

Abstract
Oxidative metabolism is needed for sustained skeletal muscle function. A key component of such metabolism is cytochrome c oxidase, the 13-subunit terminal complex of the mitochondrial electron transport chain. We used mice null for one of the two isoforms of Cox subunit 7a, heart/skeletal muscle-specific Cox7a1, to examine the cellular and functional responses of muscle adaptation in response to mitochondrial dysfunction. Specifically we determined if deletion of Cox7a1 would (1) limit exercise capacity, and (2) alter genes responsible for skeletal muscle capillarity and mitochondrial biogenesis. Sixteen male mice (Cox7a1 null mice, n = 8, and littermate controls, n = 8) performed incremental and run-to-exhaustion treadmill tests. The hindlimb muscles for both groups were analysed. The results indicated that capillary indices were reduced (by 30.7–44.9%) in the Cox7a1 null mice relative to controls. In addition, resting ATP levels and Cox specific activity were significantly reduced (>60%) in both glycolytic and oxidative muscle fibre types despite an increase in a major regulator of mitochondrial biogenesis, PGC-1β. These changes in the skeletal muscle resulted in exercise intolerance for the Cox7a1 null mice. Thus, our data indicate that deletion of the Cox7a1 isoform results in reduced muscle bioenergetics and hindlimb capillarity, helping to explain the observed impairment of muscle structure and function.
Key points
Cytochrome c oxidase (Cox) is the proposed rate-limiting enzyme of aerobic metabolism in mammals.
This study examined a mouse model that has a heart/skeletal muscle-specific isoform of Cox deleted and represents a mild form of mitochondrial myopathy.
The results indicated that endurance capacity was severely limited in the knockout animals compared with controls.
The observed exercise intolerance was accompanied by decreased capillarity in the hindlimb muscle as well as reduced energy production.
The results suggest that mild mitochondrial myopathy results in adverse structural and functions changes in muscle bioenergetics.
Introduction
The ability of working muscles to maintain continuous exercise results from the integrative response between central and peripheral factors to deliver oxygen to the mitochondria (Wagner, 1996). The utilization of oxygen by the mitochondria to generate energy is under the control of cytochrome c oxidase (complex IV or Cox) which consists of 13 subunits (10 encoded in the nuclear genome, 3 encoded in the mitochondrial genome) (Hüttemann et al. 2012a). It has been proposed that Cox is the rate-limiting enzyme of the electron transport chain (ETC) (Villani et al. 1998; Acin-Perez et al. 2003) and studies have revealed six isoforms of Cox subunits that are tissue specific and that potentially regulate energy production (Hüttemann et al. 2012a). Deficiencies in Cox-related genes have been implicated in various clinical conditions such as Leigh syndrome (Tiranti et al. 1998).
Both delivery of oxygen to mitochondria (and removal of byproducts) and the ability of mitochondria to use oxygen effectively are important in considering the basis of muscle fatigue. Accordingly, exercise physiology studies examining muscle fatigue have primarily focused on alterations to skeletal muscle angiogenesis and regulators of mitochondrial biogenesis. Angiogenesis requires VEGF (vascular endothelial growth factor) and Olfert et al. (2009) found severe exercise limitations in muscle-specific deletion of the VEGF gene that was not reversed with 6 weeks of endurance training (Olfert et al. 2010). Conversely, Malek & Olfert (2009) reported that mice with global deletion of another component, the anti-angiogenic growth factor thrombospodin-1 (TSP-1), had significant capillary proliferation in cardiac and skeletal muscle, resulting in higher endurance capacity. Kaur et al. (2010) have suggested an interplay between VEGF-A, TSP-1 and their receptors (VEGFR2 and CD47) in regulating angiogenesis. For mitochondrial proliferation, various transgenic rodent model studies have shown that PGC-1α (Leone et al. 2005; Geng et al. 2010; Leick et al. 2010) and PGC-1β (Shao et al. 2010; Rowe et al. 2011) are critical for mitochondrial increase, which functionally influences exercise tolerance. Furthermore, studies have shown that receptor interacting protein 1 (RIP140) is potentially a transcriptional repressor of PGC-1, therefore reducing mitochondrial enzyme expression (Seth et al. 2007; Williams et al. 2009; Frier et al. 2011).
Radford and colleagues (Radford et al. 2002) showed that knockout of the heart/skeletal muscle-specific subunit VIa in mice leads to cardiac dysfunction via reduced diastolic performance. Tissue energy levels, however, were not affected. Recently, Hüttemann et al. (2012b) generated Cox7a1 null mice to examine the role of the Cox gene on cardiac function. This particular isoform was selected because it is primarily expressed in cardiac and skeletal muscle, and it was proposed to adapt Cox activity to tissue-specific energy demand in these muscles. The investigators found that Cox7a1 null mice had reduced Cox activity (53%) in the heart. In addition, these transgenic animals developed dilated cardiomyopathy at 6 weeks of age, which stabilized at 6 months (Hüttemann et al. 2012b). In contrast to these relatively mild phenotypes studied under unstressed conditions, Diaz et al. (2005) developed Cox10 null mice, which focuses on the protohaem:haem-O-farnesyl transferase required for incorporation of haem a into Cox subunit I, as a model of severe mitochondrial disease. Indeed, Cox10 null mice died at 5 to 7 months despite <5% reduction in Cox activity (Diaz et al. 2005; Wenz et al. 2009).
Investigating the cellular and functional responses in the hindlimb muscles of Cox7a1 null mice may provide further valuable information about muscle adaptation resulting from mitochondrial dysfunction. Therefore, the purposes of this study were to determine if deletion of Cox7a1 would (1) limit exercise capacity, and (2) alter expression of genes responsible for skeletal muscle capillarity and mitochondrial biogenesis. We hypothesized that exercise tolerance would be limited in Cox7a1 null mice compared with littermate controls and that this would be associated with lower Cox activity in both glycolytic and oxidative hindlimb muscles. Furthermore, we hypothesized that the reduced capillarity in the Cox7a1 null mice would be associated with an increase in the anti-angiogenic response.
Methods
Animals and ethical approval
We studied 6-month-old male Cox7a1 null mice (n = 8) and littermate controls (n = 8). In each cage, four animals per group were maintained (21°C with 12 h light:dark cycles) and given standard rodent chow and water. All experimental procedures were approved by the Wayne State University Animal Care and Use Committee.
Experimental approach and design
The main objective of the present investigation was to determine the effects of Cox7a1 deletion on exercise performance, skeletal muscle morphology and oxidative phosphorylation. Therefore, both Cox7a1 null and littermate control mice performed incremental and run-to-exhaustion treadmill tests. The quadriceps and soleus muscles were analysed.
Genotyping of Cox7a1 null mice
Detailed descriptions of generating these transgenic animals have been previously reported (Hüttemann et al. 2012b). Chimeric animals were obtained after electroporation of our knockout construct into mouse R1 embryonic stem cells, replacing all three exons with the neo cassette as described in detail, and when Cox7a1 transcripts could no longer be detected by qPCR (Hüttemann et al. 2012b). Animals were bred for more than 10 generations using C57BL/6J mice. Genotyping of both Cox7a1 null mice and littermate controls were conducted using ear and tail tissues with the Wizard Genomic DNA Purification Kit (Promega). Two separate PCRs were performed to identify the wild-type and the recombinant alleles using primer pairs PA/PB and PA/PC, respectively: forward primer PA (5′-CGCCCATTTCACATTCTCAGCACTGGAG-3′), wild-type reverse primer PB located in Cox7a1 exon I (5′-AAGAGCTTCTGCTTCTCTGCCAC-3′), recombinant-specific reverse primer PC located in the Neo gene (5′-ACGGTATCGCCGCTCCCGATTCGCAG-3′). PCR conditions were 2 min initial denaturation at 93°C; 13 cycles: 18 s, 93°C; 40 s, 54°C; 2 min, 69°C; 22 cycles: 18 s, 93°C; 35 s, 55°C; 2 min, 69°C plus 4 s per each new cycle. PCR products were loaded and separated on a 1% agarose gel containing ethidium bromide, and wild-type and recombinant PCR products were observed at about 0.7 and 1 kb, respectively.
Incremental and run-to-exhaustion treadmill test
Both Cox7a1 null mice and littermate controls were familiarized with the rodent treadmill (1055MSD Exer-6M, Columbus, OH, USA) at 5 m min-1 and 5 deg incline for ≤10 min two times in the week prior to the testing date. For the incremental test, mice started at 4 m min-1 for 2 min with an increase in speed of 2 m min-1. Each lane was equipped with a shock grid set at 0.2 mA to prevent sitting on the grid. When animals were unable to maintain running on the treadmill via frequent contacts or sitting on the shock grid as previously performed in our laboratory (Malek & Olfert, 2009; Malek et al. 2010; Nogueira et al. 2011; Hüttemann et al. 2012c) the test was terminated.
To determine endurance capacity Cox7a1 null mice ran on the treadmill at 20 m min-1 and 5 deg incline, whereas littermate controls ran at 23 m min-1 until exhaustion. These two speeds represented 70% of maximal running speed for each group. This test was performed 4 days after the incremental test. For both exercise tests, indices of performance such as distance, work and power were calculated as previously described (Handschin et al. 2007).
Hindlimb muscle preparation
After anaesthetizing (sodium pentobarbital, 60 mg kg-1, i.p.) the mice, the right quadriceps femoris muscle was dissected and a transverse section was taken and frozen in precooled isopentane and stored at –80°C for analyses. All animals were killed by removal of the heart. For capillarity analyses, transverse 10 μm serial sections were cut on a cryotome (Leica CM 1950; Buffalo Grove, IL, USA) at –20°C and mounted on slides. The left quadriceps femoris and soleus muscles were prepared and stored at –80°C until enzyme/metabolite and molecular analyses were performed.
Capillary staining and indices
The Rosenblatt method (Rosenblatt et al. 1987) was used to stain for capillarity of the quadriceps muscle. Each muscle section was viewed under a digital microscope (20× magnification, Leica DMD108). Capillarity was quantified using the method of Hepple et al. (1997) that determines the number of capillaries around a fibre (NCAF), the capillary-to-fibre ratio on an individual-fibre basis (C/Fi), and capillary density (CD) which was calculated by using the fibre area as the reference space (Hepple & Mathieu-Costello, 2001). Capillary-to-fibre perimeter exchange index (CFPE) was calculated as an estimate of the capillary-to-fibre surface area (Hepple, 1997). We also used the method of Snyder (Snyder, 1990) to estimate maximal diffusion distance (R95). Fibre cross-sectional area (FCSA) and perimeter (FP) were measured with the image analysis system and commercial software (SigmaScan Pro v. 5.0, Systat Software, Inc., Point Richmond, CA, USA).
Cytochrome c oxidase specific activity measurements
Cox activity was analysed with a micro Clark-type oxygen electrode in a closed chamber (Oxygraph system, Hansatech, Norfolk, UK). Skeletal muscle tissue (quadriceps and soleus muscle) was dissected and solubilized in 10 mm K-Hepes (pH 7.4), 40 mm KCl, 1% Tween 20, 1 μm oligomycin, 1 mm PMSF, 10 mm KF, 2 mm EGTA and 1 mm sodium vanadate, as described previously (Lee et al. 2005). Cox activity was measured in the presence of 20 mm ascorbate and by addition of increasing amounts of substrate cytochrome c from cow heart (Sigma). Protein concentration was determined with the DC protein assay kit (Bio-Rad, Hercules, CA, USA), whereas analysis of oxygen consumption was performed with the Oxygraph software. Cox specific activity is defined as consumed O2 (μmmin-1) standardized to GAPDH, as determined by Western analysis using a monoclonal anti-GAPDH antibody (G8795, Sigma-Aldrich, St Louis, MO, USA) as described previously (Hüttemann et al. 2012b). Cox activity is presented in arbitrary units (AU), set to 100% at maximal turnover for control animals.
ATP measurements
ATP levels were determined via the boiling method using frozen quadriceps and soleus tissues using the ATP bioluminescence assay kit HS II (Roche Applied Science) as described previously (Lee et al. 2005; Hüttemann et al. 2012b). Briefly, data were standardized to the protein concentration; 100 μl of a 20% SDS solution were added to each boiled sample containing precipitated proteins and cell debris, and ultrasonicated for a total time of 8 min. Protein concentration was determined as described above.
Western blotting
Western blot analysis was performed as described in Hüttemann et al. (2012c). Briefly, 50 mg of the quadriceps femoris muscle were homogenized in a glass tissue grinder with RIPA buffer (Sigma-Aldrich) and protease and phosphatase inhibitor cocktails (PhosSTOP Phosphatase and Complete Protease Inhibitor Cocktail, Roche Applied Science). Homogenates were passed through an insulin syringe five times, centrifuged for 10 min at 4°C, and the supernatant was collected, aliquoted and stored at −80°C. The BCA protein assay kit (Bio-Rad) was used to quantify protein concentration for each sample.
Protein samples (40 μg) were loaded onto 7.5% (TSP-1, PGC-1α, VEGFR2, Phospho-VEGFR2 at Tyr1175 and PGC-1β) or 12% TGX pre-cast gels (Bio-Rad) for 1 h at 160 V and transferred to polyvinylidene fluoride membranes (PVDF-FL, Immobilon transfer membranes; Millipore, Billerica, MA, USA) with a semi-dry blotting apparatus (12 V, 50 min, Bio-Rad). After 1 h of blocking (Li-Cor Biosciences, Lincoln, NE, USA) in the laboratory, the primary antibody was added and the membrane stored in a 4°C room on a rocker. The secondary antibody was added and the membrane was incubated at room temperature for 1 h on the second day. Following secondary antibody incubation, the membrane was washed four more times (5 min per wash) in Tris-buffered saline with Tween in the laboratory. Care was taken not to expose the membrane to light.
The monoclonal primary antibodies used were TSP-1 (1:500; sc-59886), PGC-1β (1:1000; sc-373771), CD47 (1:500; 3847-1, Epitomics), Anti-slow skeletal myosin heavy chain (NOQ7.5.4D; ab11083, Abcam), Anti-fast myosin skeletal heavy chain antibody (MY-32, ab51263, Abcam) and GAPDH (1:2000; ab9484). The polyclonal primary antibodies used were VEGF (1:500; sc-507), VEGFR2 (1:500; 2479, Cell Signalling), phospho-VEGFR2Tyr1175 (1:500; 2478, Cell Signalling), PGC-1α (1:1000; AB3242, Millipore), RIP140 (1:200; sc-8997) and α-tubulin (1:2000; 2144, Cell Signalling). The secondary antibodies used were goat anti-mouse IRDye (1:30,000) and goat anti-rabbit IRDye (1:30,000) purchased from Li-Cor Biosciences. All blots were scanned and quantified using the Odyssey infrared imaging system.
Statistical analyses
All data are presented as means ± SEM. Separate one-way ANOVAs were performed to compare the relevant group means for each dependent variable. For the Cox specific activity, a 2 (group: littermate control or Cox7a1 null mice) × 9 (cytochrome c concentrations: 0, 1, 2, 3, 5, 10, 15, 20 and 30 μm) mixed factorial ANOVA was performed. When appropriate post hoc Tukey's HSD was used to determine which means were significantly different from each other (Keppel & Wickens, 2004). Statistical significance was set at P ≤ 0.05, and data were analysed using the Statistical Package for the Social Sciences software (v. 19.0, IBM SPSS, Armonk, NY, USA).
Results
Animals
Cox7a1 null mice had greater body mass than littermate controls (Table 1), but there were no differences in quadriceps muscle mass between the two groups. For the soleus muscle, however, the Cox7a1 null mice had smaller muscle masses (absolute and relative) than the controls (Table 1).
Table 1.
Body and muscle masses
| Groups | ||
|---|---|---|
| Littermate controls n = 8 | Cox7a1 null mice n = 8 | |
| Body mass (g) | 28.1 | 30.3 ± 0.7* |
| Quadriceps femoris mass (mg) | 186 ± 4.3 | 191 ± 2.2 |
| Quadriceps femoris/body mass (mg g−1) | 6.6 ± 0.1 | 6.3 ± 0.2 |
| Soleus (mg) | 7.3 ± 0.5 | 5.4 ± 0.5** |
| Soleus/body mass (mg g−1) | 0.26 ± 0.02 | 0.18 ± 0.02** |
Values are mean ± SEM.
P < 0.05;
P < 0.01.
Exercise performance
For the incremental treadmill exercise, the control mice were able to achieve a greater maximal running speed, which corresponded to a greater distance and work (ranging from 22 to 33.8%), than for the Cox7a1 null mice (Table 2). Littermate controls were able to run 37.7% longer than Cox7a1 null mice during the submaximal run-to-exhaustion treadmill test (Table 3). As a result, control mice were able to achieve a greater distance and, therefore, perform more work.
Table 2.
Performance on incremental treadmill test
| Mice | Time (s) | Maximal speed (m min−1) | Distance (m) | Work (J) | Maximal power (W) |
|---|---|---|---|---|---|
| Littermate controls | 662 ± 10 | 23 ± 0.40 | 191 ± 40 | 133 ± 5 | 0.006 ± 0.00020 |
| Cox7a1 null mice | 543 ± 9* | 20 ± 0.3* | 149 ± 3* | 88 ± 4* | 0.004 ± 0.0001* |
| Difference (%) | −17.9% | −13.0% | −22.0% | −33.8% | −33.3% |
Values are mean ± SEM.
P < 0.001.
Table 3.
Results of treadmill submaximal run-to-exhaustion
| Mice | Time (min) | Distance (m) | Work (J) |
|---|---|---|---|
| Littermate controls | 61 ± 2 | 925 ± 35 | 825 ± 36 |
| Cox7a1 null mice | 38 ± 1* | 488 ± 18* | 436 ± 21* |
| Difference (%) | −37.7% | −47.2% | −47.2% |
Values are mean ± SEM.
P < 0.001.
Capillary profile of the hindlimb
In the quadriceps muscle there were significantly higher capillary indices (NCAF, C/Fi, CD and CFPE) in the control mice than in Cox7a1 null mice (Fig. 1). In addition, there were statistical differences between the two groups for FCSA in the quadriceps muscle (control, 3998 ± 81 μm2 vs. Cox7a1 null, 4684 ± 126 μm2, P = 0.01). Similarly, there were significant differences in the mean fibre perimeter between the two groups in the same muscle (control, 265 ± 2.0 μm vs. Cox7a1 null, 294 ± 4.6 μm, P = 0.004). The maximal diffusion distance (R95) was significantly shorter for the control group compared with the Cox7a1 null mice in the quadriceps muscle (39.1 ± 0.7 vs. 49.6 ± 0.7 μm, P = 0.0004). In addition, we examined myosin heavy chain (MHC) composition in the quadriceps muscle between the two groups using monoclonal specific antibodies. As shown in Fig. 1, there was no difference in slow-MHC protein expression between the two groups. There was, however, an increase (∼180%) in fast-MHC protein expression compared with littermate controls (Fig. 1).
Figure 1. Capillarity is lower in the quadriceps femoris muscle of Cox7a1 null mice, with an increase in fast-myosin heavy chain composition protein expression.

Histological representation of capillary profile in Cox7a1 and littermate controls at 20× magnification of the quadriceps muscle (top panels; scale bar, 100 μm). Comparison of NCAF (the number of capillaries around a fibre, A), C/Fi (capillary-to-fibre ratio on an individual-fibre basis, B), CD (capillary density, C) and CFPE (capillary-to-fibre perimeter exchange, D) between the two groups (n = 3 per group). Representative Western blot results for fast- or slow-myosin heavy chain composition in the quadriceps muscle (E; n = 7 per group). Values are mean ± SEM; *P < 0.001.
Protein expression for angiogenesis in hindlimb
To examine the basis for the differences in capillarity, we measured protein expression of VEGF-A and TSP-1, as well as their receptors (VEGFR2 and CD47), in the quadriceps muscles for control and Cox7a1 null mice (Fig. 2). The protein content of VEGF-A did not differ between the two groups, whereas Cox7a1 null mice showed an ∼68% increase in TSP-1 protein expression compared with littermate controls. Interestingly, we found no differences (P > 0.05) in VEGFR2 or CD47, but an ∼44% reduction in phosphorylation of VEGFR2 at Tyr1175 in Cox7a1 null mice (Fig. 2).
Figure 2.
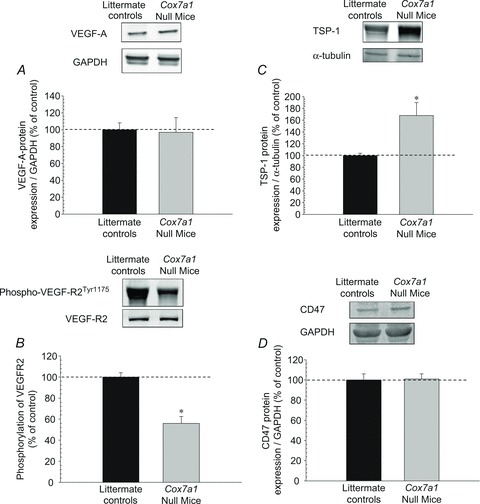
The effects of heart/muscle-specific subunit 7a1 deletion on protein expression of angiogenic and anti-angiogenic factors Representative Western blot results from the quadriceps femoris muscle are shown. Western blotting was carried out as described in Methods, using 40 μg muscle homogenate protein loaded onto 7.5% (TSP-1) or 12% (VEGF-A, VEGF-R2, CD47) precast polyacrylamide gels, run for 1 h at 160 V, and electrotransferred onto PVDF membranes. After antibody detection, images of blots were acquired with the Odyssey infrared imaging system and quantitated with its software program (Li-Cor Biosciences). *P < 0.05 (mean ± SEM; 5–6 per group).
Protein expression of regulators for mitochondrial biogenesis in hindlimb
We examined two key regulators of mitochondrial biogenesis in the quadriceps muscle of Cox7a1 null mice and littermate controls. PGC-1β was ∼214% greater in the Cox7a1 null mice compared with controls, although PGC-1α protein expression was unchanged. To further explore the difference in expression between PGC-1 forms, we probed the metabolic co-regulator RIP140 (Fritah et al. 2010), which can interact functionally with and repress PGC-1α (Hallberg et al. 2008). RIP140 protein expression was ∼156% greater in Cox7a1 null mice (Fig. 3).
Figure 3. Protein expression of mitochondrial regulators PGC-1 proteins and RIP140.
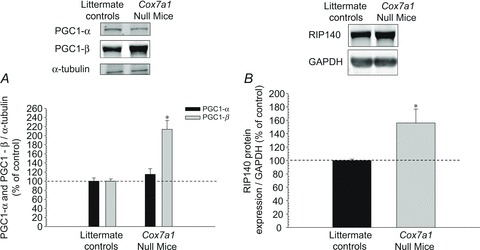
Representative Western blot results from the quadriceps femoris muscle were generated as in Fig. 2. PGC-1 (A) was separated on 7.5% gels and RIP140 (B) on 12%. *P < 0.01 (mean ± SEM; 5–8 per group).
Cox specific activity and ATP assay
We examined the effects of deleting the Cox7a1 isoform on mitochondrial energy metabolism in primary glycolytic (quadriceps) and oxidative (soleus) muscles by measuring resting ATP concentration and Cox specific activity. For both muscle types, the analyses revealed a significantly lower ATP concentration in the Cox7a1 null mice when compared with controls (Fig. 4). In addition, Cox specific activity in both muscles was significantly lower in the Cox7a1 null mice than in the control group (Fig. 5).
Figure 4. Basal ATP levels in both glycolytic and oxidative muscles are reduced in Cox7a1 null mice.

Resting ATP concentrations in the quadriceps femoris (A) and soleus (B) muscles were determined from frozen tissue as described in Methods using the boiling method and an ATP bioluminescence kit. *P < 0.001 (mean ± SEM; 8 per group).
Figure 5. Cytochrome c oxidase activity is decreased in quadriceps and soleus muscle of mice lacking heart/muscle-specific subunit 7a1.

Cox specific activity was determined in Cox7a1 null mice and littermate controls using the polarographic method by increasing the amount of substrate cytochrome c. Quadriceps muscle, which is more glycolytic (A), and the more oxidative soleus muscle (B), were analysed. Cox specific activity is defined as consumed O2 (μmol min-1) standardized to GAPDH levels. Cox activity is presented in arbitrary units (AU, set to 100% at maximal turnover for littermate controls) (mean ± SEM, n = 8 each, *P < 0.001). Note, KCN-insensitive respiration both in the wild-type and the knockout were similar (12.1 and 10.7%, respectively) which is in agreement with values from the literature.
Discussion
The main and unique finding of the present investigation is that loss of the heart/skeletal muscle isoform Cox7a1 resulted both in decreased capillarity and decreased mitochondrial energy production in the hindlimb muscles of the null mice. The decreased capillarity in Cox7a1 null mice may be due, in part, to an increase in TSP-1, an angiogenesis inhibitor, and to a reduction in phosphorylation of VEGFR2 at Tyr1175, which activates angiogenesis. Furthermore, we found that Cox7a1 null mice had higher protein expression of fast-MHC protein expression. The reduction in mitochondrial energy production in Cox7a1 null mice was the result of decreased Cox activity in the hindlimb muscles, regardless of fibre type, which manifested as decreased resting ATP concentrations. It should be noted that mitochondrial energy production decrease in Cox7a1 null mice occurred despite an increase in PGC-1β protein expression. These deficiencies in capillarity and mitochondrial energy production, taken together, resulted in substantially reduced exercise capacity.
The relationship between VEGF-A, TSP-1 and CD47 in skeletal muscle capillary maintenance
In the current investigation, we examined capillarity in the quadriceps femoris muscle of Cox7a1 and control mice. As shown in Fig. 1 (panels A–C), muscle capillarity of the Cox7a1 null mice was reduced compared with littermate controls. In addition, we measured CFPE (capillary-to-fibre perimeter exchange), which provides information regarding capillary supply of a given fibre relative to the fibre perimeter and, therefore, estimates resistance to oxygen flux (Hepple, 1997). Furthermore, we estimated maximal diffusion distance (R95), another index of the capillary-fibre interface (Hepple, 1997; Hepple & Mathieu-Costello, 2001). In Cox7a1 null mice CFPE was reduced by ∼41% (Fig. 1D), whereas R95 increased by ∼27% relative to control. Furthermore, we found that Cox7a1 null mice had significant increases in fast-MHC protein expression. Typically, skeletal muscle with a predominantly high fast-MHC composition are glycolytic with low aerobic capacity (Bloemberg & Quadrilatero, 2012). Collectively, these findings suggest the reduced capillarity observed in Cox7a1 null mice may result in impaired oxygen flux to the working muscle, thus limiting exercise performance (Tables 2 and 3). We cannot rule out, however, that reduced oxygen consumption of Cox in the transgenic mice (Fig. 5) triggers a feedback response, leading to reduction of skeletal muscle capillarity.
Interestingly, Taivassalo et al. (2012) reported increased capillarity in the vastus lateralis muscle of mitochondrial myopathy patients compared with controls. It may be that the nature of the defect determines the capillarity signalling pathway that prevails. Two notable differences between the present results and those of Taivassalo et al. are that (i) our Cox7a1 null model results from a nuclear DNA encoded Cox subunit, probably affecting oxidative phosphorylation in a different manner than the mtDNA mutations in their study, and (ii) we are explicitly lacking a Cox subunit whereas the patients in the Taivassalo et al. (2012) study, despite containing Cox negative fibres, have mutations either in tRNAs or in subunits of other complexes (5 of 9), or have deletions in an unstated position (3 of 9), or have an unidentified mutation (1 of 9). None of the mutations is in a Cox subunit. Furthermore, the patients all have ragged red fibres, typical of disease resulting from mtDNA mutations or nuclear DNA mutations that affect mtDNA content but not other nuclear DNA mutations that affect mitochondrial function (Van Hove et al. 2009; Berardo et al. 2010), suggesting different signalling pathways are operating. Lastly, tRNA mutations, potentially affecting the translation of all subunits, are expected to have the greatest multi-complex effects and two of the three tRNA patients are among the three showing the most capillarity coupled with the most Cox-deficient fibres (Taivassalo et al. 2012, their Fig. 4B).
Olfert and Birot (2011) have recently suggested that, although capillary maintenance in skeletal muscle involves a complex series of events, the (im-)balance between two primary candidate proteins, VEGF-A and TSP-1, determines angiogenesis. VEGF-A is a 45 kDa heparin-binding homodimeric glycoprotein that promotes angiogenesis (Ferrara & Davis-Smyth, 1997), whereas TSP-1 is a 450 kDa multifunctional homotrimeric matrix glycoprotein that attenuates angiogenesis (Iruela-Arispe et al. 1996; Kaur et al. 2010). Studies using siRNA against TSP-1 have shown increases in VEGF-A expression (Greenaway et al. 2007). Transgenic models (Malek & Olfert, 2009; Olfert et al. 2009, 2010), endurance training (Malek et al. 2010; Nogueira et al. 2011; Hüttemann et al. 2012c) and/or detraining (Malek et al. 2010; Roudier et al. 2010; Hüttemann et al. 2012c) protocols have shown that changes in VEGF-A and TSP-1 protein expression alter hindlimb capillarity, which influences endurance exercise capacity at the functional level. For example, Tang et al. (2004) used intramuscular injection of a viral vector to inactivate VEGF-A in regions of the gastrocnemius muscle of mice. The investigators reported a 64% decrease in capillarity of the targeted regions that was not reversed 8 weeks after the cessation of the treatment. Olfert et al. (2009) produced cardiac-skeletal muscle VEGF-A null mice and reported significant reductions in both cardiac and skeletal muscle capillarity, which corresponded to an ∼81% decrease in submaximal run-to-exhaustion treadmill test compared with controls. Furthermore, 6 weeks of endurance training did not increase hindlimb capillarity or exercise tolerance in the VEGF-A null mice (Olfert et al. 2010).
As expected, the reverse effect is found for skeletal muscle capillarity when TSP-1 or CD47 is deleted (Malek & Olfert, 2009; Roudier et al. 2010; Frazier et al. 2011). Malek and Olfert (2009) reported that TSP-1 null mice have significantly greater capillarity in both oxidative and glycolytic muscles when compared with controls. Furthermore, the resting VEGF-A protein concentration was ∼56% greater in TSP-1 null mice than controls. It should be noted that resting oxidative enzyme activity (citrate synthase and β-HAD) and MHC composition were not different in the muscles of TSP-1 null mice and controls. More recently, Hüttemann et al. (2012c) reported that 5 weeks of moderate intensity exercise (60% of maximal running speed) reduced TSP-1 protein expression by ∼66% compared with controls, whereas 15 days of detraining returned TSP-1 protein expression to control levels. Using hindlimb suspension, an unloading procedure designed to mimic weightlessness, Roudier et al. (2010) reported that TSP-1 protein expression became significantly elevated at 7 and 9 days of the intervention in the soleus muscle of rats compared with controls. However, no change was seen in TSP-1 protein expression for the plantaris muscle (Roudier et al. 2010). The differences in TSP-1 expression between the two types of muscles may be due to the baseline levels in the muscle since resting TSP-1 protein expression in the plantaris muscle was greater than 300% compared with the soleus muscle (Roudier et al. 2010).
A new anti-angiogenic mechanism of TSP-1 has been proposed that focuses on the interaction of its receptor (CD47) and the VEGF receptor, VEGFR2 (Kaur et al. 2010; Kaur & Roberts, 2011). Briefly, VEGFR2 is a tyrosine kinase receptor that is activated by ligand binding and autophosphorylation at Tyr1175, triggering various downstream signalling processes (Zachary, 2003). Studies with rodent models have shown that deletion of VEGFR2 disrupts normal vascular development (Shalaby et al. 1995; Zachary, 2003). Similarly, Lloyd and colleagues (2004) reported significant reductions in exercise-induced capillarity of rat glycolytic muscle when VEGFR2 was partially blocked by a receptor tyrosine kinase inhibitor. Recently, through a series of experiments using human umbilical vein and dermal microvascular endothelial cells, Kaur and colleagues (Kaur et al. 2010; Kaur & Roberts, 2011) reported that CD47 and VEGFR2 interact functionally and, therefore, the binding of TSP-1 to CD47 inhibits the phosphorylation of VEGFR2 at Tyr1175.
The observed increase in basal TSP-1 protein expression and the decrease in VEGFR2 phosphorylation at Tyr1175 (Fig. 2), on a background of unchanged protein expression of VEGF-A, VEGFR2 and CD47 in the quadriceps muscle, may account for the reduction in skeletal muscle capillarity observed in the Cox7a1 null mice (Fig. 1). To our knowledge, this is the first study to show the interaction between VEGF-A, TSP-1 and CD47 for maintenance of skeletal muscle capillarity using a transgenic murine model.
Reductions in skeletal muscle oxidative capacity of Cox7a1 null mice
The ability of the mitochondria to generate energy in the working muscle is critical for sustained activity, and therefore any disruptions may lead to reduced energy production (Wagner, 1996) with resultant premature muscle fatigue. Recently studies have shown that the PGC-1 proteins are strong regulators of mitochondria biogenesis (St-Pierre et al. 2003; Arany et al. 2007; Chinsomboon et al. 2009; Srivastava et al. 2009; Rowe et al. 2011). For example, Arany et al. (2007) found that PGC-1α protein was expressed at a higher level in oxidative muscle fibre types such as the soleus, but at a lower level in glycolytic muscle fibre types such as the quadriceps femoris. Arany et al. (2008), however, reported that increases in PGC-1α due to the lack of nutrients and oxygen is independent of hypoxia inducible factor. With regard to PGC-1β protein expression, however, the investigators found the reverse effect (Arany et al. 2007). In addition, they reported that the glycolytic muscle of PGC-1β overexpressed transgenic mice resembled that of oxidative muscle (Arany et al. 2007). Srivastava et al. (2009) reported that upregulation of both PGC-1α and PGC-1β partially increased mitochondrial respiration in cells taken from patients with mitochondrial dysfunction. The authors suggested that increasing expression of PGC-1 proteins in all cells would improve ATP production as a result of increasing the number of healthy mitochondria (Srivastava et al. 2009). St-Pierre and colleagues (2003) concluded that stimulation of mitochondrial biogenesis and respiration via PGC-1β is greater than PGC-1α. It has been suggested that PGC-1α is more likely to be upregulated during conditions that stimulate metabolism such as exercise, whereas PGC-1β is responsible for mitochondrial maintenance during basal conditions (Meirhaeghe et al. 2003; Liesa et al. 2008). In the current investigation, we examined the quadriceps femoris muscle of both Cox7a1 null mice and littermate controls and found that PGC-1α protein expression was not different (P > 0.05) between the two groups, whereas PGC-1β protein expression increased by ∼214% in the Cox7a1 null mice (Fig. 3). These findings provide further support for the notion that PGC-1β regulates mitochondrial maintenance during basal conditions.
It has recently been suggested that RIP140 represses PGC-1α in both skeletal muscle and adipose tissue (Seth et al. 2007; Hallberg et al. 2008; Williams et al. 2009; Frier et al. 2011). For example, Hallberg et al. (2008) concluded that PGC-1α and RIP140 may regulate mitochondrial biogenesis and respiration. Seth and colleagues (2007) reported that glycolytic hindlimb muscles of RIP140 null mice had significantly greater oxidative capacity than controls. Not surprisingly, the investigators also reported that deletion of RIP140 in the soleus muscle, a predominantly oxidative muscle, had little effect on the oxidative capacity of the muscle. Conversely, they overexpressed RIP140 in the soleus muscle and reported a dramatic reduction in oxidative capacity of the muscle which resembled the phenotype of a glycolytic muscle (Seth et al. 2007). In the present study, we found that protein expression of RIP140 was 56% greater in the quadriceps femoris muscle of Cox7a1 null mice relative to littermate controls (Fig. 3). Therefore, the lack of increase in PGC-1α protein expression that we observed in the transgenic animals (Fig. 3) may, in part, be explained by the increase in RIP140 (Fig. 3). Future studies, however, are needed to determine if RIP140 differentially affects PGC-1α and PGC-1β in regulating mitochondrial signalling.
Endurance training (Booth et al. 1998; Hüttemann et al. 2012c) or detraining (Hüttemann et al. 2012c) have been shown to alter ETC complexes as well as Cox activity and protein expression in the working muscle. Only one study (Wenz et al. 2009) to date has examined the effect of deleting Cox-associated protein on skeletal muscle function. Wenz and colleagues (2009) reported a reduction of 80% in resting ATP production in the biceps femoris muscle of Cox10 null mice at 6 months. The transgenic model used by Wenz et al. (2009), however, is an extreme model of mitochondrial myopathy as indicated by the ∼35% survival rate of their mice at the age of 5 months.
In the current investigation the Cox7a1 null mice had 100% survival at 6 months. Nevertheless, resting ATP concentration in the hindlimb muscles was reduced in the Cox7a1 null mice relative to littermate controls, independent of muscle fibre type (Fig. 4). Therefore, despite the high oxidative capacity of the soleus muscle, the deletion of this Cox isoform resulted in a ∼60% decrease of ATP concentration. Interestingly, when examining cardiac muscle in Cox7a1 null mice we showed increased ATP levels despite reduced Cox activity (Hüttemann et al. 2012b). It was proposed that increased glycolytic activity served as a compensatory mechanism. Similar to data reported in this study, Wenz et al. (2009) reported decreased ATP levels in their Cox10 null mice for the hamstring muscle, which is also primarily a glycolytic fibre.
We also examined resting Cox specific activity between the two types of muscle fibres. We found that Cox specific activity was reduced in the Cox7a1 null mice relative to littermate controls regardless of muscle fibre type (Fig. 5). At 5 μm cytochrome c substrate concentration Cox activity was reduced by 62% and 65%, whereas at maximal turnover (30 μm) Cox specific activity was reduced by 60% and 67% for the quadriceps and soleus muscles, respectively (Fig. 5). These results indicate that bioenergetic performance of Cox7a1 null mice was severely compromised in skeletal muscle.
A potential limitation of this study relates to the fact that the whole-body Cox7a1 mouse model develops early cardiomyopathy that could contribute to the observed exercise intolerance (Hüttemann et al. 2012b). Although we have shown that the hindlimb muscles of Cox7a1 null mice had reduced capillarity and oxidative capacity, future studies are needed to develop a skeletal-muscle-specific knockout of Cox7a1, which will allow us to determine structure and function of the hindlimb muscles without cardiac impairment.
In summary, we found that deletion of the Cox7a1 isoform resulted in significant loss of exercise tolerance. The exercise intolerance resulted both from reduction in skeletal muscle capillarity and from reduction of resting ATP concentration and Cox specific activity. A potential mechanism for the decrease in capillarity may be via the relationship among VEGF-A, TSP-1 and CD47. Furthermore, we found that PGC-1β, but not PGC-1α, was upregulated in the Cox7a1 null mice. Collectively, these results indicate that deletion of the heart/skeletal muscle-specific Cox subunit 7a results in impaired muscle function.
Acknowledgments
This work was supported by start-up funds (M.H.M.) from Wayne State University and funding from the National Institutes of Health (NIH) (GM089900, M.H.). Parts of this work were presented at 10th Annual Society for Heart and Vascular Metabolism Meeting in Merton College, Oxford as well as the 2012 American Society for Biochemistry and Molecular Biology Special Symposia Series (Mitochondria: Energy, Signals and Homeostasis) in East Lansing, Michigan.
Glossary
- CD
capillary density
- C/Fi
capillary-to-fibre ratio on an individual-fibre basis
- CFPE
capillary-to-fibre perimeter exchange index
- Cox
cytochrome c oxidase
- ETC
electron transport chain
- FCSA
fibre cross-sectional area
- FP
fibre perimeter
- NCAF
number of capillaries around a fibre
- VEGF
vascular endothelial growth factor
Author contributions
The study was conducted at WSU. Conception and design of study were by M.H.M. Conception of animal model by M.H. and L.I.G. All authors contributed to collection, analysis and interpretation of data and drafting the manuscript. All authors approved the final version of the manuscript for publication.
References
- Acin-Perez R, Bayona-Bafaluy MP, Bueno M, Machicado C, Fernandez-Silva P, Perez-Martos A, et al. An intragenic suppressor in the cytochrome c oxidase I gene of mouse mitochondrial DNA. Hum Mol Genet. 2003;12:329–339. doi: 10.1093/hmg/ddg021. [DOI] [PubMed] [Google Scholar]
- Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- Arany Z, Lebrasseur N, Morris C, Smith E, Yang W, Ma Y, Chin S, Spiegelman BM. The transcriptional coactivator PGC-1β drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab. 2007;5:35–46. doi: 10.1016/j.cmet.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Berardo A, DiMauro S, Hirano M. A diagnostic algorithm for metabolic myopathies. Curr Neurol Neurosci Rep. 2010;10:118–126. doi: 10.1007/s11910-010-0096-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemberg D, Quadrilatero J. Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS One. 2012;7:e35273. doi: 10.1371/journal.pone.0035273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth FW, Tseng BS, Fluck M, Carson JA. Molecular and cellular adaptation of muscle in response to physical training. Acta Physiol Scand. 1998;162:343–350. doi: 10.1046/j.1365-201X.1998.0326e.x. [DOI] [PubMed] [Google Scholar]
- Chinsomboon J, Ruas J, Gupta RK, Thom R, Shoag J, Rowe GC, et al. The transcriptional coactivator PGC-1α mediates exercise-induced angiogenesis in skeletal muscle. Proc Natl Acad Sci U S A. 2009;106:21401–21406. doi: 10.1073/pnas.0909131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz F, Thomas CK, Garcia S, Hernandez D, Moraes CT. Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum Mol Genet. 2005;14:2737–2748. doi: 10.1093/hmg/ddi307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara NM, Davis-Smyth T. The biology of vascular endothelial growth factor. Endo Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- Frazier EP, Isenberg JS, Shiva S, Zhao L, Schlesinger P, Dimitry J, et al. Age-dependent regulation of skeletal muscle mitochondria by the thrombospondin-1 receptor CD47. Matrix Biol. 2011;30:154–161. doi: 10.1016/j.matbio.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frier BC, Hancock CR, Little JP, Fillmore N, Bliss TA, Thomson DM, Wan Z, Wright DC. Reductions in RIP140 are not required for exercise- and AICAR-mediated increases in skeletal muscle mitochondrial content. J Appl Physiol. 2011;111:688–695. doi: 10.1152/japplphysiol.00279.2011. [DOI] [PubMed] [Google Scholar]
- Fritah A, Christian M, Parker MG. The metabolic coregulator RIP140: an update. Am J Physiol Endocrinol Metab. 2010;299:E335–E340. doi: 10.1152/ajpendo.00243.2010. [DOI] [PubMed] [Google Scholar]
- Geng T, Li P, Okutsu M, Yin X, Kwek J, Zhang M, Yan Z. PGC-1α plays a functional role in exercise-induced mitochondrial biogenesis and angiogenesis but not fiber-type transformation in mouse skeletal muscle. Am J Physiol Cell Physiol. 2010;298:C572–C579. doi: 10.1152/ajpcell.00481.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenaway J, Lawler J, Moorehead R, Bornstein P, Lamarre J, Petrik J. Thrombospondin-1 inhibits VEGF levels in the ovary directly by binding and internalization via the low density lipoprotein receptor-related protein-1 (LRP-1) J Cell Physiol. 2007;210:807–818. doi: 10.1002/jcp.20904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg M, Morganstein DL, Kiskinis E, Shah K, Kralli A, Dilworth SM, et al. A functional interaction between RIP140 and PGC-1α regulates the expression of the lipid droplet protein CIDEA. Mol Cell Biol. 2008;28:6785–6795. doi: 10.1128/MCB.00504-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C, Chin S, Li P, Liu F, Maratos-Flier E, Lebrasseur NK, Yan Z, Spiegelman BM. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1α muscle-specific knock-out animals. J Biol Chem. 2007;282:30014–30021. doi: 10.1074/jbc.M704817200. [DOI] [PubMed] [Google Scholar]
- Hepple RT. A new measurement of tissue capillarity: the capillary-to-fibre perimeter exchange index. Can J Appl Physiol. 1997;22:11–22. doi: 10.1139/h97-002. [DOI] [PubMed] [Google Scholar]
- Hepple RT, Mackinnon SL, Goodman JM, Thomas SG, Plyley MJ. Resistance and aerobic training in older men: effects on VO2peak and the capillary supply to skeletal muscle. J Appl Physiol. 1997;82:1305–1310. doi: 10.1152/jappl.1997.82.4.1305. [DOI] [PubMed] [Google Scholar]
- Hepple RT, Mathieu-Costello O. Estimating the size of the capillary-to-fiber interface in skeletal muscle: a comparison of methods. J Appl Physiol. 2001;91:2150–2156. doi: 10.1152/jappl.2001.91.5.2150. [DOI] [PubMed] [Google Scholar]
- Hüttemann M, Helling S, Sanderson TH, Sinkler C, Samavati L, Mahapatra G, et al. Regulation of mitochondrial respiration and apoptosis through cell signaling: Cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim Biophys Acta. 2012a;1817:598–609. doi: 10.1016/j.bbabio.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttemann M, Klewer S, Lee I, Pecinova A, Pecina P, Liu J, et al. Mice deleted for heart-type cytochrome c oxidase subunit 7a1 develop dilated cardiomyopathy. Mitochondrion. 2012b;12:294–304. doi: 10.1016/j.mito.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttemann M, Lee I, Malek MH. (–)-Epicatechin maintains endurance training adaptation in mice after 14 days of detraining. FASEB J. 2012c;26:1413–1422. doi: 10.1096/fj.11-196154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iruela-Arispe ML, Porter P, Bornstein P, Sage EH. Thrombospondin-1, an inhibitor of angiogenesis, is regulated by progesterone in the human endometrium. J Clin Invest. 1996;97:403–412. doi: 10.1172/JCI118429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Martin-Manso G, Pendrak ML, Garfield SH, Isenberg JS, Roberts DD. Thrombospondin-1 inhibits VEGF receptor-2 signaling by disrupting its association with CD47. J Biol Chem. 2010;285:38923–38932. doi: 10.1074/jbc.M110.172304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Roberts DD. CD47 applies the brakes to angiogenesis via vascular endothelial growth factor receptor-2. Cell Cycle. 2011;10:10–12. doi: 10.4161/cc.10.1.14324. [DOI] [PubMed] [Google Scholar]
- Keppel G, Wickens TD. Design and Analysis: a Researcher's Handbook. Upper Saddle River, NJ: Pearson Prentice Hall; 2004. [Google Scholar]
- Lee I, Salomon AR, Ficarro S, Mathes I, Lottspeich F, Grossman LI, Hüttemann M. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J Biol Chem. 2005;280:6094–6100. doi: 10.1074/jbc.M411335200. [DOI] [PubMed] [Google Scholar]
- Leick L, Lyngby SS, Wojtasewski JF, Pilegaard H. PGC-1α is required for training-induced prevention of age-associated decline in mitochondrial enzymes in mouse skeletal muscle. Exp Gerontol. 2010;45:336–342. doi: 10.1016/j.exger.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, et al. PGC-1α deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M, Borda-d’Agua B, Medina-Gomez G, Lelliott CJ, Paz JC, Rojo M, et al. Mitochondrial fusion is increased by the nuclear coactivator PGC-1β. PLoS One. 2008;3:e3613. doi: 10.1371/journal.pone.0003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd PG, Prior BM, Li H, Yang HT, Terjung RL. VEGF receptor antagonism blocks arteriogenesis, but only partially inhibits angiogenesis, in skeletal muscle of exercise trained rats. Am J Physiol Heart Circ Physiol. 2004;288:H759–H768. doi: 10.1152/ajpheart.00786.2004. [DOI] [PubMed] [Google Scholar]
- Malek MH, Olfert IM. Global deletion of thrombospondin-1 increases cardiac and skeletal muscle capillarity and exercise capacity in mice. Exp Physiol. 2009;94:749–760. doi: 10.1113/expphysiol.2008.045989. [DOI] [PubMed] [Google Scholar]
- Malek MH, Olfert IM, Esposito F. Detraining losses of skeletal muscle capillarization are associated with vascular endothelial growth factor protein expression in rats. Exp Physiol. 2010;85:359–368. doi: 10.1113/expphysiol.2009.050369. [DOI] [PubMed] [Google Scholar]
- Meirhaeghe A, Crowley V, Lenaghan C, Lelliott C, Green K, Stewart A, et al. Characterization of the human, mouse and rat PGC1β (peroxisome-proliferator-activated receptor-γ co-activator 1β) gene in vitro and in vivo. Biochem J. 2003;373:155–165. doi: 10.1042/BJ20030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira L, Ramirez-Sanchez I, Perkins GA, Murphy A, Taub PR, Ceballos G, et al. (–)-Epicatechin enhances fatigue resistance and oxidative capacity in mouse muscle. J Physiol. 2011;589:4615–4631. doi: 10.1113/jphysiol.2011.209924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olfert IM, Birot O. Importance of anti-angiogenic factors in the regulation of skeletal muscle angiogenesis. Microcirculation. 2011;18:316–330. doi: 10.1111/j.1549-8719.2011.00092.x. [DOI] [PubMed] [Google Scholar]
- Olfert IM, Howlett RA, Tang K, Dalton ND, Gu Y, Peterson KL, Wagner PD, Breen EC. Muscle-specific VEGF deficiency greatly reduces exercise endurance in mice. J Physiol. 2009;587:1755–1767. doi: 10.1113/jphysiol.2008.164384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olfert IM, Howlett RA, Wagner PD, Breen EC. Myocyte vascular endothelial growth factor is required for exercise-induced skeletal muscle angiogenesis. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1059–R1067. doi: 10.1152/ajpregu.00347.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford NB, Wan B, Richman A, Szczepaniak LS, Li JL, Li K, et al. Cardiac dysfunction in mice lacking cytochrome-c oxidase subunit VIaH. Am J Physiol Heart Circ Physiol. 2002;282:H726–H733. doi: 10.1152/ajpheart.00308.2001. [DOI] [PubMed] [Google Scholar]
- Rosenblatt JD, Kuzon WM, Plyley MJ, Pynn BR, McKee NH. A histochemical method for the simultaneous demonstration of capillaries and fiber type in skeletal muscle. Stain Technol. 1987;62:85–92. doi: 10.3109/10520298709107973. [DOI] [PubMed] [Google Scholar]
- Roudier E, Gineste C, Wazna A, Dehghan K, Desplanches D, Birot O. Angio-adaptation in unloaded skeletal muscle: new insights into an early and muscle type-specific dynamic process. J Physiol. 2010;588:4579–4591. doi: 10.1113/jphysiol.2010.193243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe GC, Jang C, Patten IS, Arany Z. PGC-1β regulates angiogenesis in skeletal muscle. Am J Physiol Endocrinol Metab. 2011;301:E155–E163. doi: 10.1152/ajpendo.00681.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth A, Steel JH, Nichol D, Pocock V, Kumaran MK, Fritah A, et al. The transcriptional corepressor RIP140 regulates oxidative metabolism in skeletal muscle. Cell Metab. 2007;6:236–245. doi: 10.1016/j.cmet.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- Shao D, Liu Y, Liu X, Zhu L, Cui Y, Cui A, et al. PGC-1β-regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERRα. Mitochondrion. 2010;10:516–527. doi: 10.1016/j.mito.2010.05.012. [DOI] [PubMed] [Google Scholar]
- Snyder GK. Capillarity and diffusion distances in skeletal muscles in birds. J Comp Physiol B. 1990;160:583–591. doi: 10.1007/BF00258986. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Diaz F, Iommarini L, Aure K, Lombes A, Moraes CT. PGC-1α/β induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Hum Mol Genet. 2009;18:1805–1812. doi: 10.1093/hmg/ddp093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Lin J, Krauss S, Tarr PT, Yang R, Newgard CB, Spiegelman BM. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1α and 1β (PGC-1α and PGC-1β) in muscle cells. J Biol Chem. 2003;278:26597–26603. doi: 10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- Taivassalo T, Ayyad K, Haller RG. Increased capillaries in mitochondrial myopathy: implications for the regulation of oxygen delivery. Brain. 2012;135:53–61. doi: 10.1093/brain/awr293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K, Breen EC, Gerber HP, Ferrara NM, Wagner PD. Capillary regression in vascular endothelial growth factor-deficient skeletal muscle. Physiol Genomics. 2004;18:63–69. doi: 10.1152/physiolgenomics.00023.2004. [DOI] [PubMed] [Google Scholar]
- Tiranti V, Hoertnagel K, Carrozzo R, Galimberti C, Munaro M, Granatiero M, et al. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am J Hum Genet. 1998;63:1609–1621. doi: 10.1086/302150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hove JL, Cunningham V, Rice C, Ringel SP, Zhang Q, Chou PC, Truong CK, Wong LJ. Finding twinkle in the eyes of a 71-year-old lady: a case report and review of the genotypic and phenotypic spectrum of TWINKLE-related dominant disease. Am J Med Genet A. 2009;149A:861–867. doi: 10.1002/ajmg.a.32731. [DOI] [PubMed] [Google Scholar]
- Villani G, Greco M, Papa S, Attardi G. Low reserve of cytochrome c oxidase capacity in vivo in the respiratory chain of a variety of human cell types. J Biol Chem. 1998;273:31829–31836. doi: 10.1074/jbc.273.48.31829. [DOI] [PubMed] [Google Scholar]
- Wagner PD. Determinants of maximal oxygen transport and utilization. Annu Rev Physiol. 1996;58:21–50. doi: 10.1146/annurev.ph.58.030196.000321. [DOI] [PubMed] [Google Scholar]
- Wenz T, Diaz F, Hernandez D, Moraes CT. Endurance exercise is protective for mice with mitochondrial myopathy. J Appl Physiol. 2009;106:1712–1719. doi: 10.1152/japplphysiol.91571.2008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Williams DB, Sutherland LN, Bomhof MR, Basaraba SA, Thrush AB, Dyck DJ, Field CJ, Wright DC. Muscle-specific differences in the response of mitochondrial proteins to β-GPA feeding: an evaluation of potential mechanisms. Am J Physiol Endocrinol Metab. 2009;296:E1400–E1408. doi: 10.1152/ajpendo.90913.2008. [DOI] [PubMed] [Google Scholar]
- Zachary I. VEGF signalling: Integration and multi-tasking in endothelial cell biology. Biochem Soc Trans. 2003;31:1171–1177. doi: 10.1042/bst0311171. [DOI] [PubMed] [Google Scholar]