

Abstract
In order to get a comprehensive picture of the complex adaptations of human skeletal muscle to disuse and further the understanding of the underlying mechanisms, we participated in two bed rest campaigns, one lasting 35 days and one 24 days. In the first bed rest (BR) campaign, myofibrillar proteins, metabolic enzymes and antioxidant defence systems were found to be down-regulated both post-8 days and post-35 days BR by proteomic analysis of vastus lateralis muscle samples from nine subjects. Such profound alterations occurred early (post-8 days BR), before disuse atrophy developed, and persisted through BR (post-35 days BR). To understand the mechanisms underlying the protein adaptations observed, muscle biopsies from the second bed rest campaign (nine subjects) were used to evaluate the adaptations of master controllers of the balance between muscle protein breakdown and muscle protein synthesis (MuRF-1 and atrogin-1; Akt and p70S6K), of autophagy (Beclin-1, p62, LC3, bnip3, cathepsin-L), of expression of antioxidant defence systems (NRF2) and of energy metabolism (PGC-1α, SREBP-1, AMPK). The results indicate that: (i) redox imbalance and remodelling of muscle proteome occur early and persist through BR; (ii) impaired energy metabolism is an early and persistent phenomenon comprising both the oxidative and glycolytic one; (iii) although both major catabolic systems, ubiquitin proteasome and autophagy, could contribute to the progression of atrophy late into BR, a decreased protein synthesis cannot be ruled out; (iv) a decreased PGC-1α, with the concurrence of SREBP-1 up-regulation, is a likely trigger of metabolic impairment, whereas the AMPK pathway is unaltered.
Key points
It is still debated whether an imbalance between production and removal of reactive oxygen species is a major trigger of disuse skeletal muscle atrophy in human limb muscles and what the underlying mechanisms are.
In the bed rest model of human disuse, redox imbalance, impairment of antioxidant defence systems and metabolic derangement occurred early, before vastus lateralis muscle atrophy developed, and persisted through 35 days of bed rest.
Down-regulation of PGC-1α, a master controller of muscle metabolism, and up-regulation of SREBP-1, a master controller of lipid synthesis, are likely to have triggered disuse adaptations through mitochondrial dysfunction, whereas AMP kinase, an energy sensor pathway, was unaltered.
The present and previous results on the same subjects suggest a causal link between muscle atrophy, impaired skeletal muscle metabolism, impaired whole body oxidative metabolism, and insulin sensitivity and moderate inflammation, which are major risk factors of physical inactivity related diseases.
Introduction
Skeletal muscle is a very plastic tissue that rapidly and profoundly adapts to contractile activity, nutrient supply and hormones. Disuse is a major phenomenon shaping muscle phenotype. Probably due to the relevance of muscle weakness in limiting movement and exposing subjects to risk of falls and fractures and its straightforward relation with loss of mass, disuse studies mainly focused on muscle atrophy (Fitts et al. 2001; Adams et al. 2003; Canepari et al. 2010).
Muscle atrophy depends on an imbalance between muscle protein synthesis (MPS) and breakdown (MPB). The occurrence of muscle atrophy, impaired antioxidant defence systems and protein oxidation and/or lipid peroxidation, indirect indexes of redox imbalance, suggested a major role of oxidative stress in disuse atrophy (Lawler et al. 2003; Pellegrino et al. 2011). However, it has been very recently argued that the relevance of oxidative stress in causing disuse atrophy could vary through species, muscles and models of disuse (Pellegrino et al. 2011). In fact, whereas an oxidative stress induced increase in MPB caused by activation of the ubiquitin proteasome system has been found in small mammals (Ikemoto et al. 2001b; Powers et al. 2011), it has been suggested that in human disuse a decrease in MPS might play a major role, and that oxidative stress is dispensable (Rennie et al. 2010). Indeed, although the role of oxidative stress, ubiquitin proteasome system and MPB has been elegantly shown in the fast developing (12–18 h) and dramatic (50%) diaphragm atrophy following mechanical ventilation (Levine et al. 2008), little is known about oxidative stress and MPB in disuse limb muscle atrophy in humans (Rennie et al. 2010; Pellegrino et al. 2011) and contradictory results have been reported. Little change in oxidative stress markers and in activation of the ubiquitin proteasome system, and a decrease in MPS following leg immobilization have been reported suggesting no major role for oxidative stress (de Boer et al. 2007; Glover et al. 2008, 2010). On the contrary, it has been very recently shown, for the first time, that limb muscle atrophy following bed rest is accompanied by oxidative stress (Dalla Libera et al. 2009).
Compared to structural and functional changes, much less attention has been paid to metabolic alterations following disuse notwithstanding the high metabolic relevance of skeletal muscle, which represents 40% of body weight and is a major site of insulin action, and the growing body of information pointing to physical inactivity as a major cause of metabolic diseases (Bergouignan et al. 2011). Lower oxidative metabolism following short duration (7–9 days) BR has been described in detail by two very recent studies (Alibegovic et al. 2010b; Ringholm et al. 2011). An overall impairment of energy metabolism, comprising also glycolytic metabolism and creatine kinase, has been suggested by gene expression studies in humans (Chen et al. 2007) and mice (Dapp et al. 2004) and by a very recent proteomic study of ours in mice (Brocca et al. 2010). Interestingly, mitochondrial dysfunction can be a cause of muscle atrophy (Romanello & Sandri, 2010) and of insulin resistance (Koves et al. 2008; Bergouignan et al. 2011).
Muscle adaptations are, therefore, a complex event concerning not only the quantitative change in myofibrillar proteins and the qualitative change in their isoforms, but the whole muscle proteome from myofibrillar proteins to metabolic enzymes, structural proteins, as well as antioxidant defence proteins, all being essential to muscle function (Spangenburg & Booth, 2003). Recently, proteomic analysis which can detect differentially expressed proteins among a large number of proteins (∼800) building the muscle proteome proved to be particularly suited to address the complexity of skeletal muscle adaptations to disuse, ageing, training (Gondin et al. 2010) and diabetes (Brocca et al. 2010).
With the goal to reach a comprehensive picture of the complex adaptations of human skeletal muscle to disuse and further the understanding of the underlying mechanisms, we participated in two bed rest campaigns, one lasting 35 days and one 24 days.
In the first campaign muscle biopsy samples from each subject (n = 9) were collected before bed rest (pre-BR), following 8 days of bed rest (post-8d BR) and following 35 days of bed rest (post-35d BR). A proteomic analysis by 2D gels was performed to evaluate the time course of protein adaptations. Three major functional categories of proteins were found to be affected early and persistently: myofibrillar proteins, antioxidant defence systems and metabolic enzymes.
To understand the mechanisms underlying the protein adaptations observed, muscle biopsies from the second bed rest campaign (nine subjects) were used to evaluate the adaptations of master controllers of the balance between MPB and MPS (muscle-specific RING finger 1 protein (MuRF-1) and atrogin-1 (or muscle atrophy F-box, MAFbx); Akt and p70S6K), of autophagy (Beclin-1, sequestosome 1 (p62), microtubule-associated protein light chain 3 (LC3), Bcl-2/adenovirus E1B 19 kDa-interacting protein (bnip3), cathepsin-L), of expression of antioxidant defence systems (nuclear factor (erythroid-derived 2)-like 2; NRF2) and of energy metabolism (proliferator-activated receptor γ coactivator 1α (PGC-1α) and sterol regulatory element binding protein1 (SREBP-1)).
The results show that in limb human muscles BR causes early and persistent down-regulation of myofibrillar protein content, impairment of antioxidant defence systems and redox imbalance followed at late stage by muscle fibre atrophy. Moreover, BR causes an early derangement of energy metabolism and not just a shift from an oxidative to a glycolytic one. PCG-1α down-regulation and SREBP-1 up-regulation were found and could concur to ROS production through impaired metabolism and mitochondrial dysfunction. Finally, a moderate activation of both major catabolic systems, ubiquitin proteasome and autophagy might concur to the progression of atrophy late into BR, although a decreased protein synthesis cannot be ruled out.
Methods
Subjects
Two bed rest (BR) campaigns were organized by the Osteoporosis and Muscle Atrophy (OSMA) research group, supported by the Italian Space Agency (ASI), in July–August 2007 and in July–August 2008. Nine healthy men (age = 24.3 ± 0.8 years; body mass index = 22.5 ± 0.8 kg m−2 SD) in the first campaign (group A) and nine healthy men (age = 23.3 ± 0.7 years; body mass index = 23.5 ± 0.6 kg m−2) in the second campaign (group B) were enrolled in the study. Subjects did not have a previous history of neuromuscular or cardiac diseases, and gave written, informed consent to participate to the study. The study was approved by the ethical committee of the University of Ljubljana and conformed to the principles of the Declaration of Helsinki on human experimentation.
During the bed-rest periods, each subject received an activity-adjusted diet containing 1.2 times his resting energy expenditure to maintain stable energy balance (Biolo et al. 2008). Subjects were required to consume all food served.
The subjects belonging to group A and their muscle samples were subjected by the OSMA research groups to the following studies, relevant for the present work and used in the interpretation of the data: an in vitro study in which analysis of protein carbonylation, cross sectional area (CSA) of muscle fibres on hystological cross-cryosections, and expression of two stress proteins (HO-1 and Grp75) were determined (Dalla Libera et al. 2009); an in vitro study in which muscle fibre size and myosin concentration was determined pre-BR and post-35d BR (Borina et al. 2010); an in vivo study in which maximal oxygen consumption and muscle oxygen extraction were determined (Porcelli et al. 2010); and in vivo studies in which insulin resistance and general inflammation were determined (Mazzucco et al. 2010b).
Muscle biopsies
Needle biopsies were taken from the vastus lateralis muscle according to Bergstrom (1962). The day before starting bed rest (pre-BR), subjects underwent a biopsy from the vastus lateralis muscle. Both BR campaigns lasted 35 days. Subsequent biopsies were obtained in group A 8 days (post-8d BR) and 35 days (post-35d BR) after the beginning of BR, and in group B 24 days (post-24d BR) after the beginning of BR. Although the second BR campaign lasted 35 days as the first one, biopsies were taken 24 days after the beginning to enable access to the subjects of several research groups. A tissue sharing approach of the vastus lateralis biopsies was used to optimize, for ethical and economic reasons, the burden on the volunteers, which were subjected to a number of investigations both in vivo and in vitro. The muscle samples were therefore divided in several portions. From group A we had access to two portions: one was immediately frozen in liquid nitrogen and used for analysis of myosin heavy chain (MHC) isoform distribution and for proteomic analysis; one was divided in smaller bundles, stored at −20°C in skinning solution plus 50% glycerol and used to determine the cross sectional area (CSA) of dissected single fibres. From group B we had access to one portion, which was immediately frozen in liquid nitrogen and used for RT-PCR and Western blot analysis of intracellular regulatory pathways.
Determination of cross sectional area
At least 20 muscle fibres were dissected from each sample pre-BR, post-8d BR and post-35d BR with the help of a stereomicroscope at ×10–40 magnifications. A total of 402 fibres were studied using a procedure previously described (Borina et al. 2010).
Analysis of myosin heavy chain isoform content
The myosin heavy chain (MHC) isoform content was determined using an electrophoretic approach previously described in detail (Brocca et al. 2010). Three bands could be separated in the region of MHC isoforms. Densitometric analysis of MHC bands was performed to assess the relative proportion of the three adult MHC isoforms, MHC-1, MHC-2A and MHC-2X, in each samples (Pellegrino et al. 2003).
Proteome analysis (2-DE)
Sample preparation
The methods of proteome analysis are mostly the same as those previously used (Brocca et al. 2010). In order to perform proteome analysis, a sample mix was obtained for each experimental group (pre-BR, post-8d BR, post-35d BR). Sample mix contained an equal protein quantity taken from each muscle sample of pre-BR, post-8d BR, post-35d BR.
Two-dimensional electrophoresis
Isoelectrofocusing was carried out using IPGphor system (Ettan IPGphor isoelectric Focusing System, GE Healthcare). Immobilized pH gradient (IPG) gel strips, pH 3–11 NL (non-linear) 13 cm, were used. The 2D gels were fixed for 2 h in fixing solution (ethanol 40% (v/v), acetic acid 10% (v/v)), fluorescently stained with Flamingo Fluorescent Gel Stain (Bio-Rad) for 3 h and destained with 0.1% (w/v) Tween 20 solution for 10 min (Brocca et al. 2010).
Triplicate gels of each sample were obtained, visualized using a Typhoon laser scanner (GE Healthcare) and analysed with Platinum software (GE Healthcare). The analysis was performed comparing pre-BR gels with post-8d BR gels and then with post-35d BR gels. For each analysis one gel was chosen as the master gel, and used for the automatic matching of spots in the other 2D gels. Only spots present in all gel used for the analysis were considered (Brocca et al. 2010).
Protein identification
Electrophoresis fractionation and in situ digestion. 2D gels were loaded with 300 μg of proteins per strip and stained with Colloidal Comassie. Protein spots were excised from the gel and identified by Matrix-Assisted Laser Desorption/Ionization (MALDI MS) analysis (Brocca et al. 2010). When the identity of the proteins could not be established by peptide mass fingerprinting, the peptide mixtures were further analysed by liquid chromatography tandem mass spectrometry analysis (LCMSMS) (Brocca et al. 2010).
Immunoblot analysis
The validation of expression changes of the proteins as judged by 2-DE analysis was carried out by standard 1-D immunoblotting according to procedures previously described (Gondin et al. 2010). The membranes were probed with antibody specific to troponin I (Gene Tex, Irvine, CA, USA), Peroxiredoxin 3 (Abcam, Cambridge, MA, USA), superoxide dismutase 1 (Abcam), α,β-crystallin (Abcam), heat shock protein B1 (Abcam), heat shock protein 70 (Abcam), β-enolase (Abnova, Taipei City, Taiwain), triosephosphate isomerase (Abcam), aldolase A (Abcam), lactate dehydrogenase (Abcam), creatine kinase M (Abcam) and myoglobin (Abcam). Thereafter, the membranes were incubated in horseradish peroxidase (HRP)-conjugated secondary antibody (rabbit-anti-mouse, Dako (Carpinteria, CA, USA) or goat-anti-rabbit, Millipore (Billerica, MA, USA)). The protein bands were visualized by an enhanced chemiluminescence method. The content of each protein investigated was assessed by determining the brightness–area product (BAP) of the protein band.
Some signalling pathways involved in MPB and MPS were investigated by Western blotting analysis using specific primary antibodies against the following: Beclin1 (Cell Signaling, Danvers, MA, USA), LC3 (Cell Signaling), p62 (Sigma-Aldrich), bnip3 (Sigma-Aldrich), Akt (Millipore), p-Akt(ser473) (Millipore), p70S6K (Cel Signaling), p-p70S6K(thr389) (Cel Signaling), AMPK (Cell Signaling), p-AMPK(thr172) (Cell Signaling).
Protein content was normalized on the tubulin content.
Polyubiquitinated proteins
In order to analyse the polyubiquitinated proteins, 20 μg of muscle samples (in lysis buffer 50 mm Tris-HCl pH 7.5, 150 mm NaCl, 5 mm mgCl2, 10% glycerol and a cocktail of protease inhibitors and phosphatase inhibitors) were loaded on a 4–15% SDS-PAGE electrophoresis gel. After the run the proteins were electro-transferred to nitrocellulose membranes at 35 mA overnight. The membranes were blocked with 2% BSA in Tris-Buffered Saline with Tween (TBST) for 1 h and then were incubated in primary antibody (anti-polyubiquitin 1:2500, Enzo Life Sciences, UK) diluted with 5% milk. After the rinses with TBST and the incubation with HRP-conjugated secondary antibody (rabbit anti-mouse 1:5000, from Dako) the protein bands were visualized by an enhanced chemiluminescence (ECL Advance, GE Healthcare). The amount of polyubiquitin proteins was normalized on relative ponceau staining.
Enzyme assays
The activity of lactate dehydrogenase (LDH), a marker of glycolytic metabolism, was studied using a previously described method (Pellegrino at al. 2004). Briefly fragments of frozen muscle biopsies were finely pulverized in a steel mortar with liquid nitrogen; the powder obtained was weighed, homogenized in 100 mm Tris pH 7.5 containing protease inhibitors and incubated for 15 min in ice. The samples were centrifuged at 35,000 g for 15 min in a refrigerated centrifuge. The supernatant fluid obtained was used to determine enzyme activity and to evaluate protein concentration with a protein assay kit (DC Protein Assay, Bio-Rad). The activity of lactate dehydrogenase (LDH; EC 1.1.1.27) was recorded for 10 min with a double beam recorder–spectrophotometer (Lambda 25 UV/VIS spectrometer) after mixing in a cuvette 45 mm KH2PO4, 7.3 mm KH2PO4, 0.63 mm pyruvic acid, 13.1 mm NADH, 119 mm NaHCO3, 10% Triton X-100 and 20 μl of muscle sample. Determination of each sample was repeated three times; enzyme specific activities were expressed as nmol of substrate metabolized min−1 (mg of protein)−1.
RT-PCR analysis
To complete the analysis of signalling pathway involved in MPB and MPS we performed gene expression analysis on muscle samples from group B.
Total RNA, from skeletal samples, was extracted using the Promega SV Total RNA isolation kit; the concentration of RNA were evaluated by using NanoDrop instrument (Thermo Scientific) and 300 ng were reverse-transcribed with SuperScript III reverse transcriptase (Invitrogen) to obtain cDNA. The cDNA was analysed by RT-PCR with the SYBR Green PCR kit (Applied Biosystem) and the data were normalized to GAPDH expression. Oligonucleotide primers used for RT-PCR are listed in Table 1.
Table 1.
Oligonucleotide primers used for RT-PCR
| Forward primer | Reverse primer | |
|---|---|---|
| Atrogin-1(Fbxo 32) | 5′-GCAGCTGAACAACATTCAGATCAC-3′ | 5′-CAGCCTCTGCATGATGTTCAGT-3′ |
| MuRF-1 (Trim63) | 5′-CCTGAGAGCCATTGACTTTGG-3′ | 5′-CTTCCCTTCTGTGGACTCTTCCT-3′ |
| Beclin-1 (Becn1) | 5′-TGGAAGGGTCTAAGACGT-3′ | 5′-GGCTGTGGTAAGTAATGGA-3′ |
| SREBP-1 (ADD1) | 5′-CGTCCCTCCTGTTGTGAAAT-3′ | 5′-AGTGTTTTCACGGGACCAAG-3′ |
| p62 (Sqstm1) | 5′-GCTTCCAGGCGCACTACC-3′ | 5′-CATCCTCACGTAGGACATGG-3′ |
| Cathepsin-L (CTSL1) | 5′-GCGCGTGACTGGTTGAG-3′ | 5′-AAAGGCAGCAAGGATGAGTG-3′ |
| PGC1α (Ppargc1a) | 5′-CAGGATTTCATCTGAGTGTGGA-3′ | 5′-GCGAGAGAGAAAGGAAAAGAACAA-3′ |
| GAPDH (Gapdh) | 5′-CACCATCTTCCAGGAGCGAG-3′ | 5′-CCTTCTCCATGGTGGTGAAGAC-3′ |
| NRF2 (Nfe2l2) | 5′-CACAGAAGACCCCAACCAGT-3′ | 5′-CTGTGCTTTCAGGGTGGTTT-3′ |
Statistical analysis
Data were expressed as means ± SEM. Statistical significance of the differences between means was assessed by Student's t test or by one-way ANOVA followed by a Student–Newman–Keuls test (data in Figs 1 and 6). A probability of less than 5% was considered significant (P < 0.05).
Figure 1. The effect of BR on muscle fibre size and myosin heavy chain isoform distribution.
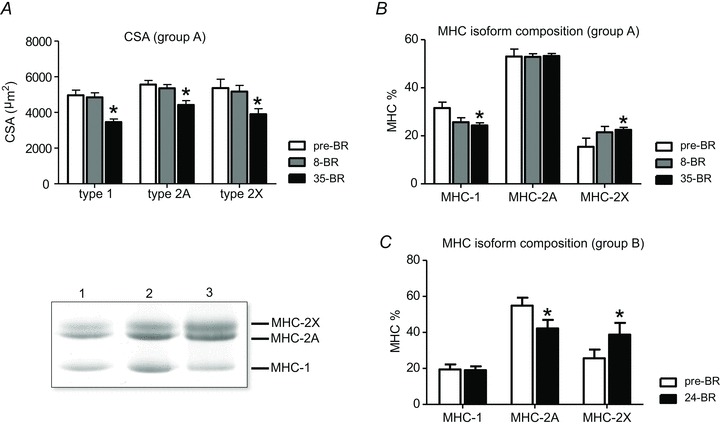
A, cross sectional area (CSA) of type 1, 2A and 2X fibres, identified on the basis of MHC isoform content, pre-BR, post-8d BR (8-BR) and post-35d BR (35-BR). B and C, MHC isoform distribution of subject group A (first BR campaign) pre-BR, post-8d BR and post-35d BR and of subject group B (second BR campaign) pre-BR and post-24d BR (24-BR). Lower left panel, representative SDS-PAGE gels used for determination of MHC isoform relative content of muscle samples.
Figure 6. The effect of 8 and 35 days of BR on lactate dehydrogenase activity of vastus lateralis muscle (subject group A).
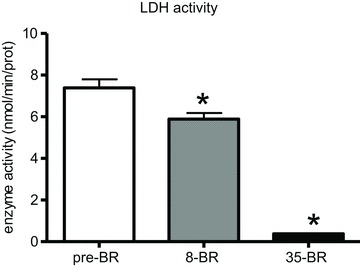
8-BR, post-8d BR; 35-BR, post-35d BR. *Significantly different from all other groups.
Results
Two populations of subjects from two bed rest (BR) campaigns were used in the present study. One population (n = 9) (group A) was subjected to three needle biopsies: pre-BR, 8 days post BR (post-8d BR) and 35 days post BR (post-35d BR). Biopsy samples were used for determination of muscle fibre CSA, of myosin heavy chain (MHC) isoform distribution, and of global protein pattern adaptations by proteomic analysis. The second population (group B) (n = 9) was subjected to two biopsies: pre-BR and post-24d BR. Muscle samples were used for analysis of MHC isoform distribution and of major components of intracellular signalling pathways controlling muscle size, metabolism and expression of antioxidant defence systems.
Bed rest and muscle fibre atrophy
From needles biopsy samples of subjects belonging to group A, a total of 143 individual muscle fibres pre-BR, 115 fibres post-8d BR and 144 fibres post-35d BR, were dissected and their CSA was very precisely determined using the approach described in Methods. Figure 1A shows the mean values of CSA of individual muscle fibres classified on the basis of MHC isoform content. For all fibre types post-35d BR, CSA was significantly reduced compared with pre-BR. The percentage loss in CSA was ∼31% for type 1, ∼21% for type 2A and ∼28% for type 2X. No significant differences were observed post-8d BR.
Bed rest and muscle phenotype
MHC isoform composition was determined in vastus lateralis muscle of both group A and group B. In group A, a significant decrease in MHC-1 content and a significant increase in MHC-2X content was found post-35d BR (Fig. 1B). In group B a significant decrease in MHC-2A content and a significant increase in MHC-2X content was found post-24d BR (Fig. 1C). The somewhat different adaptation in the relative content of MHC-1 and MHC-2A might depend on the different starting phenotype of the subjects: higher MHC-1 content in group A than in group B.
Bed rest and muscle proteome
Proteomic maps of vastus lateralis muscle, for each experimental condition, were obtained using 2D gel electrophoresis as described in Methods on muscle samples of subjects of group A. A representative 2D electrophoresis gel of vastus lateralis protein extract is reported in the online Supplemental Material (Supplemental Fig. 1) in which proteins showing statistically significant differences in content after BR and identified by MALDI-Tof are indicated by circles. Post-8d BR 45 spots, among more than 800 detected by the software, exhibited significantly different content; 33 of the spots could be identified by MALDI-Tof (details in Supplemental Table 1). Post-35d BR the differentially expressed spots were 34, all identified by MALDI-Tof (details in Supplemental Table 1). Of the differentially expressed spots most were down-regulated: 72% following 8 days and 84% following 35 days of BR.
Identified proteins were grouped on the basis of their functional role in the following categories: myofibrillar proteins, antioxidant defence systems, energy production systems (glycolytic enzymes, oxidative enzymes, creatine kinase and guanidinoacetate-N-methyltransferase), transport protein and other proteins (Supplemental Table 1). Several of the differentially expressed gene products were present as multi-spot series of similar molecular weights, but different pI.
Some general trends emerge from an overview of the data. Interestingly, proteins which were differentially expressed post-35d BR were already differentially expressed post-8d BR. Adaptations in protein content occurred early and were, with very few exceptions (4 out of 45), qualitatively the same at both time spots, e.g. for a given protein spot down-regulation was observed at both times. Moreover, the extent of protein content down-regulation was not significantly and consistently larger post-35d compared to post-8d BR, but could be either larger, smaller or similar post-35d vs. post-8d BR depending on the protein. Finally, statistics was performed post-8d vs. pre-BR and post-35d vs. pre-BR and data of a given protein were reported in the figures for both times if at least at one time statistically significant difference was found compared to pre-BR. Statistically significant differences compared to pre-BR are reported for all protein spots in figures and Supplemental Table 1.
Myofibrillar proteins
The differentially expressed myofibrillar proteins are reported in Fig. 2A. Different isoforms of myosin regulatory light chain 2 (MLC-2slow, MLC-2fast) were down-regulated. Seven spots were identified as troponin-T slow isoform and three as troponin-T fast isoform. Both the slow and fast isoforms displayed lower content, in spite of over-expression of two troponin-T slow spots. Troponin-I slow and fast isoforms and tropomyosin-α were also down-regulated. Finally, α-skeletal actin and α-cardiac actin were down-regulated.
Figure 2. Differentially expressed myofibrillar proteins of vastus lateralis muscles following 8 and 35 days of BR compared to pre-BR (subject group A).
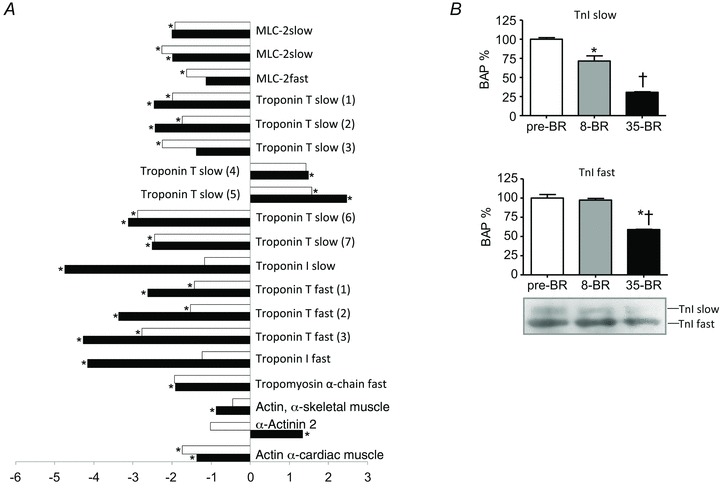
A, bar graph of volume ratios of differentially expressed proteins. The numbers on the x-axis indicate the ratio between the average volume of a given protein expressed post-8d BR (open bars) or post-35d BR (filled bars) and the average volume of the same protein pre-BR. Positive numbers (on the right) indicate up-regulation of a protein, whereas negative numbers (on the left) indicate down-regulation. *Significantly different from pre-BR. The full information on the proteins and the numerical ratios between volumes are reported in Supplemental Table 1. B, Western blot analysis of the content in troponin-I (TnI) slow and fast isoforms pre-BR, post-8d BR (8-BR) and post-35d BR (35-BR). *Significantly different from pre-BR; †significantly different from post-8 BR.
Down-regulation was statistically significant for most myofibrillar proteins post-8d BR already. The content was further decreased in some proteins (e.g. Troponin-I slow and fast), partially recovered in a few proteins (e.g. MLC-2fast) and remained unchanged in others (MLC-2slow) post-35d BR.
Only α-actinin showed an opposite adaptation at the two time points analysed, i.e. a down-regulation post-8d BR and an up-regulation post-35d BR.
Antioxidant defence systems
Seven proteins involved in cellular defence systems against reactive oxygen species or free radicals (ROS), superoxide dismutase (SOD1), carbonic anhydrase III (CAHIII), peroxiredoxin 3 (PRDX3), α,β-crystallin, heat shock protein B6 (HspB6), heat shock protein B1 (HspB1) and heat shock protein 70 (Hsp70), were down-regulated following bed rest (Fig. 3A). Adaptations occurred post-8d BR already and, with the exception of CAHIII and Hsp B6, on the whole did not progress further or even showed a recovery trend (e.g. HspB1) post-35d BR.
Figure 3. Differentially expressed antioxidant defence systems of vastus lateralis muscles following 8 and 35 days of BR compared to pre-BR (subject group A).
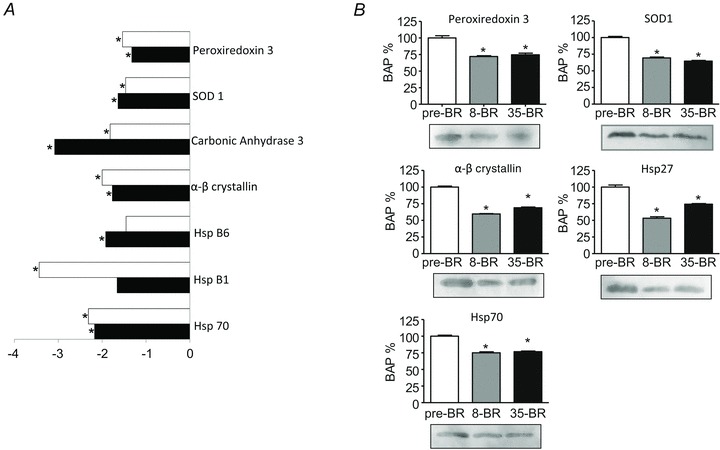
A, bar graph of volume ratios of differentially expressed proteins. The same approach to represent differentially expressed proteins was used as in Fig. 3. *Significantly different from pre-BR. The full information on the proteins and the numerical ratios between volumes are reported in Supplemental Table 1. B, Western blot analysis of the content in peroxiredoxin 3, α,β-crystallin, Hsp70, SOD1, Hsp27 pre-BR, post-8d BR (8-BR) and post-35d BR (35-BR). *Significantly different from pre-BR.
Energy production systems
Figure 4A shows the differentially expressed proteins involved in the three cellular energy production systems: glycolytic enzymes, oxidative enzymes, creatine kinase and the phosphocreatine system. The differentially expressed glycolytic enzymes, triosephosphate isomerase, lactate dehydrogenase, aldolase A and β-enolase, were all down-regulated after both 8 and 35 days of bed rest although one of the three spots corresponding to β-enolase was up-regulated after 8 days of BR. Malate dehydrogenase, a major enzyme involved in oxidative metabolism, was lower post-8d and post-35d BR compared to pre-BR. After bed rest two key enzymes involved in metabolism of creatine and phosphocreatine were differently regulated: creatine kinase, responsible for energy storage and production from phosphocreatine, showed a reduced content both post-8d and post-35d BR, while guanidinoacetate N-methyltransferase (GAMT), a key enzyme converting guanidoacetate into creatine, was higher at both 8 and 35 days of BR than pre-BR.
Figure 4. Differentially expressed energy production systems of vastus lateralis muscles following 8 and 35 days of BR compared to pre-BR (subject group A).
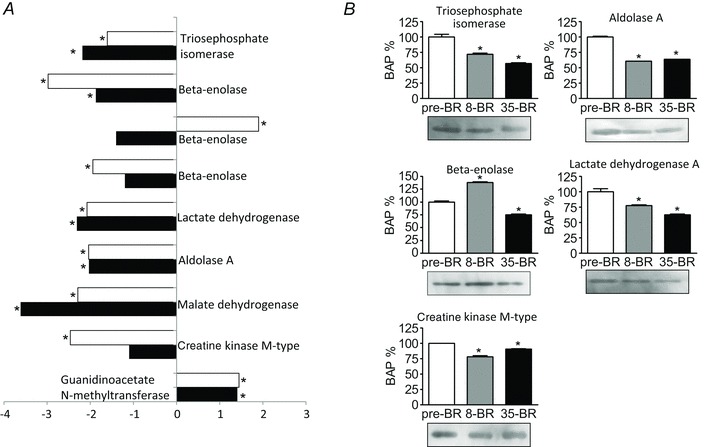
A, bar graph of volume ratios of differentially expressed proteins. The same approach to represent differentially expressed proteins was used as in Fig. 3. The full information on the proteins and the numerical ratios between volumes are reported in Supplemental Table 1. *Significantly different from pre-BR. B, Western blot analysis of the content in triosephosphate isomerase, β-enolase, creatine kinase, aldolase A, lactate dehydrogenase A post-8d BR (8-BR) and post-35d BR(35-BR). *Significantly different from pre-BR.
As for myofibrillar proteins and antioxidant defence systems, adaptations in metabolic enzymes occurred early and mostly stabilized, with the important exception of malate dehydrogenase whose down-regulation progressed further and of CK whose down-regulation was not significant post-35d BR, but only post-8d BR.
Transport proteins
Several transport proteins were differentially expressed after bed rest. As shown in Fig. 5A, three spots corresponded to myoglobin. Although one of the three spots was up-regulated after 35 days of BR, myoglobin showed lower content both post-8d BR and post-35d BR. Spots corresponding to haemoglobin, fatty acid-binding protein 3 (FABPH3) and apolipoprotein were all down-regulated, while a different expression trend was found for serum albumin, which was up-regulated. Bed rest induced an opposite effect on serotransferrin determining an up-regulation after 8 days and a down-regulation after 35 days.
Figure 5. Differentially expressed transport proteins of vastus lateralis muscles following 8 and 35 days of BR compared to pre-BR (subject group A).
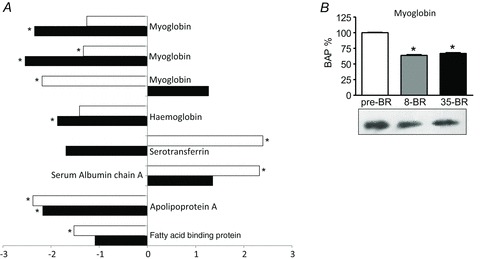
A, bar graph of volume ratios of differentially expressed proteins. The same approach to represent differentially expressed proteins was used as in Fig. 3. The full information on the proteins and the numerical ratios between volumes are reported in Supplemental Table 1. *Significantly different from pre-BR. B, Western blot analysis of myoglobin content pre-BR, post-8d BR (8-BR) and post-35d BR (35-BR). *Significantly different from pre-BR.
The adaptations in some proteins in this category (haemoglobin, apolipoprotein, serotransferrin, serum albumin) are likely to be mostly determined by the amount of blood present in muscle vessels trapped in the muscle biopsy.
Other proteins
Proteasome α-sub isoform2 was lower both post-8d BR and post-35d BR. Glycogen phosphorylase, which converts glycogen to glucose-1-phosphate, was differently regulated showing a lower content post-8d BR and higher content post-35 d BR compared to pre-BR (Supplemental Table 1).
Immunoblot analysis
As some of the differentially expressed gene products can be present as multi-spot series of similar molecular weights, but different pI, the adaptations of major differentially expressed proteins were validated by comparative immunoblotting. All results, except those referring to β-enolase, confirmed the adaptations identified by 2-DE and, interestingly, their time course (Figs 2B–5B).
The immunoblotting results confirmed the down-regulation of the proteins involved in energy production. Triosephosphate isomerase, aldolase A, LDH and creatine kinase content was lower both post-8d BR and post-35d BR (Fig. 4B). β-Enolase, which is known to generate a large number of spots in 2-DE maps, was the only protein of its category to be up-regulated post-8d BR and down-regulated post-35d BR.
Proteins involved in antioxidant defence systems, i.e. PRDX-3, α,β-crystallin, HspB1, Hsp70 and SOD1 were down-regulated at both time intervals (Fig. 3B). Troponin I slow isoform was down-regulated both post-8d and post-35d BR, while the troponin I fast isoform expression was reduced post-35d BR only (Fig. 2B). Myoglobin was reduced both post-8d BR and post-35d BR (Fig. 5B).
LDH enzyme activity
The down-regulation of glycolytic enzymes observed by proteomic analysis and confirmed by Western blotting is somewhat surprising due to the generally accepted idea that disuse causes an oxidative to glycolytic shift in energy metabolism. To further clarify the involvement of glycolytic metabolism, LDH enzyme activity was determined. A significant decrease in LDH activity was observed both post-8d and post-3d BR (Fig. 6).
Bed rest and intracellular pathways
To address the possible mechanisms underlying adaptations in muscle size and in the three major protein categories differentially expressed in the muscle proteome, myofibrillar proteins, antioxidant defence systems and proteins involved in energy metabolism, the master controllers of the major intracellular pathways potentially involved were studied. Samples were obtained from subjects belonging to group B, which were biopsied pre-BR and post-24d BR. As in group A the adaptations in the three major functional categories of proteins established within 8d BR and qualitatively did not change by 35d BR, it is very likely that at 24 days of BR they were substantially similar. However, to check that in group B adaptations in antioxidant defence systems and metabolic enzymes were actually similar to those observed in group A, Western blot analyses were performed on representative proteins. Consistent with what observed in group A, lactate dehydrogenase, aldolase, triosephosphate isomerase (metabolic enzymes), and Hsp70, PRDX3 and SOD1 (antioxidant defence systems) were significantly lower post-24d BR compared with pre-BR (Supplemental Fig. 2).
Pathways controlling muscle size
The adaptation in cell size and muscle protein breakdown (MPB) was assessed through the analysis of master controllers of the ubiquitin proteasome system (MuRF-1 and atrogin-1 ubiquitin ligases) and of the autophagic system (Beclin1, p62, bnip3, LC3), and of cathepsin-L (an endoprotease involved in lysosomal degradation). The adaptation in muscle protein synthesis (MPS) was assessed by the analysis of two master regulators of the IGF-1/Akt/mTOR pathway (Akt and p70S6K).
The mRNAs levels of MuRF-1 and atrogin-1, the two major ubiquitin ligases involved in MPB by the proteasome, and the ratios between the phosphorylated and total Akt and p70S6K, two major kinases of the IGF-1/Akt/mTOR signalling pathway, were not significantly higher post-24d BR compared to pre-BR (Fig. 7A) even if there was a trend towards an increase. The detection of polyubiquitinated proteins by Western blotting revealed a significant higher level (+15%) of ubiquitin conjugates post-24d BR compared to pre-BR (Fig. 7B).
Figure 7. The effect of 24 days of BR on the expression of two major atrogenes (MuRF-1 and atrogin-1) and on the activity of two major kinases of the Akt/mTOR pathway (Akt and p70) in vastus lateralis muscle (subject group B).
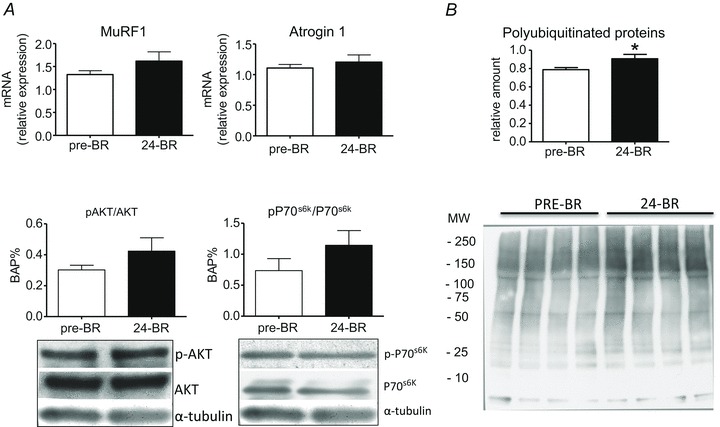
A, MuRF-1 and atrogin-1 expression was assessed by quantitative RT-PCR. The activity of Akt and p70 was assessed by Western blot analysis of the ratio between the content in the phosphorylated and total forms. 24d BR, post-24d BR. B, relative content (upper panel) and representative blot (lower panel) of polyubiquitinated proteins in vastus lateralis muscle post-24d BR (subject group B). *Significantly different from pre-BR.
To assess whether autophagy could contribute to the disuse muscle atrophy observed in this study, several markers of autophagy were determined. The mRNA levels of Beclin1, p62 and cathepsin-L were assessed by RT-PCR, and the protein content in Beclin1 and bnip3, and the ratio between the two forms of LC3 (LC3II and LC3I) were determined by Western blotting. The levels of mRNAs (Fig. 8A) and the protein content (Fig. 8B) for Beclin1 were higher post-24d BR. The increase in the ratio between the II and I form of LC3 and in p62 mRNA (Fig. 8A) and protein level (Fig. 8B) did not reach statistical significance. Cathepsin-L mRNA levels and bnip3 protein content were not statistically different post-24d BR compared to pre-BR (Fig. 8AB).
Figure 8. The effect of 24 days of BR on autophagy markers in vastus lateralis muscles (subject group B).
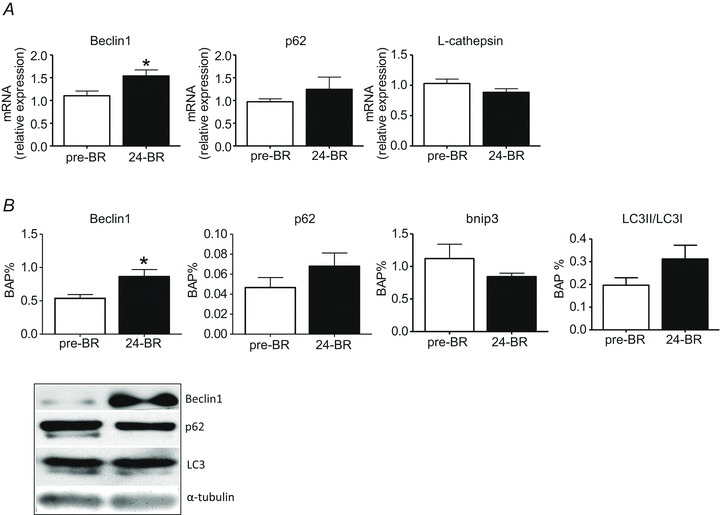
A, Beclin-1, p62 and L-cathepsin expression by quantitative RT-PCR. B, content in Beclin-1, p62, bnip3 and the ratio between the content in II and I forms of LC3 by Western blotting. C, representative Western blots of proteins in panel B. 24d BR, post-24d BR. *Significantly different from pre-BR.
Pathways controlling energy balance and metabolism
The adaptation in energy balance and metabolism was assessed by analysis of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), a master regulator of oxidative metabolism and mitochondrial biogenesis, of sterol regulatory element binding protein (SREBP-1), a master regulator of lipid metabolism, and of AMP-activated protein kinase (AMPK), the major sensor and controller of energy balance in the cell.
The ratio of the active (phosphorylated) and total form of AMPK was studied by Western blotting (Fig. 9) and was found unchanged post-24d BR.
Figure 9. The effect of 24 days of BR on PGC-1α and SREBP-1 expression determined by quantitative RT-PCR and on the ratio between the phosphorylated and total forms of AMP-kinase (AMPK) determined by Western blotting in vastus lateralis muscle (subject group B).
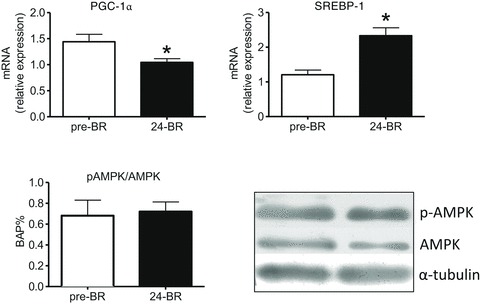
In the lower right panel, representative Western blots for pAMPK. 24d BR, post-24d BR. *Significantly different from pre-BR.
The level of PGC-1α mRNA as determined by RT-PCR was found to be lower post-24d BR (Fig. 9), whereas the level of SREBP-1 mRNA was higher post-24d BR (Fig. 9).
Control of antioxidant defence systems
Finally, the control of antioxidant defence systems was assessed by the analysis of nuclear factor E2-related factor 2 (NRF2), a sensor of cell redox balance and the major transcription factor involved in modulating expression of antioxidants (e.g. heme oxygenase-1, catalase, SOD, glutathione). The mRNA for NRF2 was found to be up-regulated post-24d BR by RT-PCR (Fig. 10).
Figure 10. The effect of 24 days of BR on NRF2 expression determined by quantitative RT-PCR in vastus lateralis muscles (subject group B).
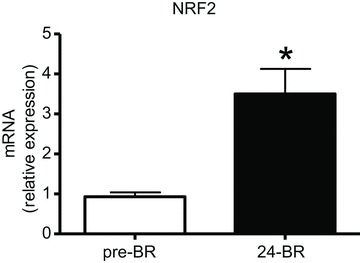
24 d BR, post-24d BR. *Significantly different from pre-BR.
Discussion
Biopsy samples obtained after 8 and 35 days of BR in a first campaign (subject population A) and after 24 days of BR (subject population B) in a second campaign were used. Based on the observations that the proteome adaptations observed in the first campaign occurred early (post-8d BR) and substantially persisted up to 35 days of BR (Figs 2–5) and that the adaptations in major metabolic enzymes and antioxidant defence systems were consistent in the two populations (Supplemental Fig. 2), the results of the analysis of the master controllers of the major intracellular signalling pathways performed on population B will be discussed together with findings of the proteome analysis performed on population A. It should be noted that the minor differences observed in the slow to fast shift in MHC isoform distribution between population A and B are unlikely to have an impact on the conclusions drawn as even the adaptations of myofibrillar proteins to BR were independent from such a shift (Fig. 2). Moreover, the conclusions are based on the overall and broad adaptations in muscle proteome, which are unlikely to depend on the subtle differences in the response to BR of the two populations.
Muscle fibre atrophy and myofibrillar protein content
Muscle fibre atrophy post-35d BR (Fig. 1A) was largely expected in group A on the basis of previous analyses performed on the same samples by Dalla Libera et al. (2009) and by us (Borina et al. 2010). The lack of muscle fibre atrophy post-8d BR in the presence of significant muscle adaptations is consistent with previous findings from short term (7–9 days) BR (Alibegovic et al. 2010b; Ringholm et al. 2011). The slow to fast shift in MHC isoform distribution (Fig. 1B and C) is expected in disuse (di Prampero & Narici, 2003; Hortobagyi & Devita, 2006), although contradictory results have been reported in humans (di Prampero & Narici, 2003).
Proteomic analysis showed an early and persistent down-regulation of thin filament proteins which, comprising both slow and fast isoforms (Fig. 2), is unlikely to be due to the slow to fast shift in muscle phenotype (Fig. 1B) and suggests a preferential loss of thin filaments. The latter finding is consistent with the reduced content at 8 and 35 days of α,β-crystallin (Fig. 3), which is known to protect actin from degradation, stabilizing actin polymers (Wang & Spector, 1995). The down-regulation of thin filament proteins in the muscle proteome is in agreement with animal studies (Jackman & Kandarian, 2004), and with human studies, which showed a decreased number of thin filaments in disuse (Riley et al. 1998) and a decreased myosin concentration in individual muscle fibres post-35d BR (Borina et al. 2010). However, it is not consistent with a previous determination of myofibrillar protein concentration on human samples following 30 and 90 days of BR (Haus et al. 2007).
Redox imbalance
The down-regulation of antioxidant defence systems (SOD1, peroxiredoxin 3, carbonic anhydrase III) and heat shock proteins (α,β-crystallin, HspB6, HspB1 and Hsp 70) (Fig. 3) suggests an early and persistent impairment of the cellular response against ROS. SOD converts the superoxide ion into hydrogen peroxide. Peroxiredoxins, such as catalase, work downstream of SOD converting hydrogen peroxide into water. Carbonic anhydrase 3 (CAHIII) protects against oxidative damage by binding free radicals (Raisanen et al. 1999). Heat shock proteins remove products induced by free radicals and have been shown to protect cells against oxidative stress (Lawler et al. 2006). Hsp70 is particularly interesting as it has been previously shown to be down-regulated in disuse atrophy (Lawler et al. 2006) and in other conditions of muscle wasting (Lecker et al. 2004) and its up-regulation prevents immobilization induced muscle atrophy in rats by inhibition of the FoxO and Nf-kB pathways (Senf et al. 2008).
As direct determination of reactive oxygen species (ROS) content is complex and exposed to errors (Palomero et al. 2008), redox balance has been mostly studied indirectly assessing protein carbonylation or lipid peroxidation (Lawler et al. 2003; Pellegrino et al. 2011). Protein carbonylation post-35d BR, but not post-8d BR, occurred in the biopsy samples used in this study as reported by Dalla Libera et al. (2009) and by Mazzucco et al. (2010a), which, in the frame of a tissue sharing program (see Methods), used portions of our same biopsies.
The up-regulation of nuclear factor E2-related factor 2 (NRF2) (Fig. 10) indicates ongoing redox imbalance and the activation of a compensatory mechanism post-24d BR. NRF2 is a constitutively active transcription factor controlling genes of antioxidant defence systems such as heme oxygenase-1, catalase, SOD, peroxiredoxins, and genes involved in glutathione synthesis and function (Baird & Dinkova-Kostova, 2011; Hur & Gray, 2011). Its activity is finely tuned by the concentration of intracellular ROS and its response is a major way to protect the cell against oxidative stress (Nguyen et al. 2009; Miyata et al. 2011). Redox imbalance post-24d BR is consistent with the down-regulation of antioxidant defence systems post-8d BR and post-35d BR (Fig. 3) and with increased carbonylation post-35d (Dalla Libera et al. 2009).
The data reported so far recapitulate, for the first time in human limb muscles, what has been previously observed in soleus muscles of small mammals following disuse, i.e. muscle atrophy, redox imbalance, protein oxidation and impaired antioxidant defence systems (Pellegrino et al. 2011), and seemingly suggest a pathogenetic role of oxidative stress, although a casual relationship with disuse atrophy could not be established. The early impairment of antioxidant defence systems and the profound alterations in muscle proteome are not fully consistent with the hypothesis, based on different determinations on the same samples, that post-8d BR over-expression of two Hsps, heme oxygenase-1 (HO-1) and glucose-regulated protein-75 (Grp75), could effectively counteract redox imbalance in the early phases (Dalla Libera et al. 2009).
Energy metabolism
The down-regulation of malate dehydrogenase, a major oxidative enzyme, of several key enzymes of glycolytic metabolism and of creatine kinase (Fig. 4), together with the large decrease of lactate dehydrogenase activity (Fig. 6) does not indicate just a shift from an oxidative towards a glycolytic metabolism, but a general impairment of energy metabolism. Furthermore, over-expression of GAMT (Fig. 4), a key enzyme in the biosynthesis of creatine (Braissant et al. 2001; Schmidt et al. 2004) could be a compensatory mechanism and be responsible for the recovery in CK content between post-8d BR and post-35d BR. Interestingly, metabolic derangement occurred early into BR (post-8d BR) and preceded, as the adaptations in myofibrillar proteins and antioxidant defence systems, the development of muscle atrophy and protein oxidation.
An overall derangement of energy metabolism is also supported by the down-regulation of myoglobin and FABPH3 (Fig. 5). Myoglobin is not only an oxygen reservoir, but can stimulate mitochondrial biogenesis and facilitate oxygen diffusion in the cell (Flogel et al. 2001; Merx et al. 2001). FABPHs are a family of proteins involved in lipid metabolism and signalling, which are expressed in many isoforms in different tissues (Makowski & Hotamisligil, 2004). Mice lacking FABPH3, the cardiac/skeletal muscle isoform of FABPHs, show a dramatic impairment of long-chain fatty acid oxidation (Binas et al. 2003). Down-regulation of FABPH3 has been previously shown in soleus muscle of hind-limb unloaded mice (Brocca et al. 2010).
Importantly, Porcelli et al. (2010) suggested an impaired skeletal muscle oxidative metabolism through an in vivo analysis of oxygen consumption and muscle oxygen extraction in the same subjects (group A) enrolled in this study.
An impairment of oxidative metabolism after 7–9 days of BR has been described in detail by two very recent studies (Alibegovic et al. 2010a; Ringholm et al. 2011) and suggested by a recent proteomic analysis of vastus lateralis following 55 days of BR (Moriggi et al. 2010). Although a shift from oxidative to glycolytic metabolism is expected in disuse, evidence for an increase in glycolytic metabolism in humans is not compelling (Acheson et al. 1995; Fitts et al. 2000; Stein & Wade, 2005). It should be noted that the overall impairment of energy metabolism comprising both the aerobic and glycolytic metabolism and creatine kinase observed in this study is consistent with gene expression studies in mice (Dapp et al. 2004) and humans (Chen et al. 2007) and with a proteomic analysis of mice soleus muscle (Brocca et al. 2010) showing a down-regulation of both oxidative and glycolytic enzymes and CK in disuse.
The lower expression of PGC-1α (Fig. 9) could be a major determinant of the metabolic derangement. Interestingly, mice lacking PGC-1α show a down-regulation of several metabolic enzymes among which are malate dehydrogenase, lactate dehydrogenase and creatine kinase (Fig. 4) (Sandri et al. 2006). In skeletal muscle, PCG-1α is a master controller of mitochondrial biogenesis and oxidative metabolism, enhancing fatty acid β-oxidation, oxidative phosphorylation and ATP production (Lin et al. 2005). Enhanced expression of PGC-1α is a major phenomenon underlying skeletal muscle adaptations to exercise (Pilegaard et al. 2003). Although a down-regulation is expected following disuse, little is known on PGC-1α expression following a decrease in neuromuscular activity in humans and contradictory results have been obtained. The down-regulation here observed is in agreement with a previous gene expression study following 9 days of BR (Alibegovic et al. 2010b), but not with two very recent studies showing no change following 7 days of BR (Ringholm et al. 2011) and 55 days of BR (Moriggi et al. 2010).
As the exercise induced increase in PGC-1α expression has been attributed to calcium mediated signalling during contraction (Irrcher et al. 2003), it can be hypothesized that the decrease in neuromuscular activity causes lower PCG-1α expression due to a decreased intracellular calcium signalling. However, whether intracellular calcium increases (Ingalls et al. 1999) or decreases (Fraysse et al. 2003) in disuse is still debated.
Pathways involved in protein synthesis and degradation
Muscle atrophy ultimately depends on an imbalance between muscle protein synthesis (MPS) and breakdown (MPB). Two major ATP-dependent systems are involved in MPB: the ubiquitin-proteasome system and the autophagy system (Sandri, 2008).
MuRF-1 and atrogin-1, two muscle specific ubiquitin ligases, are key components of the ubiquitin proteasome system and widely considered among master genes of muscle atrophy. The role of MuRF-1 and atrogin-1 in breaking down myofibrillar proteins in disuse in small mammals (Ikemoto et al. 2001a) and following mechanical ventilation in humans (Levine et al. 2008) is rather well established. On the contrary, activation of MuRF-1 and atrogin-1 has been observed only transiently and at early times (2–5 days) in disuse atrophy of human limb muscle (de Boer et al. 2007; Urso et al. 2007; Gustafsson et al. 2010). Indeed, evidence showing up-regulation of such atrogenes at later times when muscle atrophy is observed is lacking. Consequently, their causal role has been questioned and the alternative view that disuse atrophy in human limb muscles is caused by decreased protein synthesis has been suggested (de Boer et al. 2007; Agostini et al. 2008; Rennie et al. 2010). The lack of up-regulation of MuRF-1 and atrogin-1 (Fig. 9) is, therefore, not surprising and could support the latter conclusion.
However, expression of MuRF-1 and atrogin-1 is known to be transient and transcriptional activation might be followed by a delayed activation of translation (Urso et al. 2007), i.e. activities of ubiquitin ligases could be higher at times when their expression is back to normal. Interestingly, a moderately higher level (+15%) of polyubiquitinated proteins (Fig. 7B) was found, consistent with the only previous analysis performed late in bed rest (Ogawa et al. 2006). Increased polyubiquitination of proteins can be related to an increase in the activity of the ubiquitin proteasome system (Berthon et al. 2007; Baptista et al. 2010) and, therefore, to increased MPB. Even a moderate increase (+15%) in the ubiquitin proteasome activity (Fig. 7B) lasting weeks could account for significant atrophy (Fig. 1). As it cannot be excluded that a decreased activity of the proteasome (Tawa et al. 1997) and altered regulation of de-ubiquitination processes (Lecker et al. 2004) are involved in determining the level of ubiquitinated proteins, further analyses are required to definitely settle this issue.
The autophagy system is constitutively active in skeletal muscle to remove old and damaged organelles and protein aggregates, and is important for cell survival (Sandri, 2010b). Moreover, it has been recently understood that an increase in activity of the autophagy system can play a relevant role in muscle atrophy (Sandri, 2010a). Interestingly, an activation of the autophagy system has been observed in human diaphragm atrophy following mechanical ventilation (Hussain et al. 2010). In this study, several markers of autophagy have been studied (Sandri, 2010b): Beclin-1 is a major protein involved in autophagosome formation; LC3 is involved in autophagosome expansion and completion; p62 delivers proteins to the autophagosome; bnip3 is involved in autophagy of damaged mitochondria (mitophagy) (Zhang & Ney, 2009); cathepsin-L is a major lysosome enzyme. The up-regulation of Beclin-1 suggests an increased autophagosome formation and therefore a higher activity of the macro-autophagy system post-24d BR, notwithstanding the lack of statistically significant up-regulation of p62, LC3II/I ratio and cathepsin-L. As bnip3 was not up-regulated, it is unlikely that an activation of mitophagy occurred. Although we cannot define the role of autophagy at early stages of BR, the present findings suggest that an increased activity of macroautophagy could contribute to the progression of muscle atrophy at least at late stages of BR.
Several evidences have suggested a major role of decreased protein synthesis in causing disuse atrophy in humans (de Boer et al. 2007; Glover et al. 2010; Rennie et al. 2010). The lack of changes in the phosphorylation level of Akt and p70S6K, two key enzymes of the IGF-1/Akt/mTOR pathway, which is a major controller of MPS (Blaauw et al. 2009), does not support decreased protein synthesis playing a major role in the late phases of BR. Interestingly, the same subjects from which biopsies were taken had insulin resistance, increased insulin and normal glucose in blood (Mazzucco et al. 2010a,b). The high blood levels of insulin, which is a major controller of the activity of the IGF-1/Akt/mTOR, could compensate for disuse induced insulin resistance maintaining normal muscle glucose uptake, blood glucose at normal levels and normal activity of the IGF-1/Akt/mTOR pathways and therefore normal protein synthesis. However, it has been strongly argued that normal activation of Akt/mTOR can coexist with a significant decrease in MPS and disuse muscle atrophy (de Boer et al. 2007; Glover et al. 2008, 2010) and that decreased MPS is the major determinant of disuse muscle atrophy (Biolo et al. 2008; Rennie et al. 2010). Therefore, the decreased MPS in the late stages of BR atrophy cannot be ruled out in the absence of a direct determination of the rate of MPS and further analysis is required to definitely conclude on this matter.
Triggers of altered balance between protein synthesis and breakdown
The findings reported in this study prompt to us consider several potential triggers controlling the systems involved in MPS and MPB: redox imbalance, metabolic alterations through PGC-1α and SREBP-1 altered mitochondrial function.
ROS could enhance protein degradation through the ubiquitin proteasome system stimulating ubiquitin conjugation by transcriptional regulation of E2 and E3 (MuRF-1 and atrogin-1) enzymes (Li et al. 2003). ROS are also known to activate FoxO, which in turn can activate MuRF-1 and atrogin-1 (de Keizer et al. 2011) and autophagy (Sandri, 2010a). As no up-regulation of MuRF-1 and atrogin-1 was observed post-24d BR (Fig. 9), whereas redox imbalance was present through BR (Fig. 3, 10), it appears that ROS do not necessarily stably activate MuRF-1 and atrogin-1 expression.
Metabolic alterations are potential candidates in determining muscle atrophy (Greer et al. 2007; Sandri, 2008). Indeed, several metabolic enzymes can be considered ‘atrogenes’, as well as MuRF-1 and atrogin-1, as they have been shown to be consistently down-regulated in most conditions of muscle wasting (systemic diseases, fasting and disuse) (Sacheck et al. 2007). Four of the seven metabolic enzymes down-regulated both post-8d and post-35d BR (Fig. 4) belong to atrogenes (Sacheck et al. 2007): triosephosphate isomerase, malate dehydrogenase, lactate dehydrogenase and creatine kinase. Interestingly, metabolic impairment occurred before muscle atrophy together with alterations of myofibrillar proteins and antioxidant defence systems. A potential link between metabolic alterations and muscle mass could be an impairment of energy balance and activation of AMPK (Greer et al. 2007; Sandri, 2008).
AMPK is an energy sensor, which is activated by metabolic stress, namely an increase in cellular AMP/ATP ratio. To restore energy balance AMPK would inhibit protein synthesis and stimulate protein degradation through inhibition of the Akt/mTOR and activation of the FoxO pathways (Hardie et al. 2006). The lack of activation of AMPK (Fig. 9), consistent with normal Akt/mTOR activation (Fig. 7A), indicates that the AMP/ATP ratio is normal in muscle fibres during human BR suggesting that the lower energy requirements due to disuse allow normal energy levels in muscle cells even in the presence of impaired metabolism (Fig. 4). The link between impaired metabolism and disuse muscle atrophy appears more complex than the activation of catabolic signals due to energy imbalance.
Several pieces of evidence in mice indicate that lower PGC-1α expression can be associated with muscle wasting (Sandri et al. 2006; Sacheck et al. 2007). As PGC-1α inhibits the FoxO proteolytic pathway (Sandri et al. 2006), its down-regulation could cause activation of both the ubiquitin proteasome system and autophagy. However, the lack of increased MuRF-1 and atrogin-1 expression (Fig. 7) does not support a PGC-1α role in increasing the ubiquitin proteasome activity in BR atrophy. Moreover, lower PGC-1α expression, causing lower number and impaired function of mitochondria and fatty acid accumulation, can alter the ratio between ATP production and oxygen consumption causing excessive mitochondrial production of superoxide (Kim et al. 2008). ROS production could therefore follow metabolic alterations and further damage mitochondria creating a vicious cycle. Finally, a PGC1-α-dependent ROS production could be a potential link between metabolism and inflammation (Handschin, 2009). Interestingly, the subjects used in this study showed not only insulin resistance, but also moderate general inflammation in response to BR (Mazzucco et al. 2010a).
SREBPs (SREBP-1 and -2) are transcription factors controlling lipid and cholesterol synthesis and accumulation (Horton et al. 2002). SREBP-1, which stimulates lipogenesis and triglyceride deposition, has been suggested to play an important role in regulating lipid metabolism in skeletal muscle (Horton et al. 2002; Commerford et al. 2004). Interestingly, the increased SREBP-1 expression (Fig. 9), a novel finding in disuse, could cause lipid synthesis and intramuscular lipid accumulation, which are known features of disuse (Bergouignan et al. 2011) and would contribute to mitochondrial dysfunction.
PGC-1α and SREBP-1 through mitochondrial dysfunction and lipid accumulation in muscle fibres could cause insulin resistance even in non-diabetic subjects (Phillips et al. 1996). Importantly, the subjects enrolled in this study did develop insulin resistance and hyperinsulinaemia during BR (Mazzucco et al. 2010a). Interestingly, the increase in SREBP-1 expression (Fig. 9) might depend on hyperinsulinaemia as SREBP-1 is stimulated by insulin (Porstmann et al. 2008).
Conclusions
This study shows that in disuse of human limb muscles, down-regulation of myofibrillar proteins, redox imbalance and impairment of antioxidant defence systems occur early and are followed by protein carbonylation and muscle atrophy, recapitulating what happens in slow muscles of small mammals and suggesting a role of redox imbalance in disuse atrophy although a causal relationship was not established.
It is hypothesized that the progression of muscle atrophy late into BR could be sustained by a moderate activation of both major catabolic systems, the ubiquitin proteasome system and autophagy. It is also shown that the Akt/mTOR anabolic pathway is not impaired consistently with the hyper-insulinaemia of the subjects. However, only further specific analyses could definitely establish the relative role of each pathway in disuse atrophy in humans.
It is suggested that altered PGC-1α and SREBP-1 expression could trigger metabolic impairment and cause ROS production, through mitochondrial dysfunction, whereas the AMPK pathway was unaltered.
Translational perspective
Most disuse studies have focused on the loss of muscle mass, whereas less attention has been paid to metabolic adaptations notwithstanding the metabolic relevance of skeletal muscle and the major role of physical inactivity in causing metabolic diseases. This study addresses the complexity of muscle adaptations to disuse by a proteomic approach and the underlying mechanisms by analysis of intracellular signalling pathways. Using the bed rest (BR) model of human disuse, we report that (i) redox imbalance, impairment of antioxidant defence systems and metabolic derangement occurred early, before muscle atrophy developed, and persisted through BR; (ii) both major catabolic systems, ubiquitin proteasome and autophagy, could contribute to the progression of atrophy late into BR, although the role of decreased protein synthesis could not be ruled out; (iii) down-regulation of PGC-1α, a master controller of metabolism, and up-regulation of SREBP-1, a master controller of lipid synthesis, are likely to have triggered disuse adaptations through mitochondrial dysfunction, whereas the AMP kinase, an energy sensor pathway, was unaltered. Consistently with previously published in vivo findings in the same subjects and BR campaigns, it can be hypothesized that disuse induced mitochondrial dysfunction plays a central role in the overall adaptations to physical inactivity. Mitochondrial dysfunction could, in fact, link adverse phenomena at muscle level, i.e. impaired metabolism and muscle atrophy, to profound metabolic and endocrine alterations at the whole body level such as impaired oxygen consumption, and insulin resistance and systemic inflammation, which are the key risk factors of physical inactivity-induced chronic diseases.
Acknowledgments
The authors wish to thank the Italian Space Agency for the grants to R.B. and (project OSMA ‘Osteoporosis and Muscle Atrophy’) and the European Commission for MYOAGE (no. 223576) funded under FP7 to R.B. The authors wish to thank Mr Luigi Guidotti for excellent technical help during all experiments related to muscle disuse. We thank the volunteers, who gave their time and effort to ensure the success of this project. We acknowledge the excellent assistance of the entire staff of the Valdoltra Orthopaedic Hospital of Ankaran (Koper, Slovenia) and of Prof. Rado Pisot, University of Primorska, Slovenia.
Glossary
- AMPK
5′-adenosine monophosphate-activated protein kinase
- bnip3
Bcl-2/adenovirus E1B 19 kDa-interacting protein
- BR
bed rest
- CAHIII
carbonic anhydrase III
- CK
creatine kinase
- CSA
cross sectional area
- FABPH3
fatty acid-binding protein 3
- GAMT
guanidinoacetate N-methyltransferase
- HspB6
heat shock protein B6
- HspB1
heat shock protein B1
- Hsp70
heat shock protein 70
- LDH
lactate dehydrogenase
- LC3
microtubule-associated protein light chain 3
- MPB
muscle protein breakdown
- MPS
muscle protein synthesis
- MHC
myosin heavy chain
- MLC
myosin light chain
- MuRF-1
muscle-specific RING finger 1 protein
- NRF2
nuclear factor (erythroid-derived 2)-like 2
- PGC-1α
proliferator-activated receptor γ coactivator 1α
- PRDX3
peroxiredoxin 3
- ROS
reactive oxygen species
- p62/SQSTM1
sequestosome 1
- Tn
troponin
- SOD1
superoxide dismutase1
- SREBP-1
sterol regulatory element binding protein1
Author contributions
The experiments were performed in R.B.'s (proteomic, RT-PCR and Western blot analyses) and M.S.'s laboratories (RT-PCR and Western blot). Bed rest was performed under the supervision of G.B. Conception and design of the experiments: R.B., M.A.P. and M.S. Collection, analysis and interpretation of data: L.B., J.C., L.C. and G.B. Drafting the article or revising it critically for important intellectual content: R.B., M.S., M.A.P., G.B. and L.B.
Supplementary material
Supplemental Fig. 1
Supplemental Table 1
Supplemental Fig. 2
References
- Acheson KJ, Decombaz J, Piguet-Welsch C, Montigon F, Decarli B, Bartholdi I, Fern EB. Energy, protein, and substrate metabolism in simulated microgravity. Am J Physiol Regul Integr Comp Physiol. 1995;269:R252–260. doi: 10.1152/ajpregu.1995.269.2.R252. [DOI] [PubMed] [Google Scholar]
- Adams GR, Caiozzo VJ, Baldwin KM. Skeletal muscle unweighting: spaceflight and ground-based models. J Appl Physiol. 2003;95:2185–2201. doi: 10.1152/japplphysiol.00346.2003. [DOI] [PubMed] [Google Scholar]
- Agostini F, Heer M, Guarnieri G, Biolo G. Physical inactivity decreases whole body glutamine turnover independently from changes in proteolysis. J Physiol. 2008;586:4775–4781. doi: 10.1113/jphysiol.2008.153783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alibegovic AC, Hojbjerre L, Sonne MP, van Hall G, Alsted TJ, Kiens B, Stallknecht B, Dela F, Vaag A. Increased rate of whole body lipolysis before and after 9 days of bed rest in healthy young men born with low birth weight. Am J Physiol Endocrinol Metab. 2010a;298:E555–564. doi: 10.1152/ajpendo.00223.2009. [DOI] [PubMed] [Google Scholar]
- Alibegovic AC, Sonne MP, Hojbjerre L, Bork-Jensen J, Jacobsen S, Nilsson E, Faerch K, Hiscock N, Mortensen B, Friedrichsen M, Stallknecht B, Dela F, Vaag A. Insulin resistance induced by physical inactivity is associated with multiple transcriptional changes in skeletal muscle in young men. Am J Physiol Endocrinol Metab. 2010b;299:E752–763. doi: 10.1152/ajpendo.00590.2009. [DOI] [PubMed] [Google Scholar]
- Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol. 2011;85:241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- Baptista IL, Leal ML, Artioli GG, Aoki MS, Fiamoncini J, Turri AO, Curi R, Miyabara EH, Moriscot AS. Leucine attenuates skeletal muscle wasting via inhibition of ubiquitin ligases. Muscle Nerve. 2010;41:800–808. doi: 10.1002/mus.21578. [DOI] [PubMed] [Google Scholar]
- Bergouignan A, Rudwill F, Simon C, Blanc S. Physical inactivity as the culprit of metabolic inflexibility: Evidences from bed-rest studies. J Appl Physiol. 2011;111:1201–1210. doi: 10.1152/japplphysiol.00698.2011. [DOI] [PubMed] [Google Scholar]
- Bergstrom J. Muscle electrolytes in man. Scand J Clin Lab Invest Suppl. 1962;68:1–110. [Google Scholar]
- Berthon P, Duguez S, Favier FB, Amirouche A, Feasson L, Vico L, Denis C, Freyssenet D. Regulation of ubiquitin-proteasome system, caspase enzyme activities, and extracellular proteinases in rat soleus muscle in response to unloading. Pflugers Arch. 2007;454:625–633. doi: 10.1007/s00424-007-0230-6. [DOI] [PubMed] [Google Scholar]
- Binas B, Han XX, Erol E, Luiken JJ, Glatz JF, Dyck DJ, Motazavi R, Adihetty PJ, Hood DA, Bonen A. A null mutation in H-FABP only partially inhibits skeletal muscle fatty acid metabolism. Am J Physiol Endocrinol Metab. 2003;285:E481–489. doi: 10.1152/ajpendo.00060.2003. [DOI] [PubMed] [Google Scholar]
- Biolo G, Agostini F, Simunic B, Sturma M, Torelli L, Preiser JC, Deby-Dupont G, Magni P, Strollo F, di Prampero P, Guarnieri G, Mekjavic IB, Pisot R, Narici MV. Positive energy balance is associated with accelerated muscle atrophy and increased erythrocyte glutathione turnover during 5 wk of bed rest. Am J Clin Nutr. 2008;88:950–958. doi: 10.1093/ajcn/88.4.950. [DOI] [PubMed] [Google Scholar]
- Blaauw B, Canato M, Agatea L, Toniolo L, Mammucari C, Masiero E, Abraham R, Sandri M, Schiaffino S, Reggiani C. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J. 2009;23:3896–3905. doi: 10.1096/fj.09-131870. [DOI] [PubMed] [Google Scholar]
- Borina E, Pellegrino MA, D’Antona G, Bottinelli R. Myosin and actin content of human skeletal muscle fibers following 35 days bed rest. Scand J Med Sci Sports. 2010;20:65–73. doi: 10.1111/j.1600-0838.2009.01029.x. [DOI] [PubMed] [Google Scholar]
- Braissant O, Henry H, Loup M, Eilers B, Bachmann C. Endogenous synthesis and transport of creatine in the rat brain: an in situ hybridization study. Brain Res Mol Brain Res. 2001;86:193–201. doi: 10.1016/s0169-328x(00)00269-2. [DOI] [PubMed] [Google Scholar]
- Brocca L, Pellegrino MA, Desaphy JF, Pierno S, Camerino DC, Bottinelli R. Is oxidative stress a cause or consequence of disuse muscle atrophy in mice? A proteomic approach in hindlimb-unloaded mice. Exp Physiol. 2010;95:331–350. doi: 10.1113/expphysiol.2009.050245. [DOI] [PubMed] [Google Scholar]
- Canepari M, Pellegrino MA, D’Antona G, Bottinelli R. Skeletal muscle fibre diversity and the underlying mechanisms. Acta Physiol (Oxf) 2010;199:465–476. doi: 10.1111/j.1748-1716.2010.02118.x. [DOI] [PubMed] [Google Scholar]
- Chen YW, Gregory CM, Scarborough MT, Shi R, Walter GA, Vandenborne K. Transcriptional pathways associated with skeletal muscle disuse atrophy in humans. Physiol Genomics. 2007;31:510–520. doi: 10.1152/physiolgenomics.00115.2006. [DOI] [PubMed] [Google Scholar]
- Commerford SR, Peng L, Dube JJ, O’Doherty RM. In vivo regulation of SREBP-1c in skeletal muscle: effects of nutritional status, glucose, insulin, and leptin. Am J Physiol Regul Integr Comp Physiol. 2004;287:R218–227. doi: 10.1152/ajpregu.00377.2003. [DOI] [PubMed] [Google Scholar]
- Dalla Libera L, Ravara B, Gobbo V, Tarricone E, Vitadello M, Biolo G, Vescovo G, Gorza L. A transient antioxidant stress response accompanies the onset of disuse atrophy in human skeletal muscle. J Appl Physiol. 2009;107:549–557. doi: 10.1152/japplphysiol.00280.2009. [DOI] [PubMed] [Google Scholar]
- Dapp C, Schmutz S, Hoppeler H, Fluck M. Transcriptional reprogramming and ultrastructure during atrophy and recovery of mouse soleus muscle. Physiol Genomics. 2004;20:97–107. doi: 10.1152/physiolgenomics.00100.2004. [DOI] [PubMed] [Google Scholar]
- de Boer MD, Selby A, Atherton P, Smith K, Seynnes OR, Maganaris CN, Maffulli N, Movin T, Narici MV, Rennie MJ. The temporal responses of protein synthesis, gene expression and cell signalling in human quadriceps muscle and patellar tendon to disuse. J Physiol. 2007;585:241–251. doi: 10.1113/jphysiol.2007.142828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Keizer PL, Burgering BM, Dansen TB. Forkhead box O as a sensor, mediator, and regulator of redox signaling. Antioxid Redox Signal. 2011;14:1093–1106. doi: 10.1089/ars.2010.3403. [DOI] [PubMed] [Google Scholar]
- di Prampero PE, Narici MV. Muscles in microgravity: from fibres to human motion. J Biomech. 2003;36:403–412. doi: 10.1016/s0021-9290(02)00418-9. [DOI] [PubMed] [Google Scholar]
- Fitts RH, Riley DR, Widrick JJ. Physiology of a microgravity environment invited review: microgravity and skeletal muscle. J Appl Physiol. 2000;89:823–839. doi: 10.1152/jappl.2000.89.2.823. [DOI] [PubMed] [Google Scholar]
- Fitts RH, Riley DR, Widrick JJ. Functional and structural adaptations of skeletal muscle to microgravity. J Exp Biol. 2001;204:3201–3208. doi: 10.1242/jeb.204.18.3201. [DOI] [PubMed] [Google Scholar]
- Flogel U, Merx MW, Godecke A, Decking UK, Schrader J. Myoglobin: A scavenger of bioactive NO. Proc Natl Acad Sci U S A. 2001;98:735–740. doi: 10.1073/pnas.011460298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraysse B, Desaphy JF, Pierno S, De Luca A, Liantonio A, Mitolo CI, Camerino DC. Decrease in resting calcium and calcium entry associated with slow-to-fast transition in unloaded rat soleus muscle. FASEB J. 2003;17:1916–1918. doi: 10.1096/fj.02-1012fje. [DOI] [PubMed] [Google Scholar]
- Glover EI, Phillips SM, Oates BR, Tang JE, Tarnopolsky MA, Selby A, Smith K, Rennie MJ. Immobilization induces anabolic resistance in human myofibrillar protein synthesis with low and high dose amino acid infusion. J Physiol. 2008;586:6049–6061. doi: 10.1113/jphysiol.2008.160333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover EI, Yasuda N, Tarnopolsky MA, Abadi A, Phillips SM. Little change in markers of protein breakdown and oxidative stress in humans in immobilization-induced skeletal muscle atrophy. Appl Physiol Nutr Metab. 2010;35:125–133. doi: 10.1139/H09-137. [DOI] [PubMed] [Google Scholar]
- Gondin J, Brocca L, Bellinzona E, D’Antona G, Maffiuletti NA, Miotti D, Pellegrino MA, Bottinelli R. Neuromuscular electrical stimulation training induces atypical adaptations of human skeletal muscle phenotype: a functional and proteomic analysis. J Appl Physiol. 2010;110:433–450. doi: 10.1152/japplphysiol.00914.2010. [DOI] [PubMed] [Google Scholar]
- Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- Gustafsson T, Osterlund T, Flanagan JN, von Walden F, Trappe TA, Linnehan RM, Tesch PA. Effects of 3 days unloading on molecular regulators of muscle size in humans. J Appl Physiol. 2010;109:721–727. doi: 10.1152/japplphysiol.00110.2009. [DOI] [PubMed] [Google Scholar]
- Handschin C. Peroxisome proliferator-activated receptor-γ coactivator-1α in muscle links metabolism to inflammation. Clin Exp Pharmacol Physiol. 2009;36:1139–1143. doi: 10.1111/j.1440-1681.2009.05275.x. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase: development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haus JM, Carrithers JA, Carroll CC, Tesch PA, Trappe TA. Contractile and connective tissue protein content of human skeletal muscle: effects of 35 and 90 days of simulated microgravity and exercise countermeasures. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1722–1727. doi: 10.1152/ajpregu.00292.2007. [DOI] [PubMed] [Google Scholar]
- Hortobagyi T, Devita P. Mechanisms responsible for the age-associated increase in coactivation of antagonist muscles. Exerc Sport Sci Rev. 2006;34:29–35. doi: 10.1097/00003677-200601000-00007. [DOI] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: transcriptional mediators of lipid homeostasis. Cold Spring Harb Symp Quant Biol. 2002;67:491–498. doi: 10.1101/sqb.2002.67.491. [DOI] [PubMed] [Google Scholar]
- Hur W, Gray NS. Small molecule modulators of antioxidant response pathway. Curr Opin Chem Biol. 2011;15:162–173. doi: 10.1016/j.cbpa.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Hussain SN, Mofarrahi M, Sigala I, Kim HC, Vassilakopoulos T, Maltais F, Bellenis I, Chaturvedi R, Gottfried SB, Metrakos P, Danialou G, Matecki S, Jaber S, Petrof BJ, Goldberg P. Mechanical ventilation-induced diaphragm disuse in humans triggers autophagy. Am J Respir Crit Care Med. 2010;182:1377–1386. doi: 10.1164/rccm.201002-0234OC. [DOI] [PubMed] [Google Scholar]
- Ikemoto M, Nikawa T, Takeda S, Watanabe C, Kitano T, Baldwin KM, Izumi R, Nonaka I, Towatari T, Teshima S, Rokutan K, Kishi K. Space shuttle flight (STS-90) enhances degradation of rat myosin heavy chain in association with activation of ubiquitin-proteasome pathway. FASEB J. 2001a;15:1279–1281. doi: 10.1096/fj.00-0629fje. [DOI] [PubMed] [Google Scholar]
- Ikemoto M, Nikawa T, Takeda S, Watanabe C, Kitano T, Baldwin KM, Izumi R, Nonaka I, Towatari T, Teshima S, Rokutan K, Kishi K. Space shuttle flight (STS-90) enhances degradation of rat myosin heavy chain in association with activation of ubiquitin-proteasome pathway. FASEB J. 2001b;15:1279–1281. doi: 10.1096/fj.00-0629fje. [DOI] [PubMed] [Google Scholar]
- Ingalls CP, Warren GL, Armstrong RB. Intracellular Ca2+ transients in mouse soleus muscle after hindlimb unloading and reloading. J Appl Physiol. 1999;87:386–390. doi: 10.1152/jappl.1999.87.1.386. [DOI] [PubMed] [Google Scholar]
- Irrcher I, Adhihetty PJ, Sheehan T, Joseph AM, Hood DA. PPARgamma coactivator-1alpha expression during thyroid hormone- and contractile activity-induced mitochondrial adaptations. Am J Physiol Cell Physiol. 2003;284:C1669–1677. doi: 10.1152/ajpcell.00409.2002. [DOI] [PubMed] [Google Scholar]
- Jackman RW, Kandarian SC. The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol. 2004;287:C834–843. doi: 10.1152/ajpcell.00579.2003. [DOI] [PubMed] [Google Scholar]
- Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102:401–414. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Lawler JM, Song W, Demaree SR. Hindlimb unloading increases oxidative stress and disrupts antioxidant capacity in skeletal muscle. Free Radic Biol Med. 2003;35:9–16. doi: 10.1016/s0891-5849(03)00186-2. [DOI] [PubMed] [Google Scholar]
- Lawler JM, Song W, Kwak HB. Differential response of heat shock proteins to hindlimb unloading and reloading in the soleus. Muscle Nerve. 2006;33:200–207. doi: 10.1002/mus.20454. [DOI] [PubMed] [Google Scholar]
- Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- Levine S, Nguyen T, Taylor N, Friscia ME, Budak MT, Rothenberg P, Zhu J, Sachdeva R, Sonnad S, Kaiser LR, Rubinstein NA, Powers SK, Shrager JB. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med. 2008;358:1327–1335. doi: 10.1056/NEJMoa070447. [DOI] [PubMed] [Google Scholar]
- Li YP, Chen Y, Li AS, Reid MB. Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am J Physiol Cell Physiol. 2003;285:C806–812. doi: 10.1152/ajpcell.00129.2003. [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Makowski L, Hotamisligil GS. Fatty acid binding proteins – the evolutionary crossroads of inflammatory and metabolic responses. J Nutr. 2004;134:2464S–2468S. doi: 10.1093/jn/134.9.2464S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzucco S, Agostini F, Biolo G. Inactivity-mediated insulin resistance is associated with upregulated pro-inflammatory fatty acids in human cell membranes. Clin Nutr. 2010a;29:386–390. doi: 10.1016/j.clnu.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Mazzucco S, Agostini F, Mangogna A, Cattin L, Biolo G. Prolonged inactivity up-regulates cholesteryl ester transfer protein independently of body fat changes in humans. J Clin Endocrinol Metab. 2010b;95:2508–2512. doi: 10.1210/jc.2009-2561. [DOI] [PubMed] [Google Scholar]
- Merx MW, Flogel U, Stumpe T, Godecke A, Decking UK, Schrader J. Myoglobin facilitates oxygen diffusion. FASEB J. 2001;15:1077–1079. doi: 10.1096/fj.00-0497fje. [DOI] [PubMed] [Google Scholar]
- Miyata T, Takizawa S, van Ypersele de Strihou C. Hypoxia. 1. Intracellular sensors for oxygen and oxidative stress: novel therapeutic targets. Am J Physiol Cell Physiol. 2011;300:C226–231. doi: 10.1152/ajpcell.00430.2010. [DOI] [PubMed] [Google Scholar]
- Moriggi M, Vasso M, Fania C, Capitanio D, Bonifacio G, Salanova M, Blottner D, Rittweger J, Felsenberg D, Cerretelli P, Gelfi C. Long term bed rest with and without vibration exercise countermeasures: effects on human muscle protein dysregulation. Proteomics. 2010;10:3756–3774. doi: 10.1002/pmic.200900817. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa T, Furochi H, Mameoka M, Hirasaka K, Onishi Y, Suzue N, Oarada M, Akamatsu M, Akima H, Fukunaga T, Kishi K, Yasui N, Ishidoh K, Fukuoka H, Nikawa T. Ubiquitin ligase gene expression in healthy volunteers with 20-day bedrest. Muscle Nerve. 2006;34:463–469. doi: 10.1002/mus.20611. [DOI] [PubMed] [Google Scholar]
- Palomero J, Pye D, Kabayo T, Spiller DG, Jackson MJ. In situ detection and measurement of intracellular reactive oxygen species in single isolated mature skeletal muscle fibers by real time fluorescence microscopy. Antioxid Redox Signal. 2008;10:1463–1474. doi: 10.1089/ars.2007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MA, Canepari M, Rossi R, D’Antona G, Reggiani C, Bottinelli R. Orthologous myosin isoforms and scaling of shortening velocity with body size in mouse, rat, rabbit and human muscles. J Physiol. 2003;546:677–689. doi: 10.1113/jphysiol.2002.027375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MA, D’Antona G, Bortolotto S, Boschi F, Pastoris O, Bottinelli R, Polla B, Reggiani C. Clenbuterol antagonizes glucocorticoid-induced atrophy and fibre type transformation in mice. Exp Physiol. 2004;89:89–100. doi: 10.1113/expphysiol.2003.002609. [DOI] [PubMed] [Google Scholar]
- Pellegrino MA, Desaphy JF, Brocca L, Pierno S, Camerino DC, Bottinelli R. Redox homeostasis, oxidative stress and disuse muscle atrophy. J Physiol. 2011;589:2147–2160. doi: 10.1113/jphysiol.2010.203232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips DI, Caddy S, Ilic V, Fielding BA, Frayn KN, Borthwick AC, Taylor R. Intramuscular triglyceride and muscle insulin sensitivity: evidence for a relationship in nondiabetic subjects. Metabolism. 1996;45:947–950. doi: 10.1016/s0026-0495(96)90260-7. [DOI] [PubMed] [Google Scholar]
- Pilegaard H, Saltin B, Neufer PD. Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J Physiol. 2003;546:851–858. doi: 10.1113/jphysiol.2002.034850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcelli S, Marzorati M, Lanfranconi F, Vago P, Pisot R, Grassi B. Role of skeletal muscles impairment and brain oxygenation in limiting oxidative metabolism during exercise after bed rest. J Appl Physiol. 2010;109:101–111. doi: 10.1152/japplphysiol.00782.2009. [DOI] [PubMed] [Google Scholar]
- Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Smuder AJ, Criswell DS. Mechanistic links between oxidative stress and disuse muscle atrophy. Antioxid Redox Signal. 2011;15:2519–2528. doi: 10.1089/ars.2011.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisanen SR, Lehenkari P, Tasanen M, Rahkila P, Harkonen PL, Vaananen HK. Carbonic anhydrase III protects cells from hydrogen peroxide-induced apoptosis. FASEB J. 1999;13:513–522. doi: 10.1096/fasebj.13.3.513. [DOI] [PubMed] [Google Scholar]
- Rennie MJ, Selby A, Atherton P, Smith K, Kumar V, Glover EL, Philips SM. Facts, noise and wishful thinking: muscle protein turnover in aging and human disuse atrophy. Scand J Med Sci Sports. 2010;20:5–9. doi: 10.1111/j.1600-0838.2009.00967.x. [DOI] [PubMed] [Google Scholar]
- Riley DA, Bain JL, Thompson JL, Fitts RH, Widrick JJ, Trappe SW, Trappe TA, Costill DL. Disproportionate loss of thin filaments in human soleus muscle after 17-day bed rest. Muscle Nerve. 1998;21:1280–1289. doi: 10.1002/(sici)1097-4598(199810)21:10<1280::aid-mus6>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Ringholm S, Bienso RS, Kiilerich K, Guadalupe-Grau A, Aachmann-Andersen NJ, Saltin B, Plomgaard P, Lundby C, Wojtaszewski JF, Calbet JA, Pilegaard H. Bed rest reduces metabolic protein content and abolishes exercise-induced mRNA responses in human skeletal muscle. Am J Physiol Endocrinol Metab. 2011;301:E649–658. doi: 10.1152/ajpendo.00230.2011. [DOI] [PubMed] [Google Scholar]
- Romanello V, Sandri M. Mitochondrial biogenesis and fragmentation as regulators of muscle protein degradation. Curr Hypertens Rep. 2010;12:433–439. doi: 10.1007/s11906-010-0157-8. [DOI] [PubMed] [Google Scholar]
- Sacheck JM, Hyatt JP, Raffaello A, Jagoe RT, Roy RR, Edgerton VR, Lecker SH, Goldberg AL. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 2007;21:140–155. doi: 10.1096/fj.06-6604com. [DOI] [PubMed] [Google Scholar]
- Sandri M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 2008;23:160–170. doi: 10.1152/physiol.00041.2007. [DOI] [PubMed] [Google Scholar]
- Sandri M. Autophagy in health and disease. 3. Involvement of autophagy in muscle atrophy. Am J Physiol Cell Physiol. 2010a;298:C1291–1297. doi: 10.1152/ajpcell.00531.2009. [DOI] [PubMed] [Google Scholar]
- Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010b;584:1411–1416. doi: 10.1016/j.febslet.2010.01.056. [DOI] [PubMed] [Google Scholar]
- Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, Spiegelman BM. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci U S A. 2006;103:16260–16265. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Marescau B, Boehm EA, Renema WK, Peco R, Das A, Steinfeld R, Chan S, Wallis J, Davidoff M, Ullrich K, Waldschutz R, Heerschap A, De Deyn PP, Neubauer S, Isbrandt D. Severely altered guanidino compound levels, disturbed body weight homeostasis and impaired fertility in a mouse model of guanidinoacetate N-methyltransferase (GAMT) deficiency. Hum Mol Genet. 2004;13:905–921. doi: 10.1093/hmg/ddh112. [DOI] [PubMed] [Google Scholar]
- Senf SM, Dodd SL, McClung JM, Judge AR. Hsp70 overexpression inhibits NF-κB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J. 2008;22:3836–3845. doi: 10.1096/fj.08-110163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenburg EE, Booth FW. Molecular regulation of individual skeletal muscle fibre types. Acta Physiol Scand. 2003;178:413–424. doi: 10.1046/j.1365-201X.2003.01158.x. [DOI] [PubMed] [Google Scholar]
- Stein TP, Wade CE. Metabolic consequences of muscle disuse atrophy. J Nutr. 2005;135:1824S–1828S. doi: 10.1093/jn/135.7.1824S. [DOI] [PubMed] [Google Scholar]
- Tawa NE, Jr, Odessey R, Goldberg AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest. 1997;100:197–203. doi: 10.1172/JCI119513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urso ML, Chen YW, Scrimgeour AG, Lee PC, Lee KF, Clarkson PM. Alterations in mRNA expression and protein products following spinal cord injury in humans. J Physiol. 2007;579:877–892. doi: 10.1113/jphysiol.2006.118042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KY, Spector A. α-Crystallin can act as a chaperone under conditions of oxidative stress. Invest Ophth Vis Sci. 1995;36:311–321. [PubMed] [Google Scholar]
- Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.