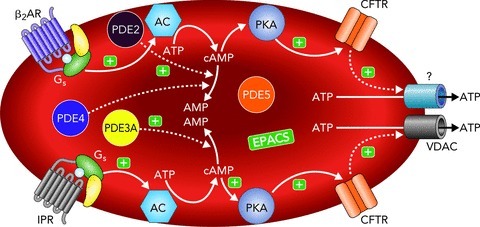
Binding of selective agonists to the IPR or βAR results in activation of Gs and, subsequently, AC leading to increases in cAMP. In the IPR pathway, increases in cAMP are regulated by PDE3 while in the βAR pathway, cAMP is hydrolysed by PDEs 2 and 4. In both pathways increases in cAMP activate PKA and, subsequently, CFTR. The final conduit for ATP release in the IPR signalling pathway is VDAC while the distinct channel responsible for ATP release in the βAR signalling pathway remains to be determined. Abbreviations: β2AR, β2-adrenergic receptor; IPR, prostacyclin receptor; Gs, heterotrimeric G protein; PDE3A, phosphodiesterase 3A; PDE2, phosphodiesterase 2; PDE4, phosphodiesterase 4; cAMP, cyclic adenosine monophosphate; AMP, adenosine monophosphate; AC, adenylyl cyclase; PKA, protein kinase A; CFTR, cystic fibrosis transmembrane conductance regulator; VDAC, voltage dependent anion channel; ATP, adenosine triphosphate; (+), activation; (−), inhibition. Both phosphodiesterase 5 (PDE5) (Adderley et al. 2011b) and exchange proteins activated by cAMP (EPACs) (Adderley et al. 2011a) are present in erythrocytes and their interaction with the IPR and βAR signalling pathways is under investigation.
In many intracellular signalling pathways, protein kinase A (PKA) is phosphorylated (activated) by cAMP. Activation of both PKA and PKC occurs in the IPR signalling pathway in rabbit erythrocytes (Adderley et al. 2010b). Although PKA and PKC have numerous intracellular targets, their role in the activation of CFTR is crucial for IPR-mediated ATP release from human erythrocytes (Sprague et al. 1998; Sridharan et al. 2012). Although previously thought to be an ATP conduit, more recent evidence supports the hypothesis that CFTR regulates the movement of anions such as chloride into and out of cells (Gadsby et al. 2006). The influx of an anion is essential for the release of the highly negatively charged ATP to maintain the ionic balance of the erythrocyte (Bergfeld & Forrester, 1992). Recently, the conduit for ATP release from human erythrocytes in response to IPR activation has been identified as the voltage-dependent anion channel (VDAC) (Sridharan et al. 2012).
Comparison of the low O2- and prostacyclin-induced pathways for ATP release from erythrocytes
Although the release of ATP in response to endothelial-derived PGI2 would amplify a local vasodilatation, such a response would be less effective in directing microvascular perfusion to regions of O2 need in skeletal muscle. Indeed, computational modelling has confirmed that general vasodilatation, such as that produced by PGI2, is less effective in providing appropriate amounts of O2 to skeletal muscle when compared with low O2-induced ATP release (Sprague et al. 2010). The incorporation of a functional regulatory system that directs blood flow only to areas of need provides optimal tissue oxygenation by eliminating the likely oversupply of O2 to regions which were already adequately supplied, a theoretical consequence of non-directed general vasodilatation. However, the observation that erythrocytes release ATP in response to both low O2 exposure (directed vasodilatation) and prostacyclin (general vasodilatation) places these cells in a unique position to contribute to the dramatic increase in total skeletal muscle blood flow during exercise and, simultaneously, direct that flow to specific tissue regions. Importantly, the dual mechanisms provide for a redundant control mechanism should one of the signalling pathways be impaired, as can occur under pathological conditions (Sprague & Ellsworth, 2012).
Although these two mechanisms for ATP release from erythrocytes can work together to augment blood flow to skeletal muscle, the signalling pathways responsible for that release are distinct (Table 1). In spite of the fact that in both pathways ATP release requires increases in cAMP and activation of CFTR (Sprague et al. 1998, 2008; Sridharan et al. 2010), the G protein involved in initiating the response is different. In the low O2 pathway Gi activation is required (Sprague et al. 2002; Olearczyk et al. 2004b) while binding of an agonist to IPR activates Gs (Olearczyk et al. 2001). In addition, it is now clear that the final conduits for ATP release in response to low O2 exposure and IPR activation are likewise distinct having been identified as pannexin 1 (Sridharan et al. 2010a) and VDAC (Sridharan et al. 2012), respectively.
Table 1.
Distinguishing characteristics of discrete signalling pathways for ATP release from erythrocytes
| Signalling pathway | Heterotrimeric G protein | Phosphodiesterase(s) | Protein kinases | Final ATP conduit |
|---|---|---|---|---|
| Low O2 Mechanical Stress | Gi | PDE3 (B?) | PKA | Pannexin 1 |
| IPR | Gs | PDE3 (A?) | PKA and PKC | VDAC |
| βAR | Gs | PDE2 and PDE4 | PKA | Unknown |
Abbreviations: IPR, prostacyclin receptor; βAR, β-adrenergic receptor; VDAC, voltage dependent anion channel.
The mechanism of receptor-mediated activation of Gs in the IPR signalling pathway has been well defined (Nobles et al. 2005). In contrast, the mechanism by which exposure to low O2 stimulates Gi is less well understood. It is clear that mechanical deformation of erythrocytes results in ATP release by activation of a signalling pathway that appears to be identical to the pathway activated by exposure of erythrocytes to low O2 (Olearczyk et al. 2004a,b). This finding led to the hypothesis that upon exposure of erythrocytes to low O2, the desaturation of the haemoglobin molecules associated with the erythrocyte membrane produces a local membrane deformation (Wan et al. 2008) which results in the activation of Gi. A similar mechanism for Gi activation has been described in smooth muscle cells (Li & Xu, 2000). Studies in which low O2-induced ATP release was prevented by either decreased membrane deformability (Sridharan et al. 2010b) or by incubation of erythrocytes with carbon monoxide to hold haemoglobin in the ‘oxygenated’ conformation (Jagger et al. 2001) provide strong support for this mechanism.
The IPR and low O2 signalling pathways are also distinct in their response to exogenous NO. Exposure of erythrocytes to the NO donor spermine NONOate results in inhibition of low O2-induced ATP release (Olearczyk et al. 2004b). Although the exact mechanism responsible for this inhibition has yet to be confirmed, it was suggested that NO might stimulate ADP-ribosylation of the α subunit of Gi (Olearczyk et al. 2004b) thereby preventing its activation and thus initiation of the signalling cascade for ATP release. In contrast, NO does not inhibit IPR-mediated ATP release from erythrocytes (Olearczyk et al. 2004a).
Receptor mediated ATP release from erythrocytes: β-adrenergic activation
In addition to pathways for ATP release that are activated by exposure to low O2, mechanical deformation, or binding of an agonist to the IPR, activation of β-adrenergic receptors (βARs) on erythrocytes also stimulates increases in cAMP and ATP release (Fig. 2). It has been known for some time that erythrocytes possess βARs and available evidence suggests that the predominant receptor expressed is β2AR (Horga et al. 2000). The βAR signalling pathway for ATP release from erythrocytes is distinct from the other well characterized signalling pathways in that the PDEs that regulates increases in cAMP levels are PDEs 2 and 4 (Adderley et al. 2009). It is important to note that amounts of ATP released in response to incubation of erythrocytes with the βAR agonists isoproterenol or adrenaline are not as great as that associated with activation of the IPR signalling pathway (Olearczyk et al. 2001). Moreover, it is not clear what physiological role βAR-mediated ATP release from erythrocytes plays. It has been suggested that βAR signalling in mature erythrocytes represents residual activity that remains from signalling pathways that were important for erythrocyte maturation prior to their release into the circulation (Kaiser et al. 1974; Rodan et al. 1975).
Perspective
The distribution of a ‘vital essence’ first suggested by Ionian philosophers in the 6th century BC has become universally accepted as a basic physiological concept (Cournand, 1982). Barcroft, in the early 20th century, suggested that the tissue must send out a ‘call for oxygen’ which the circulation provides (1914). Recent research suggests that the erythrocyte, first described in the 1600s (Bessis & Delpech, 1981), serves an important role in helping to answer that call. This mobile cell, by virtue of its ability to respond to both physiological and pharmacological stimuli by releasing ATP into the vasculature, enhances flow to specific tissue regions. The release of ATP in tissue regions with low O2 tension directs flow to that specific region via the initiation of a conducted vasodilator response. An increase in wall shear stress induces the release of PGI2 from the endothelial cells, which via its direct action on smooth muscle cells, induces a local dilatation. Importantly, this released PGI2 also augments the release of ATP from the erythrocyte further contributing to ATP-induced increases in blood flow. This redundancy, a hallmark of important physiological processes, suggests that the erythrocyte, although not the sole controller of total perfusion and its distribution within skeletal muscle, does serve a critical role possibly as a mechanism to fine tune the distribution of flow to meet local tissue need.
Acknowledgments
The authors thank Kelly Paralis of Penumbra Design, Inc. for creating the figures and Dr Hans H. Dietrich for assistance in translating the German papers. Original work from the authors’ laboratories was supported by the National Institutes of Health, the American Diabetes Association and United Therapeutics. The authors thank the many students, research assistants and colleagues who contributed to the author's research presented here. In addition, we thank J. L. Sprague for inspiration.
References
- Adderley SP, Dufaux EA, Sridharan M, Bowles EA, Hanson MS, Stephenson AH, Ellsworth ML, Sprague RS. Iloprost- and isoproterenol-induced increases in cAMP are regulated by different phosphodiesterases in erythrocytes of both rabbits and humans. Am J Physiol Heart Circ Physiol. 2009;296:H1617–H1624. doi: 10.1152/ajpheart.01226.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adderley SP, Sprague RS, Stephenson AH, Hanson MS. Regulation of cAMP by phosphodiesterases in erythrocytes. Pharmacol Rep. 2010a;62:475–482. doi: 10.1016/s1734-1140(10)70303-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adderley SP, Sridharan M, Bowles EA, Stephenson AH, Sprague RS, Ellsworth ML. Inhibition of ATP release from erythrocytes: a role for EPACs and PKC. Microcirculation. 2011a;18:128–135. doi: 10.1111/j.1549-8719.2010.00073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adderley SP, Sridharan M, Bowles EA, Stephenson AH, Ellsworth ML, Sprague RS. Protein kinases A and C regulate receptor-mediated increases in cAMP in rabbit erythrocytes. Am J Physiol Cell Physiol. 2010b;298:C587–C593. doi: 10.1152/ajpheart.00975.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adderley SP, Thuet KM, Sridharan M, Bowles EA, Stephenson AH, Ellsworth ML, Sprague RS. Identification of cytosolic phosphodiesterases in the erythrocyte: a possible role for PDE5. Med Sci Monit. 2011b;17:CR241–CR247. doi: 10.12659/MSM.881763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcroft J. The Respiratory Function of the Blood. Cambridge: Cambridge University Press; 1914. [Google Scholar]
- Bergfeld GR, Forrester T. Release of ATP from human erythrocytes in response to a brief period of hypoxia and hypercapnia. Cardiovasc Res. 1992;26:40–47. doi: 10.1093/cvr/26.1.40. [DOI] [PubMed] [Google Scholar]
- Bessis M, Delpech Discovery of the red blood cell with notes on priorities and credits of discoveries, past, present and future. Blood Cells. 1981;7:447–480. [PubMed] [Google Scholar]
- Collins DM, McCullough WT, Ellsworth ML. Conducted vascular responses: communication across the capillary bed. Microvasc Res. 1998;56:43–53. doi: 10.1006/mvre.1998.2076. [DOI] [PubMed] [Google Scholar]
- Cournand A. Air and blood. In: Fishman AP, Richards DW, editors. Circulation of the Blood: Men and Ideas. Bethesda, MD: American Physiological Society; 1982. p. 3. [Google Scholar]
- Dietrich HH, Ellsworth ML, Sprague RS, Dacey RG., Jr Red blood cell regulation of microvascular tone through adenosine triphosphate. Am J Physiol Heart Circ Physiol. 2000;278:H1294–H1298. doi: 10.1152/ajpheart.2000.278.4.H1294. [DOI] [PubMed] [Google Scholar]
- Ellsworth ML. The red blood cell as an oxygen sensor: what is the evidence? Acta Physiol Scand. 2000;168:551–559. doi: 10.1046/j.1365-201x.2000.00708.x. [DOI] [PubMed] [Google Scholar]
- Ellsworth ML, Forrester T, Ellis CG, Dietrich HH. The erythrocyte as a regulator of vascular tone. Am J Physiol Heart Circ Physiol. 1995;269:H2155–H2161. doi: 10.1152/ajpheart.1995.269.6.H2155. [DOI] [PubMed] [Google Scholar]
- Ellsworth ML, Ellis CG, Goldman D, Stephenson AH, Dietrich HH, Sprague RS. Erythrocytes: oxygen sensors and modulators of vascular tone. Physiology (Bethesda) 2009;24:107–116. doi: 10.1152/physiol.00038.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsyth AM, Wan J, Owrutsky PD, Abkarian M, Stone HA. Multiscale approach to link red blood cell dynamics, shear viscosity, and ATP release. Proc Natl Acad Sci U S A. 2011;108:10986–10991. doi: 10.1073/pnas.1101315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby DC, Vergani P, Csanády L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440:477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MS, Stephenson AH, Bowles EA, Sridharan M, Adderley S, Sprague RS. Phosphodiesterase 3 is present in rabbit and human erythrocytes and its inhibition potentiates iloprost-induced increases in cAMP. Am J Physiol Heart Circ Physiol. 2008;295:H786–H793. doi: 10.1152/ajpheart.00349.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey W. In: Motion of the Heart and Blood in Animals. Bowie A, translator. London: George Bell and Sons; 1889. [Google Scholar]
- Hoppe-Seyler F. Über die oxydation in lebendem blute. Medizinisch-chemische Untersuchungen. 1866;1:133–140. [Google Scholar]
- Horga JF, Gisbert J, Agustin D, Hernandez M, Zapater P. A beta-2 adrenergic receptor activates adenylate cyclase in human erythrocyte membranes at physiological calcium plasma concentrations. Blood Cells Mol Dis. 2000;26:223–228. doi: 10.1006/bcmd.2000.0299. [DOI] [PubMed] [Google Scholar]
- Hünefeld FL. Die Chemismus in der thierischen Organization. F.A. Brockhaus, Leipzig; 1840. [Google Scholar]
- Jagger JE, Bateman RM, Ellsworth ML, Ellis CG. Role of erythrocyte in regulating local O2 delivery mediated by hemoglobin oxygenation. Am J Physiol Heart Circ Physiol. 2001;280:H2833–H2839. doi: 10.1152/ajpheart.2001.280.6.H2833. [DOI] [PubMed] [Google Scholar]
- Kaiser G, Quiring K, Gauger D, Palm D, Becker H, Schoeppe W. Occurrence of adenyl cyclase activity in human erythrocytes. Blut. 1974;29:115–122. doi: 10.1007/BF01633835. [DOI] [PubMed] [Google Scholar]
- Koller A, Kaley G. Endothelium regulates skeletal muscle microcirculation by a blood flow velocity-sensing mechanism. Am J Physiol Heart Circ Physiol. 1990;258:H916–H920. doi: 10.1152/ajpheart.1990.258.3.H916. [DOI] [PubMed] [Google Scholar]
- Krogh A. The Anatomy and Physiology of Capillaries. New York: Hafner Publishing Company; 1959. Lecture II (1929): The distribution and number of capillaries in selected organs; pp. 22–46. [Google Scholar]
- Li C, Xu Q. Mechanical stress-initiated signal transductions in vascular smooth muscle. Cell Signal. 2000;12:435–445. doi: 10.1016/s0898-6568(00)00096-6. [DOI] [PubMed] [Google Scholar]
- McCullough WT, Collins DM, Ellsworth ML. Arteriolar responses to extracellular ATP in striated muscle. Am J Physiol Heart Circ Physiol. 1997;272:H1886–H1891. doi: 10.1152/ajpheart.1997.272.4.H1886. [DOI] [PubMed] [Google Scholar]
- Nobles M, Benians A, Tinker A. Heterotrimeric G proteins precouple with G protein-coupled receptors in living cells. Proc Natl Acad Sci U S A. 2005;102:18706–18711. doi: 10.1073/pnas.0504778102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngai CY, Yao X. Vascular responses to shear stress: the involvement of mechanosensors in endothelial cells. Open Circ Vasc J. 2010;3:85–94. [Google Scholar]
- Olearczyk JJ, Stephenson AH, Lonigro AJ, Sprague RS. Receptor- mediated activation of the heterotrimeric G-protein Gs results in ATP release from erythrocytes. Med Sci Monit. 2001;7:669–674. [PubMed] [Google Scholar]
- Olearczyk JJ, Stephenson AH, Lonigro AJ, Sprague RS. Heterotrimeric G protein Gi is involved in a signal transduction pathway for ATP release from erythrocytes. Am J Physiol Heart Circ Physiol. 2004a;286:H940–H945. doi: 10.1152/ajpheart.00677.2003. [DOI] [PubMed] [Google Scholar]
- Olearczyk JJ, Stephenson AH, Lonigro AJ, Sprague RS. NO inhibits signal transduction pathway for ATP release from erythrocytes via its action on heterotrimeric G protein Gi. Am J Physiol Heart Circ Physiol. 2004b;287:H748–H754. doi: 10.1152/ajpheart.00161.2004. [DOI] [PubMed] [Google Scholar]
- Osanai T, Fujita N, Fujiwara N, Nakano T, Takahashi K, Guan W, Okumura K. Cross talk of shear-induced production of prostacyclin and nitric oxide in endothelial cells. Am J Physiol Heart Circ Physiol. 2000;278:H233–H238. doi: 10.1152/ajpheart.2000.278.1.H233. [DOI] [PubMed] [Google Scholar]
- Poole DC, Copp SW, Hirai DM, Musch TI. Dynamics of muscle microcirculatory and blood-myocyte O2 flux during contractions. Acta Physiol (Oxf) 2011;202:293–310. doi: 10.1111/j.1748-1716.2010.02246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodan SB, Rodan GA, Sha’Afi RI. Demonstration of adenylate cyclase activity in human red blood cell ghosts. Biochim Biophys Acta. 1975;428:509–515. doi: 10.1016/0304-4165(76)90059-3. [DOI] [PubMed] [Google Scholar]
- Sprague RS, Bowles EA, Achilleus D, Ellsworth ML. Erythrocytes as controllers of perfusion distribution in the microvasculature of skeletal muscle. Acta Physiol (Oxf) 2011;202:285–292. doi: 10.1111/j.1748-1716.2010.02182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague RS, Bowles EA, Hanson MS, DuFaux E, Sridharan M, Adderley S, Ellsworth ML, Stephenson AH. Prostacyclin analogues stimulate receptor-mediated cAMP synthesis and ATP release from rabbit and human erythrocytes. Microcirculation. 2008;15:461–471. doi: 10.1080/10739680701833804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague RS, Bowles EA, Olearczyk JJ, Stephenson AH, Lonigro AJ. The role of G protein β subunits in the release of ATP from human erythrocytes. J Physiol Pharmacol. 2002;53:667–674. [PubMed] [Google Scholar]
- Sprague RS, Ellsworth ML. Erythrocyte-derived ATP and perfusion distribution: Role of intracellular and intercellular communication. Microcirculation. 2012 doi: 10.1111/j.1549-8719.2011.00158.x. DOI: 10.1111/j.1549-8719.2012.00158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague RS, Ellsworth ML, Stephenson AH, Kleinhenz ME, Lonigro AJ. Deformation-induced ATP release from red blood cells requires cystic fibrosis transmembrane conductance regulator activity. Am J Physiol Heart Circ Physiol. 1998;275:H1726–H1732. doi: 10.1152/ajpheart.1998.275.5.H1726. [DOI] [PubMed] [Google Scholar]
- Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ. Participation of cAMP in a signal-transduction pathway relating erythrocyte deformation to ATP Release. Am J Physiol Cell Physiol. 2001;281:C1158–C1164. doi: 10.1152/ajpcell.2001.281.4.C1158. [DOI] [PubMed] [Google Scholar]
- Sprague RS, Goldman D, Bowles EA, Achilleus D, Stephenson AH, Ellis CG, Ellsworth ML. Divergent effects of low O2 tension and iloprost on ATP release from erythrocytes of humans with type 2 diabetes: Implications for O2 supply to skeletal muscle. Am J Physiol Heart Circ Physiol. 2010;299:H566–H573. doi: 10.1152/ajpheart.00430.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan M, Adderley SP, Bowles EA, Egan TM, Stephenson AH, Ellsworth ML, Sprague RS. Pannexin 1 is the conduit for low oxygen tension-induced ATP release from human erythrocytes. Am J Physiol Heart Circ Physiol. 2010a;299:H1146–H1152. doi: 10.1152/ajpheart.00301.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan M, Bowles EA, Richards JP, Krantic M, Davis KL, Dietrich KA, Stephenson AH, Ellsworth ML, Sprague RS. Prostacyclin receptor-mediated ATP release from erythrocytes requires the voltage-dependent anion channel (VDAC) Am J Physiol Heart Circ Physiol. 2012;302:H553–H559. doi: 10.1152/ajpheart.00998.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan M, Sprague RS, Adderley SP, Bowles EA, Ellsworth ML, Stephenson AH. Diamide decreases deformability of rabbit erythrocytes and attenuates low oxygen tension-induced ATP release. Exp Biol Med (Maywood) 2010b;235:1142–1148. doi: 10.1258/ebm.2010.010118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein JC, Ellsworth ML. Capillary oxygen transport during severe hypoxia: role of hemoglobin oxygen affinity. J Appl Physiol. 1993;75:1601–1607. doi: 10.1152/jappl.1993.75.4.1601. [DOI] [PubMed] [Google Scholar]
- Wan J, Ristenpart WD, Stone HA. Dynamics of shear-induced ATP release from red blood cells. Proc Natl Acad Sci U S A. 2008;105:16432–16437. doi: 10.1073/pnas.0805779105. [DOI] [PMC free article] [PubMed] [Google Scholar]