

Abstract
When a polyadenylated nonstop transcript is fully translated, a complex consisting of the ribosome, the nonstop mRNA, and the C-terminally polylysine-tagged protein is generated. In Saccharomyces cerevisiae, a 3-step quality control system prevents formation of such dead-end complexes. Nonstop mRNA is rapidly degraded, translation of nonstop mRNA is repressed, and finally, nonstop proteins are cotranslationally degraded. Nonstop mRNA degradation depends on Ski7 and the exosome; nonstop protein degradation depends on the ribosome-bound E3 ligase Ltn1 and the proteasome. However, components which mediate translational repression of nonstop mRNA have previously not been identified. Here we show that the ribosome-bound chaperone system consisting of the ribosome-associated complex (RAC) and the Hsp70 homolog Ssb is required to stabilize translationally repressed ribosome-polylysine protein complexes, without affecting the folding or the degradation of polylysine proteins. As a consequence, in the absence of RAC/Ssb, polylysine proteins escaped translational repression and subsequently folded into their native conformation. This active role of RAC/Ssb in the quality control of polylysine proteins significantly contributed to the low level of expression of nonstop transcripts in vivo.
INTRODUCTION
Quality control at the level of the mRNA prevents translation of truncated or otherwise nonfunctional transcripts, which may generate translation products with dominant-negative effects. Degradation of nonfunctional mRNA is mediated by conserved nucleases, which digest mRNA from either the 5′ or the 3′ end (5, 14, 28, 51). Depending on the type of defect within the mRNA, recognition involves different surveillance machineries. mRNA containing premature termination codons is degraded via Upf1-dependent nonsense-mediated decay (NMD) (5, 14, 28), mRNA which forms an impassable secondary structure is degraded in an Hbs1/Dom34-dependent manner via no-go decay (NGD) (5, 13, 41, 50), and mRNA which lacks in-frame stop codons is degraded in a Ski7-dependent manner via nonstop decay (NSD) (5, 14, 28, 51, 54). Ski7 is a member of the eRF3 protein family, which also includes the NGD factor Hbs1 and the translation termination factor Sup35 (5). In vivo, incorrect processing and polyadenylation within open reading frames (ORFs) are major sources of nonstop transcripts. The current model predicts that in the absence of a stop codon the ribosome continues translation to the very end of the transcript (15, 27, 53). In this situation, Ski7 binds to the empty A site and recruits the exosome, which then degrades the nonstop mRNA, including the poly(A) tail (51, 53). Recent evidence revealed that Hbs1/Dom34 not only is involved in NGD but also stimulates the release of stalled ribosomes at the 3′ end of nonstop mRNA during NSD (52).
The fate of nascent polypeptides generated from nonstop transcripts (here termed nonstop proteins) which have escaped NSD is only incompletely understood. The level of expression of nonstop proteins is very low, and it was recognized early that NSD does not fully account for the low level of nonstop protein (27, 29). The current model suggests that a polylysine tag at the C terminus of nonstop proteins which results from the translation of the poly(A) tail causes translational repression as well as enhanced cotranslational degradation of the nascent peptide (12, 29, 54). Translational repression involves stalling of nascent polypeptides due to electrostatic interactions between the positively charged polylysine stretches and the negatively charged walls of the ribosomal tunnel (6, 12, 29, 35). Moreover, when the ribosome reaches the 3′ end of a poly(A) tail, the poly(A) binding protein Pab1 is released (27). Pab1 might be directly dislodged by the ribosome or might be released because Sup35, which is involved in Pab1 binding, is not recruited to complexes containing nonstop mRNA (16, 23, 24, 30). In any case, the absence of Pab1 results in the repression of further rounds of translation (37). Nonstop proteins which have escaped translational repression are removed via cotranslational degradation. Again, the polylysine tag is thought to serve as the signal. Two ribosome-bound E3 ligases, Not4 (12) and Ltn1 (6), have been implicated in the polyubiquitination of polylysine-tagged nascent polypeptides, which are subsequently delivered to the proteasome.
Saccharomyces cerevisiae possesses a ribosome-bound chaperone triad consisting of the Hsp70 homologs Ssb (38) and Ssz1 (18) and the J-domain protein Zuo1 (60). Zuo1 and Ssz1 form a stable complex termed the ribosome-associated complex (RAC) (18), which acts as a J-domain partner of Ssb (25). Consistently, the two genes encoding Ssb (SSB1 and SSB2), SSZ1, and ZUO1 genetically interact (19, 26, 43). Ssb directly binds to ribosomes and interacts with nascent chains. The interaction of Ssb with nascent chains but not with the ribosome requires the presence of RAC (18, 19, 38, 43, 44). A general role of RAC/Ssb in the early steps of protein biogenesis is highly suggestive; however, it is only poorly understood (43). Recent evidence suggests that RAC/Ssb is specifically involved in the folding and assembly of ribosomal proteins (2, 31). In addition, RAC/Ssb is thought to be connected to diverse cellular functions, including protein degradation (39, 40, 43).
Here we have investigated whether the RAC/Ssb chaperone system affects the biogenesis of nonstop proteins. To that end, we have analyzed the effects of RAC/Ssb on the biogenesis of C-terminally polylysine-tagged proteins (here termed polylysine proteins) as well as on nonstop proteins. We found that in the absence of RAC/Ssb the expression of enzymatically active polylysine proteins as well as nonstop proteins was significantly increased. The major consequence of loss of RAC/Ssb function was a release from translational repression. Beyond that, our data reveal that translation of nonstop mRNA does not in general result in the production of polylysine proteins. Only in the absence of Ski7 was a significant fraction of nonstop mRNA translated into species containing C-terminal polylysine.
MATERIALS AND METHODS
Yeast strains and plasmids.
S. cerevisiae MH272-3fa/α (ura3/ura3 leu2/leu2 his3/his3 trp1/trp1 ade2/ade2) (22) is the parental wild-type strain for all haploid mutant strains used in this study. Deletion strains lacking SSB1 and SSB2 (Δssb1 Δssb2) and ZUO1 (Δzuo1) have been described previously (11, 18). The Δski7 strain (ski7::LEU2) was constructed by replacing a 1,847-bp StuI/XbaI fragment in the SKI7-coding region with the LEU2 open reading frame ±300 bp up- and downstream. The Δltn1 strain was constructed by replacing LTN1 with the ltn1::kanMX4 deletion cassette amplified by PCR from strain Y00834 (Euroscarf); the Δupf1 strain was constructed by replacing UPF1 with the upf1::kanMX4 deletion cassette from Y16214 (Euroscarf); the Δhbs1 strain was constructed by replacing HBS1 with the hbs1::kanMX4 deletion cassette from strain Y06000 (Euroscarf). The Δerg6 strain was constructed by replacing ERG6 with the erg6::kanMX4 deletion cassette from strain Y00568 (Euroscarf) in the diploid wild-type strain. As Δerg6 strains are incapable of tryptophan uptake (17), the diploid ERG6/erg6::kanMX4 strain was transformed with the empty pYCPlac22 plasmid, which contains the TRP1 gene (20). All haploid Δerg6 single-deletion strains derived from the diploid contain pYCPlac22. Strains containing multiple deletions (Δzuo1 Δski7, Δzuo1 Δupf1, Δzuo1 Δhbs1, Δzuo1 Δltn1, Δzuo1 Δerg6, Δssb1 Δssb1 Δski7, and Δhbs1 Δski7) were generated via mating followed by dissection and tetrad analysis.
For constitutive expression of HIS3 and luciferase constructs, the respective genes were cloned into the low-copy-number plasmid pYCPlac33 (CEN URA3) or the multicopy plasmid pYEPlac195 (2μ URA3) (20). The HIS3 open reading frame plus 237 bp upstream and 266 bp downstream of it was amplified from genomic DNA and was cloned into the SphI/BamHI sites of pYCPlac33, resulting in pYCPlac33-His3-stop. The HIS3 5′ region and the HIS3 ORF were then replaced by his3-nonstop, which was amplified from genomic DNA with primers deleting the T residue within the first stop codon as previously described (53). This created an open reading frame extending past all previously mapped polyadenylation sites (36, 53). The plasmid was termed pYCPlac33-His3-nonstop. The parental vector for the luciferase constructs was pYCPlac33-Zuo1-NcoI, containing the ZUO1-coding region ±300 bp up- and downstream of it with an NcoI site at the start codon of the ZUO1 open reading frame (55). In this vector, the ZUO1-coding region was replaced with the luciferase gene amplified from pSP-Luc+ (Promega), and the ZUO1 3′ region was replaced with a fragment containing 266 bp 3′ of the HIS3 open reading frame. The resulting plasmid was termed pYCPlac33-Luc-stop. To generate Luc-nonstop, the 3′ region of HIS3 was replaced with the 3′ region of pYCPlac33-His3-nonstop (see Fig. S1 in the supplemental material). To generate the different C-terminally tagged versions of luciferase complementary oligonucleotides, coding for either polylysine, polyarginine, or polyserine were annealed and were introduced into a PstI site at the 3′ end of the Luc-stop gene. Multimers of (AAG AAG AAA)n were employed to generate the sequence encoding polylysine tags. Note that the choice of the codon coding for lysine does not influence translational repression or degradation of polylysine proteins (29). All luciferase constructs were transferred to the pYEPlac195 high-copy-number plasmid, resulting in pYEPlac195-Luc-stop, pYEPlac195-Luc-nonstop, pYEPlac195-Luc-Lys12, pYEPlac195-Luc-Lys16, pYEPlac195-Luc-Lys20, pYEPlac195-Luc-Arg12, and pYEPlac195-Luc-Ser12. On the basis of these plasmids, N-terminally FLAG-tagged Luc-Lys20 was generated via PCR technology. The resulting plasmid is termed pYEPlac195-FLAG-Luc-Lys20. For expression under the control of the GAL1 promoter, the coding region plus the 3′ region of either Luc-stop or Luc-nonstop was transferred into pESC-URA (Agilent Technologies), resulting in pESC-URA-Luc-stop and pESC-URA-Luc-nonstop. Tape measure constructs were generated on the basis of pYEPlac195-Luc-nonstop by introducing stop codons into the HIS3 3′ untranslated region (UTR) upstream of the first polyadenylation site via PCR according to a previously published strategy (27). Luc-UTR12 contains a 12-amino-acid extension, and Luc-UTR25 contains a 25-amino-acid C-terminal extension (see Fig. S1 in the supplemental material). The resulting plasmids were termed pYEPlac195-Luc-UTR12 and pYEPlac195-Luc-UTR25, respectively.
Culture conditions.
Strains were grown on rich medium containing glucose (1% yeast extract, 2% peptone, 2% glucose [YPD]) or galactose (1% yeast extract, 2% peptone, 2% galactose [YPGal]) or on minimal medium (0.67% yeast nitrogen base without amino acids [Difco], 2% glucose, containing the appropriate supplements [SD]). Liquid cultures were incubated at 30°C on a shaker at 200 rpm. The ability of the his3-nonstop allele to suppress the histidine auxotrophy of yeast strains was analyzed on SD plates lacking histidine. To that end, 10-fold serial dilutions of log-phase cultures containing the same number of cells were spotted onto SD plates either supplemented with histidine or lacking histidine. For proteasome inhibition, cells were harvested in early log phase and the medium was replaced with medium containing 50 μM MG132 (Sigma).
mRNA stability assays, RNA isolation, and Northern blotting.
Strains harboring pESC-URA-Luc-stop or pESC-URA-Luc-nonstop were grown to mid-log phase on YPGal. Transcription of the luciferase constructs was then turned off by replacing YPGal with YPD. Aliquots were removed at the time points indicated. Total RNA was isolated according to the instructions of the manufacturer (RNeasy minikit; Qiagen). Two micrograms of total RNA was separated on 1.2% agarose gels and was then blotted onto Hybond-N+ nylon membranes (Amersham) using 10× SSC (1.5 M sodium chloride plus 150 mM sodium citrate) as transfer buffer. Hybridization with radiolabeled probes was carried out in hybridization solution either with formamide (50% formamide, 5× SSC, 5× Denhardt's solution, 0.5% SDS, 0.1 mg/ml salmon sperm DNA; for the luciferase probe) or without formamide (5× SSC, 5× Denhardt's solution, 1% SDS, 0.1 mg/ml salmon sperm DNA, 10% dextran sulfate; for the ACT1 probe). Probes were radiolabeled with a Rediprime II random prime labeling system (GE Healthcare BioScience). The probe for ACT1 was prepared as described previously (9). Luciferase mRNA was detected with a 636-bp probe covering the 5′ region of the ORF. Northern blots were quantified using AIDA image analyzer software (Raytest). The ACT1 signal was employed for normalization. The half-life of transcripts was calculated via fitting of the data to exponential decay using Kaleidagraph software (Synergy Software).
Aggregation assay.
Aggregation of proteins was tested under conditions which result in the disruption of ribosomal complexes (31). To that end, cells were harvested at mid-log phase and extracts were prepared in lysis buffer (20 mM NaH2PO4, pH 6.8, 10 mM dithiothreitol [DTT], 1 mM EDTA, 0.1% Tween 20, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1× protease inhibitor mix) (31) by glass bead disruption for six 1-min intervals interrupted by 1-min cooling periods on ice. Extracts were then subjected to a preclearing spin for 1 min at 5,000 × g at 4°C, and the resulting supernatants were centrifuged for 15 min at 20,000 × g at 4°C to separate soluble proteins from insoluble material. Aliquots of the total material after the preclearing spin, supernatants, and pellets resuspended in lysis buffer were analyzed by SDS-PAGE followed by immunoblotting. The protein concentration of total extracts was determined with the Bradford assay (Bio-Rad) and was employed to adjust the loading of extracts derived from different yeast strains.
Ribosome profiles and ribosome sedimentation assays.
Yeast cells were harvested in early log phase after addition of cycloheximide to a final concentration of 100 μg/ml. Preparation of extracts was carried out by glass bead disruption in 20 mM HEPES-KOH, pH 7.4, 100 mM potassium acetate, 2 mM magnesium acetate, 100 μg/ml cycloheximide, and 0.5 mM DTT as described previously (4, 42). RNase treatment (0.25 mg/ml RNase A) was performed for 15 min on ice. Samples corresponding to 5 A260 units were loaded onto a 15 to 55% linear sucrose gradient, centrifuged for 2.5 h at 200,000 × g (TH641; Sorvall), and fractionated from top to bottom with a density gradient fractionator monitoring the A254 (Teledyne). Aliquots of the fractions and a total corresponding to 5% of the material loaded onto the gradient were analyzed via SDS-PAGE followed by immunoblotting.
For ribosome sedimentation assays, cells were harvested in mid-log phase after addition of 100 μg/ml cycloheximide. Cells were lysed by glass bead disruption in 20 mM HEPES-KOH, pH 7.4, 120 mM potassium acetate, 2 mM magnesium acetate, 2 mM DTT, 100 μg/ml cycloheximide, 1 mM PMSF, 1× protease inhibitor mix. Samples corresponding to 0.5 A260 unit in a total volume of 60 μl were loaded onto a 90-μl sucrose cushion (25% sucrose, 20 mM HEPES-KOH, pH 7.4, 120 mM potassium acetate, 2 mM magnesium acetate, 2 mM DTT, 1 mM PMSF, 1× protease inhibitor mix). Ribosomes were collected by ultracentrifugation at 350,000 × g for 30 min at 4°C (TLA-100; Beckman). Aliquots of the total material, supernatant, and pellet were analyzed via SDS-PAGE followed by immunoblotting.
Luciferase activity assay.
Luciferase activity was determined essentially as described previously (45). In brief, glass bead lysates were diluted in luciferase assay buffer (100 mM KH2PO4, pH 7.8, 1 mM EDTA, 1 mM DTT), and the activities of two different dilutions of each sample were determined after addition of luciferase reagent (500 μM d-luciferin, 20 mM Tricine, 5 mM MgCl2, 0.1 mM EDTA, 3.3 mM DTT, 270 μM coenzyme A, 500 μM ATP) using a Lumat LB 9507 device (Berthold Technologies GmbH). Relative light units were normalized to the protein concentration determined by the Bradford assay (Bio-Rad).
Denaturing FLAG-IP.
Denaturing FLAG-immunoprecipitations (FLAG-IPs) were essentially performed as described previously (6). In brief, MG132-treated yeast cells were incubated with 0.1 M NaOH for 5 min at room temperature and were subsequently collected via centrifugation at 20,000 × g. Cell pellets corresponding to a 1-ml culture with an optical density at 600 nm (OD600) of 1 were resuspended in 125 μl IP lysis buffer (50 mM Tris-HCl, pH 7.4, 5 mM EDTA, 5 mM N-ethylmaleinimide, 1% SDS, 1 mM PMSF, 1× protease inhibitor mix) and were incubated for 10 min at 95°C. In the case of strains lacking RAC/Ssb, 1.5 ml of culture with an OD600 of 1 was used in the same volume of lysis buffer (55). Lysates were cleared by centrifugation, and 400 μl of each lysate was incubated with 25 μl anti-FLAG M2 affinity matrix (FLAG-beads; Sigma) resuspended in 600 μl IP buffer (50 mM Tris-HCl, pH 7.4, 5 mM EDTA, 250 mM NaCl, 1% Triton X-100). Binding of FLAG-tagged luciferase to the beads was allowed for 2 h at 4°C. Washed FLAG-beads were boiled in SDS sample buffer, and the material released from the beads was subsequently analyzed via SDS-PAGE followed by immunoblotting.
Miscellaneous.
Protease inhibitor mix (1×) contained 1.25 μg/ml leupeptin, 0.75 μg/ml antipain, 0.25 μg/ml chymostatin, 0.25 μg/ml elastinal, and 5 μg/ml pepstatin A. Total yeast extract for immunoblot analysis was prepared as described previously (46, 59). Loading of samples of the Δzuo1 and Δssb1 Δssb2 strains was normalized as described previously (55). The relative amounts of luciferase constructs expressed in different strain backgrounds were compared on the same exposure of a single immunoblot using AIDA image analyzer software (Raytest). Polyclonal antibodies were raised in rabbits (Eurogentec). Rabbit antiluciferase antibody was from Sigma (L0159), and rabbit antiubiquitin antibody was from Dako (Z0458). Immunoblots were developed via enhanced chemiluminescence as described using horseradish peroxidase-conjugated goat anti-rabbit IgG (Pierce) as secondary antibody (46).
RESULTS
RAC/Ssb represses expression of polylysine proteins.
In order to test the effect of the RAC/Ssb system on proteins which contain C-terminal stretches of positively charged amino acids, we constructed in vivo reporters consisting of luciferase fused to either polylysine, polyarginine, or, as a control, polyserine. Wild-type luciferase (Luc-stop) served as a control. (Fig. 1A; see Fig. S1 in the supplemental material). Expression of the different luciferase constructs was first tested in a wild-type background. The steady-state level of luciferase fused to a consecutive stretch of 12 C-terminal lysines (Luc-Lys12) or arginines (Luc-Arg12) was reduced, while the steady-state level of luciferase fused to 12 consecutive serine residues (Luc-Ser12) was not (Fig. 1B). Increasing the length of the polylysine tag to 16 (Luc-Lys16) or 20 (Luc-Lys20) residues further reduced the expression level, and Luc-Lys16 and Luc-Lys20 were detected only upon long exposure of the immunoblot (Fig. 1B). These data confirmed earlier observations obtained with different polylysine reporter constructs indicating that stretches of positively charged amino acids within nascent chains result in translation arrest and enhanced degradation (6, 8, 12, 29). The use of luciferase reporters enabled us to test not only for the expression level but also for enzymatic activity.
Fig 1.

RAC/Ssb inhibits expression of polylysine proteins. (A) Schematic view of Luc-stop and Luc-tag luciferase reporter constructs. Luc-tag constructs contain sequences coding for different stretches of consecutive amino acids located 5′ of the termination codon (see also Fig. S1 in the supplemental material). (B) Expression level of Luc-stop and Luc-tag constructs in wild-type yeast. Luc-Lys constructs contain 12 (Luc-Lys12), 16 (Luc-Lys16), or 20 (Luc-Lys20) lysines at the very C terminus. Luc-Arg12 and Luc-Ser12 contain 12 arginine or serine residues, respectively. Total cell extracts corresponding to the same amount of total protein were analyzed via immunoblotting using antibodies specific for luciferase and, as a loading control, Sse1. Because the levels of expression of luciferase reporters differed significantly, a short exposure (exp.) and a long exposure of the same immunoblot are displayed. (C) The level of Luc-stop and Luc-tag constructs expressed in wild-type, Δzuo1, and Δssb1 Δssb2 strains was analyzed as described for panel B.
Luciferase reporters were then expressed in Δzuo1 and Δssb1 Δssb2 strains, both of which lack a functional RAC/Ssb system (19, 25, 26; see the introduction). We first tested if the biogenesis of Luc-stop was affected by RAC/Ssb. Neither the expression level nor the enzymatic activity of Luc-stop was altered in Δzuo1 or Δssb1 Δssb2 strains compared to wild type (see Fig. S2 in the supplemental material). Next, C-terminally tagged luciferase versions were analyzed. The level of Luc-Lys12, Luc-Lys16, and Luc-Lys20 expression in the absence of RAC/Ssb was significantly higher than that in the corresponding wild-type strain (Fig. 1C). However, the absence of RAC/Ssb did not affect the expression of Luc-Arg12 or Luc-Ser12, suggesting a specific effect of the polylysine tag (Fig. 1C).
RAC/Ssb is not required for the folding of polylysine proteins.
To test if in the absence of RAC/Ssb the polylysine tag might prevent folding and induce aggregation of luciferase, we took two approaches. First, the solubility of Luc-Lys20 was tested via high-speed centrifugation under conditions which disrupt ribosomal particles but preserve insoluble protein aggregates (31). Luc-Lys20 was fully soluble, whether or not RAC/Ssb was present (Fig. 2A). Thus, enhanced expression in Δzuo1 or Δssb1 Δssb2 strains was not due to accumulation of Luc-Lys20 in inclusion body-like structures. Second, Luc-Lys20 expression and enzymatic activity were determined side by side within the same cell extract (Fig. 2B). Compared to the level of expression in the wild type, Luc-Lys20 expression was enhanced 2.5-fold in the Δzuo1 strain and was enhanced 2.1-fold in the Δssb1 Δssb2 strain (Fig. 2B and C). At the same time, Luc-Lys20 activity was enhanced 2.6-fold in the Δzuo1 strain and was enhanced 2.1-fold in the Δssb1 Δssb2 strain (Fig. 2B and D). Thus, whether or not RAC/Ssb was present, the specific activity, i.e., the ratio of luciferase activity per number of Luc-Lys20 molecules, remained unaltered. In the absence of RAC/Ssb, not only was Luc-Lys20 expression enhanced, but also Luc-Lys20 was able to fold into its enzymatically active conformation as efficiently as in the wild-type background.
Fig 2.

RAC/Ssb does not affect the folding of polylysine proteins. (A) Solubility of Luc-Lys20 in the absence of RAC/Ssb. Total cell extracts of strains expressing Luc-Lys20 were generated under conditions disrupting ribosomal particles (see Materials and Methods). Total extracts (lanes T) were separated into a soluble supernatant (lanes S) and an insoluble pellet (lanes P) by centrifugation. Aliquots were analyzed via immunoblotting using Sse1 as a loading control and the ribosomal protein Rpl24 as a marker for the absence of ribosomes in the pellet fractions. (B) Enzymatic activity of Luc-Lys20. Total cell extracts of strains expressing Luc-Lys20 were prepared as described in Materials and Methods. Aliquots were analyzed (i) for the Luc-Lys20 expression level via densitometry (relative intensity [rel. int.]) and (ii) for the enzymatic activity of luciferase in relative light units (RLU). Sse1 served as loading control. (C) Effect of RAC/Ssb on the expression of Luc-Lys20. Graphical representation of six independent expression experiments performed as described for panel B. The changes in expression are given relative to the level of Luc-Lys20 expression in the wild-type strain. Error bars represent the standard error of the mean (SEM). (D) Effect of RAC/Ssb on the activity of Luc-Lys20. Graphical representation of six independent luciferase activity assays performed as described for panel B. The activity is given relative to the Luc-Lys20 activity obtained in the wild-type strain. Error bars represent SEMs.
Complexes of ribosomes and polylysine proteins are stabilized by RAC/Ssb.
Luc-Lys20 was associated with polysomes and translating monosomes in extracts derived from wild-type yeast (Fig. 3A; see also reference 6). Luc-Lys20 was also associated with polysomes and translating monosomes in extracts derived from a Δzuo1 strain (Fig. 3B). However, compared to the wild-type strain, a significantly larger fraction of Luc-Lys20 was recovered in the cytosol (compare fractions 1 in Fig. 3A and B). To assess the distribution of Luc-Lys20 between the ribosome-bound and the cytosolic pool more quantitatively, ribosome sedimentation assays were performed. Only about 15% of Luc-Lys20 was released from ribosomes in wild type, while more than 50% of Luc-Lys20 was released from ribosomes in the Δzuo1 strain (Fig. 3C and D). Thus, the increased level of Luc-Lys20 expression in the absence of RAC/Ssb correlated with the reduced stability of the complex between Luc-Lys20 and the ribosome. Because the interaction between polylysine stretches and the ribosomal tunnel results in translational repression as well as in proteasomal degradation (6, 12, 29, 35), this observation suggested that either of the two quality control steps was hampered in the absence of RAC/Ssb.
Fig 3.

RAC/Ssb stabilizes complexes between ribosomes and polylysine proteins. (A) Ribosome profiles of wild type expressing Luc-Lys20. Cell extracts were separated into polysomes, 80S ribosomes, and ribosomal subunits (60S and 40S) via ultracentrifugation as described in Materials and Methods. Fractionation was performed with untreated (−RNase) or RNase-treated (+RNase) cell extract. Due to RNase treatment, polysomes are destroyed and accumulate in an 80S ribosomal peak. Fractions were analyzed via immunoblotting using an antibody directed against luciferase. T, 1/20 of the cell extract loaded onto the gradient; P, material collected in the pellet after ultracentrifugation. (B) Ribosome profiles of Δzuo1 cells expressing Luc-Lys20. Analysis was performed as described for panel A. (C) Ribosome association of Luc-Lys20 in the presence or absence of RAC/Ssb. Total extracts (lanes T) of wild-type and Δzuo1 cells expressing Luc-Lys20 were separated into a cytosolic supernatant (lanes C) and a ribosomal pellet (lanes R) as described in Materials and Methods. Aliquots of the samples were loaded and analyzed via immunoblotting using antibodies directed against luciferase, the cytosolic marker Sse1, and the ribosomal marker Rpl24. Note that the material derived from the Δzuo1 strain was loaded in an amount four times less to obtain intensities similar to that of the Luc-Lys20 band (compare also Fig. 1C). For quantification, the sum of the relative intensities of the Luc-Lys20 bands in the cytosolic and ribosomal fractions was set to 100%. Given are the fraction of Luc-Lys20 which was soluble in the cytosol and the fraction which cosedimented with ribosomes. (D) Distribution of Luc-Lys20 between a cytosolic and ribosome-bound pool. The fraction of Luc-Lys20 released from ribosomes (ribos) was determined as described for panel C. Mean values representing the ribosome-associated fraction of Luc-Lys20 in the wild-type and Δzuo1 strains are based on eight independent experiments. Error bars indicate the SEMs.
RAC/Ssb is dispensable for proteasomal degradation of polylysine proteins.
In order to distinguish a role of RAC/Ssb on translational repression versus degradation of polylysine proteins, we employed several experimental approaches. Inhibition of de novo protein synthesis revealed that the half-life of Luc-Lys20 was unaffected by RAC/Ssb (Fig. 4A and B). This was consistent with a more pronounced role of RAC/Ssb in translational repression than protein degradation. To test this in more detail, we employed the Δerg6 strain background, because this mutation allows the proteasome inhibitor MG132 to efficiently enter yeast cells (17, 33). Indeed, MG132 caused accumulation of polyubiquitinated proteins in the Δerg6 strain background (Fig. 4C, top). A direct comparison revealed that the amount of polyubiquitinated proteins in the Δerg6 and Δerg6 Δzuo1 strains was similar (Fig. 4C, top). Thus, loss of RAC/Ssb did not cause bulk accumulation of misfolded, polyubiquitinated proteins targeted for degradation. RAC/Ssb also did not interfere with ubiquitination in general, because proteasome inhibition in the Δerg6 or Δerg6 Δzuo1 strain resulted in accumulation of polyubiquitinated proteins to a similar extent (Fig. 4C, top).
Fig 4.

RAC/Ssb acts independently of the proteolytic machinery. (A) Stability of Luc-Lys20 in wild-type and Δzuo1 strains. De novo protein synthesis of logarithmically growing cultures was inhibited by the addition of cycloheximide (CHX; final concentration, 100 μg/ml). The level of Luc-Lys20 expression was monitored at the time points indicated. Total cell extracts were analyzed via immunoblotting using antibodies directed against luciferase and Sse1. (B) Half-life of Luc-Lys20 in wild-type and Δzuo1 strains. Quantification of the data shown in panel A. The intensity of the Luc-Lys20 band was normalized for differences in loading using the intensity of the Sse1 band. The intensity at time zero (t0) was set equal to 1, and the data were fitted to an exponential decay function. (C) Expression of Luc-Lys20 in the Δerg6 and Δerg6 Δzuo1 strains after proteasome inhibition. The proteasome inhibitor MG132 was added to logarithmically growing cultures as indicated. Accumulation of polyubiquitinated proteins was monitored via immunoblotting using ubiquitin-specific antibody. The level of Luc-Lys20 expression was analyzed via immunoblotting using luciferase antibody. Expression of Luc-Lys20 was quantified in three independent experiments. Mean values and the SEM relative to untreated Δerg6 (lane 1) are given below a representative immunoblot. (D) Polyubiquitination of FLAG-Luc-Lys20 in Δerg6 and Δerg6 Δzuo1 strains. Total extracts of Δerg6 and Δerg6 Δzuo1 strains expressing Luc-Lys20 or FLAG-Luc-Lys20, as indicated, were subjected to affinity purification using FLAG-beads. Polyubiquitination of affinity-purified FLAG-Luc-Lys20 was analyzed via immunoblotting using ubiquitin-specific antibody (ubiquitin). The amount of isolated FLAG-Luc-Lys20 was determined using an antibody directed against luciferase. (E) Expression level of Luc-Lys20 in wild-type, Δzuo1, Δltn1, and Δzuo1 Δltn1 strains. Total cell extracts were analyzed via immunoblotting using antibodies specific for luciferase and, as a loading control, Sse1.
We next tested the effect of proteasome inhibition specifically on the expression of Luc-Lys20. Consistent with previous studies on proteins carrying polylysine extensions (see the introduction), Luc-Lys20 expression was enhanced about 3-fold when the proteasome was inhibited in the Δerg6 strain (Fig. 4C, bottom, lanes 1 and 2). The absence of RAC/Ssb caused a 4-fold increase in Luc-Lys20 expression in the Δerg6 background (Fig. 4C, bottom; compare lanes 1 and 3). Importantly, Luc-Lys20 expression in the absence of RAC/Ssb was further increased to about 8.5-fold when the proteasome was inhibited (Fig. 4C, bottom, lanes 3 and 4). The additive effect strongly suggested that RAC/Ssb was regulating Luc-Lys20 expression at a stage different from proteasomal degradation. To further test this, a FLAG-tagged version of Luc-Lys20 (FLAG-Luc-Lys20) was expressed in the presence or absence of RAC/Ssb and was subsequently affinity purified (Fig. 4D). This allowed us to test for accumulation of polyubiquitinated FLAG-Luc-Lys20, which is an indicator of inefficient degradation. As a control, untagged Luc-Lys20 was included in the analysis (Fig. 4D, lanes 1 and 2). The level of polyubiquitinated FLAG-Luc-Lys20 was low in the Δerg6 strain as well as in the Δerg6Δzuo1 strain (Fig. 4D, FLAG pull down, top, lane 3 and lane 5). Thus, increased expression of FLAG-Luc-Lys20 in the absence of RAC/Ssb (Fig. 4C and D, FLAG pull down, bottom) was not due to a delivery defect of polyubiquitinated FLAG-Luc-Lys20 to the proteasome, which would result in the accumulation of polyubiquitinated FLAG-Luc-Lys20. When the level of polyubiquitinated FLAG-Luc-Lys20 was analyzed after inhibition of the proteasome, a significant increase was observed only in the Δerg6 Δzuo1 strain and not in the Δerg6 strain (Fig. 4D, FLAG pull down, top, lane 4 and lane 6). Thus, RAC/Ssb was also not required for the polyubiquitination of FLAG-Luc-Lys20, which was easily detected when the proteasome was inhibited. This was further supported by the observation that loss of RAC/Ssb and loss of the E3 ligase Ltn1 (6) synergistically increased the level of Luc-Lys20 expression (Fig. 4E). The combined data support a model in which Luc-Lys20, which had escaped translational repression in the absence of RAC/Ssb, was subsequently polyubiquitinated in an Ltn1-dependent manner and then degraded by the proteasome.
RAC/Ssb and NSD synergistically affect nonstop protein expression.
C-terminally polylysine-tagged proteins serve as models of translation products derived from nonstop mRNA (see the introduction). In order to compare the effects of RAC/Ssb on proteins containing a polylysine tag with the effect on proteins derived from bona fide nonstop mRNA, the luciferase open reading frame lacking the stop codon was fused to the HIS3 3′ region, resulting in the Luc-nonstop construct. The HIS3 3′ region does not contain an in-frame stop codon within a sequence extending past all 13 previously mapped processing and polyadenylation sites (36, 53) (Fig. 5A; see Fig. S1 in the supplemental material). Depending on the experiment, Luc-nonstop was expressed either under the control of the constitutive ZUO1 promoter or under the control of the glucose-repressed GAL1 promoter (Fig. 5A).
Fig 5.

RAC/Ssb is required to establish translational repression of Luc-nonstop. (A) Schematic view of the Luc-nonstop reporter constructs. Luc-nonstop was expressed under the control of the constitutive ZUO1 promoter (Luc-nonstop) or the galactose-inducible GAL1 promoter (GAL-Luc-nonstop). Luc-nonstop is an in-frame fusion of the luciferase open reading frame fused to the 3′ region of the HIS3 gene. The fusion does not contain in-frame stop codons (compare with Fig. S1 in the supplemental material). (B) Expression of Luc-nonstop, Luc-stop, Luc-Lys12, and Luc-Lys20 in wild-type cells. In order to compare the luciferase constructs directly, loading was adjusted according to the different expression levels (percentage loaded). To avoid loss of highly diluted samples, total extract of a wild-type strain which did not express luciferase was employed for dilution. (C) Activity of Luc-nonstop in the absence of RAC/Ssb and Ski7. Luciferase activity was determined in total cell extracts as described in Materials and Methods. The activity determined in wild-type extract was set equal to 1. Error bars represent the SEMs of six independent experiments. (D) Solubility of Luc-nonstop in the absence of RAC/Ssb and Ski7. Total cell extracts (lanes T) were separated into a soluble supernatant (lanes S) and an insoluble pellet (lanes P) and were analyzed as described in the legend to Fig. 2A. (E) Expression of Luc-nonstop in the absence of RAC/Ssb and Ski7. As a relative molecular mass marker, 100-fold-diluted Luc-stop was loaded as described for panel B. Expression of Luc-nonstop was quantified in nine independent experiments (four independent experiments for the Δssb1 Δssb2 strain). Mean values and SEMs are given below a representative immunoblot. (F) Luc-nonstop translation products of higher molecular mass accumulate in the absence of RAC/Ssb and Ski7. Strains expressing Luc-nonstop under the control of the GAL1 promoter were grown on galactose-containing medium. Levels of expression in total cell extracts were analyzed via immunoblotting using luciferase antibody. Sse1 served as a loading control. Expression of GAL-Luc-nonstop was quantified in three independent experiments. Mean values and SEMs are given below one representative immunoblot. (G) Expression of Luc-nonstop in Δerg6 and Δerg6 Δzuo1 strains after proteasome inhibition. The proteasome inhibitor MG132 was added to logarithmically growing cultures as indicated. In order to estimate the molecular mass of Luc-nonstop translation products, a diluted sample of Luc-Lys20 was analyzed in parallel.
Luc-nonstop mRNA was destabilized about 2-fold compared to Luc-stop mRNA (see Fig. S3A and B in the supplemental material). However, on the protein level, Luc-nonstop was reduced more than 50-fold compared to Luc-stop (Fig. 5B). The decrease of Luc-nonstop expression correlated with the decrease in luciferase activity (see Fig. S4A in the supplemental material). The Luc-nonstop translation product migrated as a relatively sharp band with an apparent molecular mass similar to that of Luc-Lys12 on SDS-PAGE (Fig. 5B). This was surprising because translation is expected to proceed to the very end of the nonstop mRNA, including its poly(A) tail. Depending on the processing site within the HIS3 3′ region (36), this would add between 27 and 58 amino acids to the C terminus of luciferase, corresponding to a molecular mass increase of roughly 4 to 11 kDa (see Fig. S1 in the supplemental material). Translation of the poly(A) tail would add additional lysines, resulting in an additional molecular mass increase. In order to compare the molecular mass of the Luc-nonstop translation product more directly, molecular tape measures termed Luc-UTR12 and Luc-UTR25 were constructed by introducing stop codons into the HIS3 3′ region upstream of the first predicted polyadenylation site (see Fig. S5A in the supplemental material). The translation product of Luc-UTR12 contains an additional 12 amino acids, and that of Luc-UTR25 contains an additional 25 amino acids (see Fig. S1 in the supplemental material). Consistent with previous findings (27), shortening of the 3′ UTR resulted in decreased levels of luciferase expression (see Fig. S5B in the supplemental material). When similar amounts of luciferase translation products were analyzed, the molecular mass increase of Luc-stop < Luc-UTR12 < Luc-UTR25 was clearly resolved (see Fig. S5B, short exposure, in the supplemental material). In comparison, Luc-nonstop migrated with a molecular mass larger than that of Luc-UTR12 but at most equal to that of Luc-UTR25 (see Fig. S5B, long exposure, in the supplemental material). Thus, in the wild-type strain, translation of Luc-nonstop proceeded into the 3′ UTR; however, it did not proceed beyond the HIS3 3′ region into the poly(A) tail. Thus, the bulk of Luc-nonstop expressed in wild type did not contain a polylysine tail. Consistently, Luc-nonstop was not stably associated with ribosomes when expressed in the wild-type strain (see Fig. S4B in the supplemental material; compare the ribosome associations of Luc-Lys20 in the presence and absence of RAC/Ssb in Fig. 3C).
The RAC/Ssb system did not affect the turnover of Luc-nonstop mRNA (see Fig. S3A in the supplemental material) and also did not affect folding (Fig. 5C) or solubility (Fig. 5D) of the Luc-nonstop protein. With minor variations in different experiments, Luc-nonstop expression was not or was only slightly enhanced in the absence of RAC/Ssb (Fig. 5E and F). However, Luc-nonstop expression was strongly enhanced when RAC/Ssb was absent, and at the same time, the proteasome was inhibited (Fig. 5G). The additive effect indicated that the action of RAC/Ssb and the proteasome were required for independent, most likely successive steps of nonstop protein quality control. Of note, the molecular mass of Luc-nonstop protein, which accumulated upon proteasome inhibition, did not exceed that of Luc-Lys20 (Fig. 5G). This suggests that Luc-nonstop translation products lacking a C-terminal polylysine were also degraded by the proteasome.
To investigate the combined effects of the NSD machinery and RAC/Ssb on the expression of Luc-nonstop, strains lacking the NSD factor Ski7 were generated (15, 53; see the introduction). Interestingly, in the Δski7 strain, Luc-nonstop expression not only was enhanced, but also species with higher molecular masses were detected (Fig. 5E). The fuzzy appearance of the Luc-nonstop band in immunoblots indicated that translation products of different lengths were generated in the absence of Ski7. This is consistent with translation of the HIS3 3′ region, which contains multiple polyadenylation sites (36). When Luc-nonstop was expressed from the strong inducible GAL1 promoter, the generation of distinct Luc-nonstop species of increased length became even more clearly visible (Fig. 5F). While in the wild-type or Δzuo1 strains the higher-molecular-mass Luc-nonstop species had a low abundance, their expression was strongly increased in the absence of Ski7 (Fig. 5F). As a control, Ski7 did not affect the expression or molecular mass of Luc-stop (see Fig. S2D in the supplemental material). The data strongly suggest that in the absence of Ski7, translation of nonstop transcripts proceeded through the 3′ region into the poly(A) tail and polylysine-tagged translation products were generated. Consistent with this notion, Luc-nonstop species with higher molecular masses were more efficiently associated with ribosomes than lower-molecular-mass species in the Δski7 strain (see Fig. S4B in the supplemental material). Loss of RAC/Ssb significantly enhanced translation of the higher-molecular-mass Luc-nonstop species in the Δski7 background (Fig. 5E and F). Enhanced expression of Luc-nonstop in the Δzuo1 Δski7 or Δssb1 Δssb2 Δski7 strain was observed even without inhibition of proteasomal degradation. This observation suggests that the higher-molecular-mass Luc-nonstop translation products were less prone to degradation than the low-molecular-mass species.
In order to investigate if the effect of RAC/Ssb on the expression of nonstop proteins was functionally relevant, we employed the his3-nonstop allele (53, 57). The his3-nonstop allele enables growth of a Δhis3 strain only if the concentration of the His3-nonstop translation product is enhanced compared to that of the wild type (53). We then tested if RAC/Ssb affected growth in the presence of his3-nonstop by itself or in combination with other mutations known to affect the level of His3-nonstop. Note that in the presence of histidine, none of the triple- or quadruple-deletion strains displayed synthetic growth defects (Fig. 6A to C, + histidine).
Fig 6.
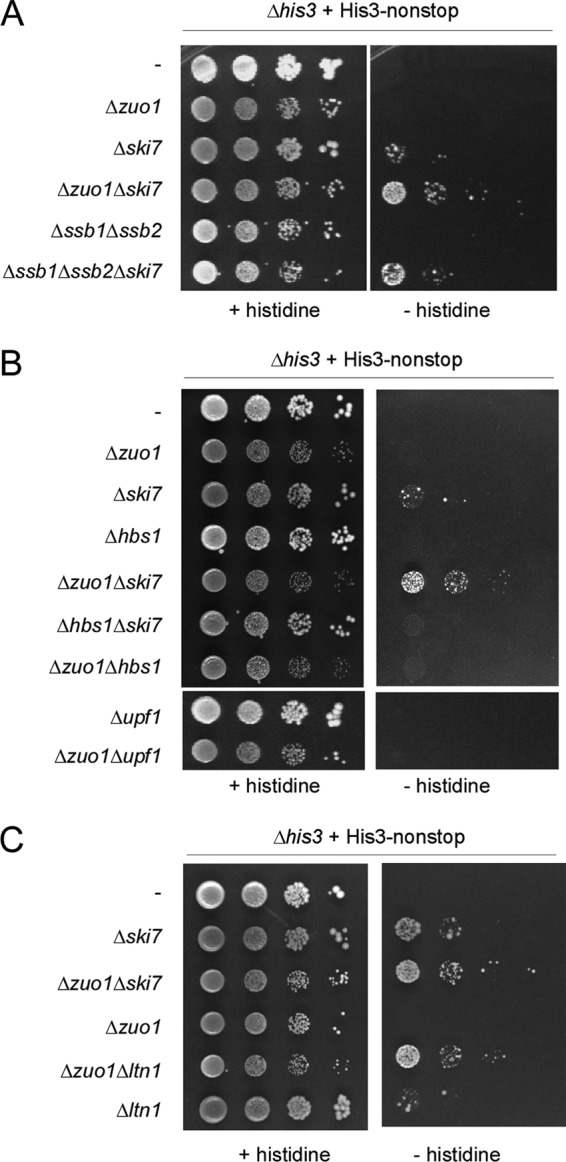
RAC/Ssb affects the level of a functional nonstop protein in vivo. (A) Loss of RAC/Ssb and Ski7 synergistically enhances growth of Δhis3 plus his3-nonstop strains. Growth of the strains containing the indicated gene disruptions was monitored in the presence or absence of histidine. Cell suspensions containing the same number of cells were spotted in 10-fold serial dilutions onto SD plates either supplemented with histidine (+ histidine) or lacking histidine (− histidine) and were incubated for 4 days at 30°C. (B) Loss of RAC/Ssb does not affect growth of Δhis3 plus his3-nonstop strains lacking either Hbs1 or Upf1. Analysis was performed as described for panel A. (C) Loss of RAC/Ssb and Ltn1 synergistically enhances growth of Δhis3 plus his3-nonstop strains. Analysis was performed as described for panel A. The Δzuo1 Δski7 strain was included, in order to allow direct comparison with the Δzuo1 Δltn1 strain.
his3-nonstop expressed in a Δski7 strain background allowed, albeit poor, growth in the absence of histidine (Fig. 6A). No growth was detectable in the Δzuo1 or Δssb1 Δssb2 strain (Fig. 6A). However, the Δzuo1 Δski7 or Δssb1 Δssb2 Δski7 strain displayed significantly enhanced growth in the presence of his3-nonstop compared to that of the Δski7 strain (Fig. 6A). The synergistic effect was observed in the absence of the Ski7 and RAC/Ssb machineries, even though the loss of RAC/Ssb resulted in a general slow-growth phenotype (19, 26). The effects are fully consistent with the levels of Luc-nonstop expression determined in the corresponding strain backgrounds (Fig. 5E). Moreover, the effect was specific for the NSD machinery, because the NGD factor Hbs1 or the principal NMD factor, Upf1, which is involved in the degradation not only of NMD substrates but also of a variety of other cellular transcripts (5, 34), did not suppress the his3-nonstop defect, whether or not RAC/Ssb was present (Fig. 6B). To test if the synergistic effect between RAC/Ssb and the proteasomal system (Fig. 4C to E and Fig. 5G) also applied to His3-nonstop, we made use of the finding that His3-nonstop supported growth of a Δltn1 strain in the absence of histidine, albeit inefficiently (53, 57). Indeed, a strain expressing His3-nonstop in a Δzuo1 Δltn1 background grew more efficiently and the growth was comparable to that of a strain expressing His3-nonstop in a Δzuo1 Δski7 background (Fig. 6C). The data indicate that loss of RAC/Ssb affected the growth of yeast when survival was limited by low-level expression of a protein encoded by nonstop mRNA. The effect of the loss of RAC/Ssb was synergistic with the loss of the NSD component Ski7 as well as with the loss of the ubiquitin ligase Ltn1. The combined data strongly support a model in which Ski7 is required to maintain the low level of nonstop mRNA, RAC/Ssb is required to maintain translational repression of nonstop- and polylysine-encoding transcripts, and Ltn1 is responsible for the polyubiquitination of polylysine and nonstop proteins which had escaped translational repression.
DISCUSSION
While several studies, including this work, revealed that nonstop proteins and polylysine proteins encoded by stop codon-containing mRNA share important characteristics (6, 12, 27, 29), the presence of C-terminal polylysine in nonstop proteins has not been directly demonstrated. We have now found that in wild type the bulk of the Luc-nonstop protein was devoid of a C-terminal polylysine. When expressed in a Δski7 background, however, the molecular mass of Luc-nonstop was significantly increased and differently sized species were generated. Because Ski7 recruits the exosome, the increase in the molecular mass of the nonstop protein was likely related to the status of the nonstop transcript. It was recently discovered that the endonuclease activity of the exosome plays an important role in NSD (49). Depending on where endonucleolytic cleavage of a nonstop transcript occurs, only slightly extended nonstop proteins might be generated. Based on the combined data, we suggest a model in that after the first round of translation, the ribosome stalls at the end of the polyadenylated nonstop mRNA, and Ski7 and the exosome are recruited. The exosome then generates cleavage products lacking part of the 3′ region and the poly(A) tail, which are translated into nonstop products extended by a relatively short stretch of additional amino acids. Our data indicate that in the wild-type background incomplete nonstop proteins are efficiently degraded by the proteasome. The exact mechanism of this process is currently unknown. In the absence of Ski7, the exosome is not recruited and degradation of the nonstop mRNA occurs mostly via 5′-3′ degradation (27). In this situation, full-length nonstop proteins contain the translated HIS3 3′ region and polylysine.
The focus of this study was to determine whether or not the ribosome-bound chaperones RAC and Ssb affect the expression of nonstop and polylysine-tagged proteins. Our findings indicate that this is the case. RAC/Ssb was required to maintain a low level of expression of nonstop as well as polylysine-tagged proteins, which are generated in vivo when a polyadenylated nonstop transcript is fully translated (see above). Loss of RAC (Δzuo1) or the loss of Ssb (Δssb1 Δssb2) exerted similar effects. As Ssb binds to nascent chains efficiently only in the presence of its J-domain partner RAC (19), the requirement for both ribosome-bound chaperones strongly suggests that RAC-mediated binding of Ssb to ribosome-bound nascent chains is important for protein quality control. This is important, as Ssb also functions in processes not directly related to the ribosome (43).
Depending on its conformation, a segment of 20 lysine residues will span from the peptidyl transferase center to approximately the middle of the ribosomal tunnel (7, 56). Thus, RAC and Ssb, both of which bind close to the tunnel exit (42, 43), cannot directly interact with the polylysine segment. This suggests that the polylysine segment not only stalls the nascent peptide and induces translation arrest but also communicates with factors bound at the tunnel exit. Such bidirectional effects of nascent peptides along the ribosomal tunnel are starting to emerge as a common theme in translational regulation (48, 56). How RAC/Ssb can stabilize the interaction between a polylysine protein and the ribosome awaits further investigation. One possibility is that direct interaction of exposed parts of the nascent chain with Ssb in combination with interactions between the polylysine and the interior of the tunnel is required for efficient translational arrest (see Fig. S6 in the supplemental material). However, other mechanisms may also be involved in the stabilization of translationally repressed complexes. It was recently shown that the mammalian homolog of Zuo1, termed MPP11 or ZRF1, contains a ubiquitin-binding domain, which is conserved in Zuo1 (47). Thus, it is possible that polyubiquitination of a nascent chain permanently recruits RAC via its Zuo1 subunit, entailing the stabilization of the Ssb nascent chain complex until the polyubiquitinated nascent chain is degraded.
Chaperones often intimately cooperate with protein degradation machineries (32). However, a comparison between RAC/Ssb and the relevant E3 ligase Ltn1 revealed that at least the major effect of the chaperones in the quality control of nonstop proteins is not at the level of protein degradation. First, RAC/Ssb was not required for the polyubiquitination of polylysine-tagged nascent chains. Second, expression of polylysine proteins was enhanced in the Δzuo1 and Δssb1 Δssb2 strains and was further enhanced upon proteasome inhibition. This distinguishes the Δzuo1 and Δssb1 Δssb2 strains from the Δnot4 or Δltn1 strains, in which inhibition of the proteasome did not further enhance expression of the respective reporter proteins (6, 12). Third, the loss of RAC/Ssb and Ltn1 resulted in an additive enhancement of Luc-Lys20 and His3-nonstop expression. Thus, RAC/Ssb had a major effect on the expression of polylysine proteins that was independent of the effect of the proteolytic system. Based on these data, we speculate that release of the nascent chain from the ribosome in the absence of RAC/Ssb allowed additional rounds of translation of nonstop transcripts. Of note, in mammalian cells the major mechanism reducing the level of expression of nonstop proteins is translational repression (1). On the basis of the work in the mammalian system, it was speculated that ribosome-bound chaperones, which interact with nascent polypeptides, may be involved in translational repression of nonstop mRNA by monitoring the termination process (1). In this context, it is interesting to recall that the RAC/Ssb system of yeast is intimately connected to the function of the translation termination machinery. Loss of RAC/Ssb enhances readthrough of stop codons, while overexpression of Ssb allows their efficient recognition (21, 45). In addition, Ssb affects the generation and maintenance of the yeast prion [PSI+], which represents a nonfunctional prion form of the translation termination factor Sup35 (3, 10, 45). It was suggested that Ski7-mediated NSD may be a specialized mechanism for counteracting the effects of increased stop codon readthrough in the presence of [PSI+] (5, 58). This idea is consistent with the fact that Ski7 is confined to a subset of yeast (5). Noteworthy is the fact that the presence of an Hsp70 homolog which, like Ssb, directly interacts with the ribosome is also confined to yeast (43). One function of RAC/Ssb on the ribosome is possibly to ensure low-level expression not only of proteins encoded by nonstop transcripts but also of proteins which result from readthrough events in the presence of [PSI+].
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by SFB 746, Forschergruppe 967, and the Excellence Initiative of the German federal and state governments (EXC 294) (to S.R.).
S.R. and M.C. conceived the project. M.C., C.C., and F.R. performed the experimental work, and S.R. wrote the paper.
We declare no conflict of interest.
Footnotes
Published ahead of print 24 September 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Akimitsu N, Tanaka J, Pelletier J. 2007. Translation of nonSTOP mRNA is repressed post-initiation in mammalian cells. EMBO J. 26: 2327–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Albanese V, Reissmann S, Frydman J. 2010. A ribosome-anchored chaperone network that facilitates eukaryotic ribosome biogenesis. J. Cell Biol. 189: 69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Allen KD, Chernova TA, Tennant EP, Wilkinson KD, Chernoff YO. 2007. Effects of ubiquitin system alterations on the formation and loss of a yeast prion. J. Biol. Chem. 282: 3004–3013 [DOI] [PubMed] [Google Scholar]
- 4. Ashe MP, De Long SK, Sachs AB. 2000. Glucose depletion rapidly inhibits translation initiation in yeast. Mol. Biol. Cell 11: 833–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Atkinson GC, Baldauf SL, Hauryliuk V. 2008. Evolution of nonstop, no-go and nonsense-mediated mRNA decay and their termination factor-derived components. BMC Evol. Biol. 8: 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bengtson MH, Joazeiro CA. 2010. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature 467: 470–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bhushan S, et al. 2010. Alpha-helical nascent polypeptide chains visualized within distinct regions of the ribosomal exit tunnel. Nat. Struct. Mol. Biol. 17: 313–317 [DOI] [PubMed] [Google Scholar]
- 8. Brooks SA. 2010. Functional interactions between mRNA turnover and surveillance and the ubiquitin proteasome system. Wiley Interdiscip Rev. RNA 1: 240–252 [DOI] [PubMed] [Google Scholar]
- 9. Carvin CD, Kladde MP. 2004. Effectors of lysine 4 methylation of histone H3 in Saccharomyces cerevisiae are negative regulators of PHO5 and GAL1-10. J. Biol. Chem. 279: 33057–33062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chernoff YO, Newnam GP, Kumar J, Allen K, Zink AD. 1999. Evidence for a protein mutator in yeast: role of the Hsp70-related chaperone ssb in formation, stability, and toxicity of the [PSI] prion. Mol. Cell. Biol. 19: 8103–8112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Conz C, et al. 2007. Functional characterization of the atypical Hsp70 subunit of yeast ribosome-associated complex. J. Biol. Chem. 282: 33977–33984 [DOI] [PubMed] [Google Scholar]
- 12. Dimitrova LN, Kuroha K, Tatematsu T, Inada T. 2009. Nascent peptide-dependent translation arrest leads to Not4p-mediated protein degradation by the proteasome. J. Biol. Chem. 284: 10343–10352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Doma MK, Parker R. 2006. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440: 561–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doma MK, Parker R. 2007. RNA quality control in eukaryotes. Cell 131: 660–668 [DOI] [PubMed] [Google Scholar]
- 15. Frischmeyer PA, et al. 2002. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science 295: 2258–2261 [DOI] [PubMed] [Google Scholar]
- 16. Funakoshi Y, et al. 2007. Mechanism of mRNA deadenylation: evidence for a molecular interplay between translation termination factor eRF3 and mRNA deadenylases. Genes Dev. 21: 3135–3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gaber RF, Copple DM, Kennedy BK, Vidal M, Bard M. 1989. The yeast gene ERG6 is required for normal membrane function but is not essential for biosynthesis of the cell-cycle-sparking sterol. Mol. Cell. Biol. 9: 3447–3456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gautschi M, et al. 2001. RAC, a stable ribosome-associated complex in yeast formed by the DnaK-DnaJ homologs Ssz1p and zuotin. Proc. Natl. Acad. Sci. U. S. A. 98: 3762–3767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gautschi M, Mun A, Ross S, Rospert S. 2002. A functional chaperone triad on the yeast ribosome. Proc. Natl. Acad. Sci. U. S. A. 99: 4209–4214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gietz RD, Sugino A. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74: 527–534 [DOI] [PubMed] [Google Scholar]
- 21. Hatin I, Fabret C, Namy O, Decatur W, Rousset JP. 2007. Fine tuning of translation termination efficiency in Saccharomyces cerevisiae involves two factors in close proximity to the exit tunnel of the ribosome. Genetics 177: 1527–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heitman J, Movva NR, Hiestand PC, Hall MN. 1991. FK 506-binding protein proline rotamase is a target for the immunosuppressive agent FK 506 in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 88: 1948–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoshino S, et al. 1999. Novel function of the eukaryotic polypeptide-chain releasing factor 3 (eRF3/GSPT) in the mRNA degradation pathway. Biochemistry (Mosc.) 64: 1367–1372 [PubMed] [Google Scholar]
- 24. Hosoda N, et al. 2003. Translation termination factor eRF3 mediates mRNA decay through the regulation of deadenylation. J. Biol. Chem. 278: 38287–38291 [DOI] [PubMed] [Google Scholar]
- 25. Huang P, Gautschi M, Walter W, Rospert S, Craig EA. 2005. The Hsp70 Ssz1 modulates the function of the ribosome-associated J-protein Zuo1. Nat. Struct. Mol. Biol. 12: 497–504 [DOI] [PubMed] [Google Scholar]
- 26. Hundley H, et al. 2002. The in vivo function of the ribosome-associated Hsp70, Ssz1, does not require its putative peptide-binding domain. Proc. Natl. Acad. Sci. U. S. A. 99: 4203–4208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Inada T, Aiba H. 2005. Translation of aberrant mRNAs lacking a termination codon or with a shortened 3′-UTR is repressed after initiation in yeast. EMBO J. 24: 1584–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Isken O, Maquat LE. 2007. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 21: 1833–1856 [DOI] [PubMed] [Google Scholar]
- 29. Ito-Harashima S, Kuroha K, Tatematsu T, Inada T. 2007. Translation of the poly(A) tail plays crucial roles in nonstop mRNA surveillance via translation repression and protein destabilization by proteasome in yeast. Genes Dev. 21: 519–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kobayashi T, Funakoshi Y, Hoshino SI, Katada T. 2004. The GTP-binding release factor eRF3 as a key mediator coupling translation termination to mRNA decay. J. Biol. Chem. 279: 45693–45700 [DOI] [PubMed] [Google Scholar]
- 31. Koplin A, et al. 2010. A dual function for chaperones SSB-RAC and the NAC nascent polypeptide-associated complex on ribosomes. J. Cell Biol. 189: 57–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kriegenburg F, Ellgaard L, Hartmann-Petersen R. 2012. Molecular chaperones in targeting misfolded proteins for ubiquitin-dependent degradation. FEBS J. 279: 532–542 [DOI] [PubMed] [Google Scholar]
- 33. Lee DH, Goldberg AL. 1998. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8: 397–403 [DOI] [PubMed] [Google Scholar]
- 34. Lelivelt MJ, Culbertson MR. 1999. Yeast Upf proteins required for RNA surveillance affect global expression of the yeast transcriptome. Mol. Cell. Biol. 19: 6710–6719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu J, Deutsch C. 2008. Electrostatics in the ribosomal tunnel modulate chain elongation rates. J. Mol. Biol. 384: 73–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mahadevan S, Raghunand TR, Panicker S, Struhl K. 1997. Characterisation of 3′ end formation of the yeast HIS3 mRNA. Gene 190: 69–76 [DOI] [PubMed] [Google Scholar]
- 37. Mangus DA, Evans MC, Jacobson A. 2003. Poly(A)-binding proteins: multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol. 4: 223 doi:10.1186/gb-2003-4-7-223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nelson RJ, Ziegelhoffer T, Nicolet C, Werner-Washburne M, Craig EA. 1992. The translation machinery and 70 kd heat shock protein cooperate in protein synthesis. Cell 71: 97–105 [DOI] [PubMed] [Google Scholar]
- 39. Ohba M. 1994. A 70-kDa heat shock cognate protein suppresses the defects caused by a proteasome mutation in Saccharomyces cerevisiae. FEBS Lett. 351: 263–266 [DOI] [PubMed] [Google Scholar]
- 40. Ohba M. 1997. Modulation of intracellular protein degradation by SSB1-SIS1 chaperon system in yeast S. cerevisiae. FEBS Lett. 409: 307–311 [DOI] [PubMed] [Google Scholar]
- 41. Passos DO, et al. 2009. Analysis of Dom34 and its function in no-go decay. Mol. Biol. Cell 20: 3025–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peisker K, et al. 2008. Ribosome-associated complex binds to ribosomes in close proximity of Rpl31 at the exit of the polypeptide tunnel in yeast. Mol. Biol. Cell 19: 5279–5288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peisker K, Chiabudini M, Rospert S. 2010. The ribosome-bound Hsp70 homolog Ssb of Saccharomyces cerevisiae. Biochim. Biophys. Acta 1803: 662–672 [DOI] [PubMed] [Google Scholar]
- 44. Pfund C, et al. 1998. The molecular chaperone Ssb from Saccharomyces cerevisiae is a component of the ribosome-nascent chain complex. EMBO J. 17: 3981–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rakwalska M, Rospert S. 2004. The ribosome-bound chaperones RAC and Ssb1/2p are required for accurate translation in Saccharomyces cerevisiae. Mol. Cell. Biol. 24: 9186–9197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Raue U, Oellerer S, Rospert S. 2007. Association of protein biogenesis factors at the yeast ribosomal tunnel exit is affected by the translational status and nascent polypeptide sequence. J. Biol. Chem. 282: 7809–7816 [DOI] [PubMed] [Google Scholar]
- 47. Richly H, et al. 2010. Transcriptional activation of polycomb-repressed genes by ZRF1. Nature 468: 1124–1128 [DOI] [PubMed] [Google Scholar]
- 48. Rospert S. 2004. Ribosome function: how to govern the fate of a nascent polypeptide. Curr. Biol. 14: R386–R388 [DOI] [PubMed] [Google Scholar]
- 49. Schaeffer D, van Hoof A. 2011. Different nuclease requirements for exosome-mediated degradation of normal and nonstop mRNAs. Proc. Natl. Acad. Sci. U. S. A. 108: 2366–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shoemaker CJ, Eyler DE, Green R. 2010. Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science 330: 369–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tomecki R, Drazkowska K, Dziembowski A. 2010. Mechanisms of RNA degradation by the eukaryotic exosome. Chembiochem. 11: 938–945 [DOI] [PubMed] [Google Scholar]
- 52. Tsuboi T, et al. 2012. Dom34:hbs1 plays a general role in quality-control systems by dissociation of a stalled ribosome at the 3′ end of aberrant mRNA. Mol. Cell 46: 518–529 [DOI] [PubMed] [Google Scholar]
- 53. van Hoof A, Frischmeyer PA, Dietz HC, Parker R. 2002. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science 295: 2262–2264 [DOI] [PubMed] [Google Scholar]
- 54. Vasudevan S, Peltz SW, Wilusz CJ. 2002. Non-stop decay—a new mRNA surveillance pathway. Bioessays 24: 785–788 [DOI] [PubMed] [Google Scholar]
- 55. von Plehwe U, et al. 2009. The Hsp70 homolog Ssb is essential for glucose sensing via the SNF1 kinase network. Genes Dev. 23: 2102–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wilson DN, Beckmann R. 2011. The ribosomal tunnel as a functional environment for nascent polypeptide folding and translational stalling. Curr. Opin. Struct. Biol. 21: 274–282 [DOI] [PubMed] [Google Scholar]
- 57. Wilson M, Meaux S, van Hoof A. 2007. A genomic screen in yeast reveals novel aspects of nonstop mRNA metabolism. Genetics 177: 773–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wilson MA, Meaux S, Parker R, van Hoof A. 2005. Genetic interactions between [PSI+] and nonstop mRNA decay affect phenotypic variation. Proc. Natl. Acad. Sci. U. S. A. 102: 10244–10249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yaffe MP, Schatz G. 1984. Two nuclear mutations that block mitochondrial protein import in yeast. Proc. Natl. Acad. Sci. U. S. A. 81: 4819–4823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yan W, et al. 1998. Zuotin, a ribosome-associated DnaJ molecular chaperone. EMBO J. 17: 4809–4817 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.