

Abstract
The zinc finger transcription factors GATA1 and GATA2 participate in mast cell development. Although the expression of these factors is regulated in a cell lineage-specific and differentiation stage-specific manner, their regulation during mast cell development has not been clarified. Here, we show that the GATA2 mRNA level was significantly increased while GATA1 was maintained at low levels during the differentiation of mast cells derived from mouse bone marrow (BMMCs). Unlike in erythroid cells, forced expression or small interfering RNA (siRNA)-mediated knockdown of GATA1 rarely affected GATA2 expression, and vice versa, in mast cells, indicating the absence of cross-regulation between Gata1 and Gata2 genes. Chromatin immunoprecipitation assays revealed that both GATA factors bound to most of the conserved GATA sites of Gata1 and Gata2 loci in BMMCs. However, the GATA1 hematopoietic enhancer (G1HE) of the Gata1 gene, which is essential for GATA1 expression in erythroid and megakaryocytic lineages, was bound only weakly by both GATA factors in BMMCs. Furthermore, transgenic-mouse reporter assays revealed that the G1HE is not essential for reporter expression in BMMCs and peritoneal mast cells. Collectively, these results demonstrate that the expression of GATA factors in mast cells is regulated in a manner quite distinct from that in erythroid cells.
INTRODUCTION
The zinc finger transcription factors GATA1 and GATA2 are essential for normal hematopoietic development. GATA1 is expressed in erythroid cells, megakaryocytes, eosinophils, mast cells, and dendritic cells of the hematopoietic system (5, 12, 21, 35, 47, 56). Gene ablation studies revealed that GATA1 is essential for erythroid cell differentiation, not only in embryonic, but also in postnatal hematopoiesis (7, 13, 43). GATA2 expression in hematopoietic cells overlaps mostly, but not completely, with GATA1 expression. GATA2 is expressed in hematopoietic stem cells and multilineage progenitors, where it plays key roles in their maintenance and proliferation (26, 27, 29, 45, 46).
Previous studies have shown the importance of GATA factor-dependent autoregulatory and cross-regulatory loops in directing the proper spatiotemporal expression of the Gata1 and Gata2 genes. Several GATA motifs within the active cis-regulatory regions of the Gata1 and Gata2 loci are highly conserved among multiple species (6, 9, 10, 22, 49). Our group and others previously identified a distal regulatory element located 3.9 kb upstream of the hematopoietic-cell-specific first exon (IE) of the mouse Gata1 gene. This element is indispensable for Gata1 gene expression in erythroid cells and megakaryocytes and was designated the Gata1 gene hematopoietic enhancer (G1HE) (also referred to as HS1 or mHS-3.5) (24, 31, 33, 50). In addition, a double GATA site (dblGATA), located 680 bp upstream of the IE, and a cluster of multiple GATA motifs in the first intron are required for full promoter activity in erythroid cells (30, 48). Sequence surveys demonstrated that the conserved GATA sites are also distributed within the Gata2 locus (22). One of them is a cluster of five GATA motifs positioned around 2.8 kb 5′ to the distal hematopoietic-cell-specific first exon (IS). Transgenic-mouse reporter assays demonstrated that these particular GATA sites are necessary for GATA2 expression in the early hematopoietic cells residing in the dorsal aortas of embryos 9.5 days postcoitum (19).
In erythroid cells, Gata1 and Gata2 expression levels are strictly controlled in a differentiation stage-specific manner. Upon commitment to an erythroid lineage, GATA2 expression declines, whereas GATA1 expression starts to increase and peaks at the late erythroid progenitor and proerythroblast stages. Chromatin immunoprecipitation (ChIP) assays using a GATA1-null proerythroblast-like cell line expressing tamoxifen-inducible GATA1 (G1E-ER-GATA1) demonstrated that GATA1 displaces GATA2 at several conserved GATA sites in the Gata2 locus and thereafter represses GATA2 transcription (9, 22). This process is referred to as the “GATA switch” model (2). GATA1 and GATA2 are involved in the development of mast cells (25, 46). A particular difference from the erythroid lineage is that GATA2 expression is not restricted to immature progenitors but is abundantly expressed in the mature mast cells residing in the skin connective tissues (14, 23). Moreover, GATA2 mRNA is clearly detected in mast cells derived from mouse bone marrow (BMMCs) and in most mast cell lines, as well (14, 23). In contrast, GATA1 mRNA expression appeared to be inactivated in the several types of mast cell lines (14). Furthermore, immunohistochemical analyses failed to detect GATA1 protein expression in the mast cells within skin connective tissues (23). Thus, the expression profiles of GATA1 and GATA2 during mast cell differentiation are quite different from those in erythroid cells. The molecular basis of Gata1 and Gata2 gene transcription during mast cell development, however, is largely unknown.
To gain more insight into GATA factor-dependent Gata1 and Gata2 gene regulation in mast cells, we examined the expression of GATA1 and GATA2 during BMMC differentiation. GATA2 mRNA levels were significantly increased during BMMC differentiation, whereas GATA1 mRNA remained at a lower level throughout this process. Unlike in erythroid cells, the small interfering RNA (siRNA)-mediated knockdown or genetic disruption of GATA1 expression barely affected GATA2 mRNA levels in cultured mast cell lines or BMMCs. We further examined the regulatory potential of the conserved GATA sites within the Gata1 and Gata2 loci by ChIP assays. We found that the histone marker in the G1HE region was negatively regulated and that neither GATA factor bound to G1HE in the mast cell lineage. Consistently, transgenic-mouse reporter assays clearly proved the G1HE to be dispensable for mast cell-specific Gata1 gene expression in vivo. Overall, our findings demonstrate that regulation of the Gata1 and Gata2 genes during erythroid and mast cell development is distinct in the two cell types.
MATERIALS AND METHODS
Mice.
Conditional Gata1 knockout mice (Gata1flox/y) were generated as previously described (13). The knock-in mice expressing a 4-hydroxytamoxifen (4-OHT)-inducible Cre recombinase gene under the control of the Rosa26 promoter (Rosa26CreERT2) were kindly provided by Anton Berns, The Netherlands Cancer Institute. Since the Gata1 gene is X linked, the Gata1 knockout phenotype was examined in hemizygous male mice (Gata1flox/y) expressing CreERT2. The mice were bred to a BDF1 background and maintained in an animal facility of Takasaki University of Health and Welfare in accordance with institutional guidelines.
Cells.
Bone marrow cells were harvested from 6- to 12-week-old wild-type or Gata1flox/y male mice and cultured with RPMI 1640 medium supplemented with 10 ng/ml recombinant murine interleukin 3 (IL-3) (Peprotech). After 2 weeks of culturing, 10 ng/ml recombinant murine stem cell factor (SCF) (Peprotech) was added. After 4 to 6 weeks of culturing, almost all (>95%) cells displayed a mast cell phenotype, as assessed by the expression of c-Kit and FcεRIα and alcian blue/safranin O staining. RBL-2H3 cells (a rat basophilic leukemia cell line) and murine erythroleukemia (MEL) cells were cultured with Eagle minimal essential medium (MEM) and RPMI 1640 medium, respectively. P815 cells (a mouse mast cell line) and J774.1 cells (a mouse macrophage cell line) were maintained in Dulbecco's modified Eagle medium (DMEM) containing 1.0 g/liter glucose. All culture media were purchased from Nacalai Tesque and supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1× penicillin-streptomycin solution (Gibco).
Plasmids and siRNAs.
The expression plasmids for GATA1, GATA2, and FOG-1 were generated as previously described (37, 38). The small interfering RNA (siRNA) duplexes for rat and mouse GATA1 and GATA2 were purchased from Thermo Scientific (rat) and Invitrogen (rat and mouse). Control siRNA (SIC-001) was purchased from Sigma.
Transfection.
RBL-2H3 cells (2.0 × 106) or BMMCs (2.5 × 106) were transfected with 2.5 μg of expression plasmid or 200 pmol of siRNA by electroporation using an Amaxa Nucleofector (Lonza). Programs X-001 and Y-001 were used for RBL-2H3 cells and BMMCs, respectively. Cells were harvested 24 to 48 h posttransfection and then subjected to the analyses.
Western blotting.
Nuclear extracts were prepared as previously described (31), and protein concentrations were quantified using a bicinchoninic acid (BCA) assay (Pierce). Nuclear proteins were resolved by 10% SDS-polyacrylamide gel electrophoresis, and Western blot analyses were performed as formerly explained (17) using anti-GATA1 (N6), anti-GATA2 (H-116), anti-FOG-1 (A-20), and anti-lamin B (M-20) antibodies. All antibodies were purchased from Santa Cruz.
Quantitative real-time PCR.
Total RNA was extracted from cells with a FastPure RNA kit (catalog no. 9190; TaKaRa) or by NucleoSpin RNA II (catalog no. 740955; TaKaRa). Reverse transcription was carried out using 1 μg of total RNA as the template and Superscript III First-Strand Synthesis Supermix (Invitrogen), as outlined by the manufacturer. Quantitative real-time PCR (qRT-PCR) was performed using Go Taq qPCR Master Mix (catalog no. A6001; Promega) and an Mx3000P real-time PCR system (Stratagene) as previously described (17). Primer sequences are shown in Table 1. For each primer set, a single PCR product was confirmed by 2% agarose gel electrophoresis and melting-curve analysis. Data were normalized to 18S rRNA levels and are shown as averages and standard deviations (SD). For some experiments, values were normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) levels. The relative abundance of GATA1 and GATA2 transcripts in BMMCs was calculated as previously described (44). In brief, the PCR efficiency (E) of each primer set was determined by PCR with a serially diluted known template. The sizes of the PCR fragments were 70 and 51 bp for GATA1 and GATA2, respectively. The value of the threshold cycle (CT) for GATA1 or GATA2 was determined by qRT-PCR in each sample. Then, the abundance of GATA1 transcripts relative to those of GATA2 was calculated by the following equation: GATA1/GATA2 = 51 × (1 + EGATA2)CT(GATA2)/70 × (1 + EGATA1)CT(GATA1).
Table 1.
Primer sequences for gene expression analysis
| Gene product | Species | Primer | Sequence |
|---|---|---|---|
| GATA1 | Mouse, rat | 5′ | CAG AAC CGG CCT CTC ATC C |
| 3′ | TAG TGC ATT GGG TGC CTG C | ||
| GATA2 | Mouse, rat | 5′ | GCA GAG AAG CAA GGC TCG C |
| 3′ | CAG TTG ACA CAC TCC CGG C | ||
| MC-CPA | Mouse | 5′ | GCT ACA CAT TCA AAC TGC CTC CT |
| 3′ | GAG AGA GCA TCC GTG GCA A | ||
| MCP5 | Mouse | 5′ | CCT GGG TTC CAG CAC CAA |
| 3′ | GGC GGG AGT GTG GTA TGC | ||
| c-Kit | Mouse | 5′ | AGC AAT GGC CTC ACG AGT TCT A |
| 3′ | CCA GGA AAA GTT TGG CAG GAT | ||
| MPO | Mouse | 5′ | CGG TTC TCC TTC TTC ACT GG |
| 3′ | CTG CCA TTG TCT TGG AAG CG | ||
| SCL/TAL1 | Mouse | 5′ | ATA GCC TTA GCC AGC CGC TC |
| 3′ | GCC GCA CTA CTT TGG TGT GA | ||
| MITF | Mouse | 5′ | GCT GGA GAT GCA GGC TAG AG |
| 3′ | TGA TGA TCC GAT TCA CCA GA | ||
| PU.1 | Mouse | 5′ | AAG TTT CAT GGA AGC CTG CCA TTG |
| 3′ | AAC TGG TAC AGG CGA ATC TTT TTC AAT CAC C | ||
| FOG-1 | Mouse, rat | 5′ | CTG ATG GTG GAT GAG AG |
| 3′ | GGC GTC ATC CTT CCT GTA GA | ||
| 18S rRNA | Mouse, rat | 5′ | ACA TCC AAG GAA GGC AGC AG |
| 3′ | TCG TCA CTA CCT CCC CGG | ||
| GAPDH | Mouse, rat | 5′ | CTT CAC CAC CAT GGA GAA GGC |
| 3′ | GGC ATG GAC TGT GGT CAT GAG |
Recombination analysis.
Cre-mediated recombination of the Gata1 gene was determined by PCR. To amplify the floxed (nondeleted) allele-derived DNA fragment, the forward primer sequence (5′-CGCCGAGCTCTGTCTAGTAA-3′; between the IE and the 5′ loxP site) and the reverse primer sequence (5′-TTCCTCTTTCTCCTCCTCCG-3′; downstream and adjacent to the 5′ loxP site) were used. To amplify the recombined DNA fragment, the forward primer and the reverse green fluorescent protein (GFP) primer (5′-GGTGCTCAGGTAGTGGTT-3′) were used.
ChIP.
A ChIP assay was performed using BMMCs and MEL cells, as reported previously (17). The antibodies used for ChIP were as follows: anti-GATA-1 (N6; Santa Cruz), anti-GATA2 (H-116; Santa Cruz), anti-SCL (C-21; Santa Cruz), anti-acetyl-histone H3 (Active Motif; 39139), anti-trimethyl-histone H3(Lys4) (Active Motif; 39159), anti-histone H3 (Active Motif; 39763), and anti-acetyl-histone H4 (Upstate; 06-866). For the GATA1 ChIP assay, anti-rat IgG rabbit antibody (Jackson ImmunoResearch) was used as a secondary antibody to precipitate the immune complex (17). Other ChIP assays were performed without the use of a secondary antibody. The DNA purified from ChIP samples was analyzed in triplicate using an Mx3000P real-time PCR system (Stratagene) with 3 μl of DNA solution and Go Taq qPCR Master Mix (catalog no. A6001; Promega). The primer sequences are shown in Table 2.
Table 2.
Primer sequences for ChIP analysis
| Gene | Locus | Primer | Sequence |
|---|---|---|---|
| Gata1 | kb −25 | 5′ | AAA AGA AAC CAG TGG GCT GA |
| 3′ | ACA GGA AGA AGG AGC AAG CA | ||
| Gata1 | G1HE | 5′ | TCA GGG AAG GAT CCA AGG AA |
| 3′ | CCG GGT TGA AGC GTC TTC T | ||
| Gata1 | dblGATA | 5′ | CCA AGA CAG CCT GTT ACT GC |
| 3′ | TGG GGT ACT AGG CCA GGA CT | ||
| Gata1 | kb +3.5 | 5′ | ACA GTC AGC CCT GAA AGG AA |
| 3′ | GGG ACA AGG GTC TGT TTT CA | ||
| Gata1 | Exon 6 | 5′ | GGT CCA GGA AAA GGC ATA AG |
| 3′ | TAC TGC CCA CCT CTA TCA GG | ||
| Gata2 | kb −27.7 | 5′ | TGC CAT GCC GGA TAT ATT TTG |
| 3′ | ACT AGC ACG TGT GGC ACA GTG | ||
| Gata2 | kb −3.9 | 5′ | GAG ATG AGC TAA TCC CGC TGT A |
| 3′ | AAG GCT GTA TTT TTC CAG GCC | ||
| Gata2 | kb −2.8 | 5′ | GCA TGG CCC TGG TAA TAG C |
| 3′ | CAG CCG CAC CTT CCC TAA | ||
| Gata2 | kb +9.5 | 5′ | ACA TCT GCA GCC GGT AGA TAA G |
| 3′ | CAT TAT TTG CAG AGT GGA GGG TAT TA |
Fluorescence-activated cell-sorting analysis.
Bone marrow cells were stained with allophycocyanin (APC)-conjugated rat anti-mouse CD117 (c-Kit; BD Pharmingen; clone 2B8) and phycoerythrin (PE)-conjugated mouse anti-mouse FcεRIα (eBioscience; clone MAR-1). Flow cytometric analysis was performed using a FACSCantoII flow cytometer (BD Biosciences).
Statistical analysis.
A comparison was made between two groups using the Student t test. Data are presented as means ± SD. For all analyses, statistical significance was defined as a P value of <0.05.
RESULTS
Distinct mRNA expression profiles of GATA1 and GATA2 during BMMC differentiation.
To examine the roles of GATA1 and GATA2 in mast cell development, we employed an in vitro culture system for mouse BMMCs. These primary cells were cultured for the first 14 days in the presence of recombinant murine IL-3 (10 ng/ml) and then switched to supplementation with IL-3 and recombinant murine SCF (10 ng/ml) thereafter. The average yield of c-Kit/FcεRIα double-positive cells increased with time and reached approximately 94% on day 28, as shown by flow cytometry (Fig. 1A). qRT-PCR analyses revealed that the mRNA levels of mouse mast cell carboxypeptidase (mMC-CPA), mouse mast cell protease 5 (mMCP-5), and c-Kit were significantly upregulated with time (Fig. 1B). In contrast, myeloperoxidase (MPO) mRNA, a marker for neutrophils and monocytes, was almost extinguished by day 21 (Fig. 1B). These results indicate that most, if not all, of the cells give rise to mast cells by 28 days of culturing. We next examined the mRNA levels of the hematopoietic transcription factors GATA1, GATA2, SCL/Tal1, MITF, and PU.1 in the differentiating BMMCs by qRT-PCR (Fig. 2). These factors have been shown to play critical roles in mast cell development (25, 28, 36, 46, 52). To compare the expression levels of these transcription factors between mast cells and erythroid cells, their mRNA levels were simultaneously examined in an erythroid cell line (MEL) and in mouse fetal liver at day 12.5 of gestation (E12.5FL), which contain abundant erythroblasts. We also examined the expression levels of these factors in mast cell lines (RBL-2H3 and P815) and in the murine macrophage-like cell line J774.1. Of note, the expression levels of GATA2, SCL, MITF, and PU.1 increased with time, whereas the GATA1 mRNA expression level was hardly changed during the culture period by day 35 (Fig. 2). Moreover, the GATA1 mRNA levels in BMMCs at all time points were lower than those in erythroid cells (E12.5 FL and MEL). A previous study reported that friend of GATA1 (FOG-1) is a negative regulator of mast cell development (3). Consistent with this observation, we found much lower levels of FOG-1 expression in BMMCs than in erythroid cells. Together, these results indicate that the expression levels of GATA2, SCL/Tal1, MITF, and PU.1 increased according to the differentiation, whereas Gata1 gene expression is likely to be regulated independently of mast cell maturation. As reported previously (56), GATA2, but not GATA1, was expressed in the murine mast cell line P815, whereas both GATA factors were expressed at high levels in rat basophilic leukemia cells, RBL-2H3. Neither GATA1 nor GATA2 mRNA expression was detected in J774.1 cells.
Fig 1.

In vitro differentiation of bone marrow-derived mast cells. (A) Flow cytometric analysis of BMMCs. Side scatter/forward scatter (SSC/FSC) dot plots and c-Kit/FcεRIα expression are shown for the indicated culture days. The numbers represent the average percentages and SD of the c-Kit/FcεRIα double-positive cells within the gates. The data are representative of 5 independent experiments. (B) The mRNA levels of mMC-CPA, mMCP5, c-Kit, and MPO in BMMCs on the specified culture days were measured by qRT-PCR. The data were obtained from 3 independent experiments.
Fig 2.

Expression of hematopoietic transcription factors during BMMC differentiation. The mRNA levels of GATA1, GATA2, SCL/Tal1, MITF, PU.1, and FOG-1 in BMMCs on the indicated culture days (D) were measured by qRT-PCR. The mRNA levels of these factors were simultaneously examined in primary hematopoietic cells derived from E12.5 fetal liver (E12.5FL) and other hematopoietic cell lines. The data were obtained from 3 independent experiments. **, P < 0.01 compared with the data from day 14.
Cross-regulatory gene expression of Gata1 and Gata2 was not observed in RBL-2H3 mast cells.
GATA2 upregulates Gata1 gene transcription in early erythroid progenitors (31, 34). In contrast, we found that GATA1 mRNA expression remained at a lower level throughout the culturing period of BMMCs, despite the significant increase in GATA2 mRNA. To further clarify if any cross-regulation of Gata1 and Gata2 gene expression takes place in mast cells, we transduced RBL-2H3 cells with GATA1 and/or GATA2 siRNAs (Fig. 3A and B). We examined three siRNAs with different sequences for each gene to minimize the possibility of off-target artifacts. Although small variations in silencing efficiency were observed, all of them significantly reduced the levels of their target mRNAs, as shown by qRT-PCR (Fig. 3A). If Gata1 and Gata2 cross-regulated each other in mast cells, as observed in erythroid cells, GATA2 expression would be increased upon GATA1 repression, whereas GATA1 expression would be downregulated by GATA2 repression. In RBL-2H3 cells, however, a reduction in GATA1 expression exerted no effect on GATA2 mRNA, and moreover, the GATA2 knockdown elicited no changes in GATA1 expression (Fig. 3A). These results were consistent with those obtained by Western blot analysis of protein levels (Fig. 3B). We then introduced an expression plasmid encoding GATA1 or GATA2 cDNA into RBL-2H3 cells. The GATA1 expression plasmid significantly increased both the mRNA and protein levels of GATA1 (Fig. 3C and D). The forcible expression of GATA2 reached around a 2.2-fold increase in the mRNA level with the modest GATA2 protein accumulation, presumably because the high level of endogenous GATA2 expression might eliminate additional GATA2 expression in RBL-2H3 cells (Fig. 3C and D). If the cross-regulation of Gata1 and Gata2 gene expression could occur in mast cells, GATA2 expression would be downregulated by GATA1 overexpression, whereas GATA1 expression would be increased by forced expression of GATA2. However, this was not the case in mast cells at both mRNA and protein levels (Fig. 3C and D). Recent studies showed that the association of FOG-1 is required for GATA1-mediated GATA2 repression in erythroid cells (16, 34). To test if this applies to mast cells, FOG-1 cDNA was ectopically expressed in RBL-2H3 cells. Neither the ectopic expression of FOG-1 alone nor the coexpression of FOG-1 with GATA1 repressed the expression of GATA2 (Fig. 3C and D). In order to confirm that experimental perturbation of GATA factor had functional consequences, we examined the mRNA levels of phospholipase C-γ1 (PLC-γ1), which is known to be regulated by the GATA factors in RBL-2H3 cells (17). Indeed, PLC-γ1 mRNA levels were significantly reduced by siRNA knockdown of GATA1 or GATA2. Furthermore, this repression was restored by reintroducing GATA1 or GATA2 cDNA into the siRNA-treated cells (Fig. 3E). Therefore, even under these correct experimental conditions, expression of Gata1 and Gata2 genes was not interdependent in RBL-2H3 cells. Moreover, the lack of GATA1-mediated Gata2 repression in these cells could not be attributed to a low level of FOG-1 expression. Last, we performed a similar set of experiments in MEL cells to compare the results with our findings in RBL-2H3 cells (Fig. 3F and G). Because the cross-regulation of Gata1 and Gata2 is controlled in a differentiation stage-specific manner, the results observed in this erythroleukemia cell line did not necessarily reproduce the cross-regulation of Gata1 and Gata2 during normal erythroid cell differentiation. For instance, GATA2 overexpression failed to further upregulate endogenous GATA1 expression (Fig. 3G), suggesting that the GATA2-mediated GATA1 activation that occurs in early erythroid progenitors in vivo (31, 34) does not take place in MEL cells. Compared to RBL-2H3 cells, exogenous expression of GATA1 resulted in a smaller increase in GATA1 mRNA levels in MEL cells, where endogenous GATA1 is abundantly expressed (Fig. 3G). Nonetheless, we did observe that GATA2 expression was significantly increased by GATA1 repression, reproducing the GATA1-mediated GATA2 repression observed during normal erythroid cell development (Fig. 3F).
Fig 3.

Lack of cross-regulatory gene transcription of Gata1 and Gata2 in RBL-2H3 cells. (A) RBL-2H3 cells were transfected with one of three different siRNAs (1, 2, and 3) targeting GATA1 or GATA2 or with control siRNA (ctr), and the mRNA levels of GATA1 and GATA2 were examined by qRT-PCR. The value from the control siRNA-transfected cells was set to 1. *, P < 0.05, and **, P < 0.01 compared with the control. (B) The protein levels of GATA1, GATA2 (arrowhead), and lamin B (loading control) in RBL-2H3 cells transfected with GATA1 and/or GATA2 siRNA were examined by Western blot analysis. The representative results from 3 independent experiments are shown. For both GATA1 and GATA2, siRNA 1 (see panel A) was used. ntf, no transfection control. (C) mRNA levels of GATA1, GATA2, and FOG-1 in RBL-2H3 cells were determined by qRT-PCR. Cells were transfected with a control vector expressing GFP (vtr), GATA1, GATA2, or FOG-1 or cotransfected with GATA1 and FOG-1 (G1/F). The value from cells transfected with the control GFP expression plasmid was set to 1. (D) Cells transfected as described for panel C were subjected to Western blot analysis using anti-GATA1, anti-GATA2, anti-FOG-1, and anti-lamin B (loading control) antibodies. The representative results from 5 independent experiments are shown. The asterisk indicates a nonspecific band, and the arrowhead identifies GATA2. (E) The mRNA levels of PLC-γ1 in RBL-2H3 cells were determined by qRT-PCR. Cells were transfected with siRNA and/or with an expression plasmid, as indicated. The value from cells transfected only with control siRNA was set to 1. For both GATA1 and GATA2, siRNA 1 (see panel A) was used. For the comparisons indicated, *, P < 0.05, and **, P < 0.01. (F) mRNA levels of GATA1 and GATA2 in MEL cells transfected with GATA1 or GATA2 siRNA were examined by qRT-PCR. The value from the control siRNA-transfected cells was set to 1. *, P < 0.05, and **, P < 0.01 compared with the control. (G) mRNA levels of GATA1, GATA2, and FOG-1 in MEL cells were determined by qRT-PCR. The cells were transfected with a vector expressing GFP (control), GATA1, GATA2, or FOG-1 or cotransfected with GATA1 and FOG-1 (G1/F). For all qRT-PCR analyses, data were obtained from at least 3 independent experiments. G1, GATA1; G2, GATA2; F, FOG-1; vtr, control vector expressing GFP.
BMMCs did not exhibit cross-regulatory gene transcription between Gata1 and Gata2.
We next examined whether our findings in RBL-2H3 cells apply to BMMCs. To delve into this issue, we utilized compound mutant mice bearing a floxed Gata1 allele and a 4-OHT-inducible Cre transgene (Gata1flox/y::RosaCreERT2 [13]). Bone marrow cells of the mutant mice were subjected to in vitro mast cell differentiation (here referred to as flox/cre BMMCs); then, the 4-OHT treatment was started on cell culture day 30 to diminish Gata1 gene expression. BMMCs isolated from Gata1flox/y mice lacking the Cre transgene were treated with 4-OHT simultaneously and used as controls (here referred to as flox BMMCs). Flow cytometry analysis demonstrated that nearly all flox and flox/cre BMMCs were positive for FcεRIα and c-Kit by 30 days of incubation (data not shown). Following 4-OHT treatment, the recombination efficiency of the floxed Gata1 allele was determined by PCR. The recombined Gata1-derived DNA fragment was exclusively amplified from the genomic DNA of the flox/cre BMMCs after 5 and 10 days of 4-OHT treatment (Fig. 4A). On day 10 of 4-OHT treatment, only a small amount of the nonrecombined allele-derived PCR product was amplified (Fig. 4A). Consistent with these data, qRT-PCR analyses showed that the GATA1 mRNA level in the flox/cre BMMCs was significantly reduced after 10 days of 4-OHT treatment (Fig. 4B). GATA1 protein was not detectable in these cells by Western blot analysis (data not shown). Importantly, the marked reduction in Gata1 gene expression observed in the flox/cre BMMCs did not affect the GATA2 mRNA level (Fig. 4B). These results made a stark contrast with our previous findings that GATA2 mRNA was increased up to 50-fold more than the control levels in the proerythroblasts and the early stages of erythroblasts from the flox/cre bone marrow cells (13). We next examined whether the overexpression of GATA1 and/or FOG-1 represses the quantities of endogenous GATA2 mRNA in wild-type BMMCs (Fig. 4C). Unlike in RBL-2H3 cells (Fig. 3C and D), the forced expression of both GATA1 and FOG-1 resulted in a statistically significant reduction in GATA2 mRNA to 60%. However, the repression of Gata2 by GATA1/FOG-1 in BMMCs was not as pronounced as in erythroid cells, where GATA2 mRNA rapidly decreased during erythroid cell differentiation and fell to an almost undetectable level by the differentiated-erythroblast stage (13).
Fig 4.

Lack of cross-regulatory gene transcription of Gata1 and Gata2 in BMMCs. (A) PCR analysis of recombination in genomic DNA isolated from the BMMCs of Gata1flox/y (flox) and Gata1flox/y::RosaCreERT2 (flox/cre) mice. Day 30 to 40 BMMCs were treated with 4-OHT. Genomic DNA was isolated at 0, 5, and 10 days following the onset of 4-OHT treatment and used for the PCR analysis. PCR amplicons of the G1HE region are shown as controls. (B) mRNA levels of GATA1 and GATA2 in the BMMCs of flox and flox/cre mice were examined by qRT-PCR. Day 30 to 40 BMMCs were treated with 4-OHT. Total RNA was isolated 0, 5, and 10 days after treatment and used in the analysis. (C) mRNA levels of GATA1, GATA2, and FOG-1 in BMMCs transfected with a vector expressing either GATA1, FOG-1, or both were determined by qRT-PCR. The data were obtained from 4 independent assays. The value from cells transfected with vector alone (vtr) was set to 1.0.
GATA1 and GATA2 play redundant roles in activating mast cell-specific genes in BMMCs.
We have shown that the mRNA level of GATA2, but not GATA1, was increased during BMMC differentiation and that expression of the Gata1 and Gata2 genes was regulated independently in these cells. In addition, we calculated that the abundance of GATA1 mRNA transcripts relative to that of GATA2 transcripts was 0.14 ± 0.03 in BMMCs (n = 3; see Materials and Methods for details). These findings raised the possibility that the representative GATA factor target genes expressed in mast cells, such as the mMC-CPA (56) and c-Kit (20) genes, are regulated predominantly by GATA2, and not by GATA1. To test this hypothesis, we examined the consequences of siRNA against GATA1 and/or GATA2 for those genes in BMMCs (Fig. 5A). Contrary to our hypothesis, the treatment with siRNA against GATA2 (and without siRNA against GATA1) barely reduced the mRNA levels of c-Kit and mMC-CPA. Similarly, the GATA1 siRNA treatment elicited no major effects on the expression of those genes. However, simultaneous introduction of GATA1 and GATA2 siRNAs reduced these mRNA levels to less than half of those observed in the control siRNA-transfected cells (Fig. 5A). We then examined whether the mRNA levels of c-Kit and mMC-CPA were affected by the overexpression of GATA1 and/or FOG-1 (Fig. 5B). Despite the observation that coexpression of GATA1 and FOG-1 resulted in a moderate but significant reduction of GATA2 mRNA levels in the BMMCs (Fig. 4C), mRNA levels of c-Kit and mMC-CPA were not affected upon the forced expression of GATA1 and/or FOG-1 in these cells. Collectively, these results suggest that GATA1 and GATA2 are redundant in their abilities to activate transcription of these genes in BMMCs.
Fig 5.
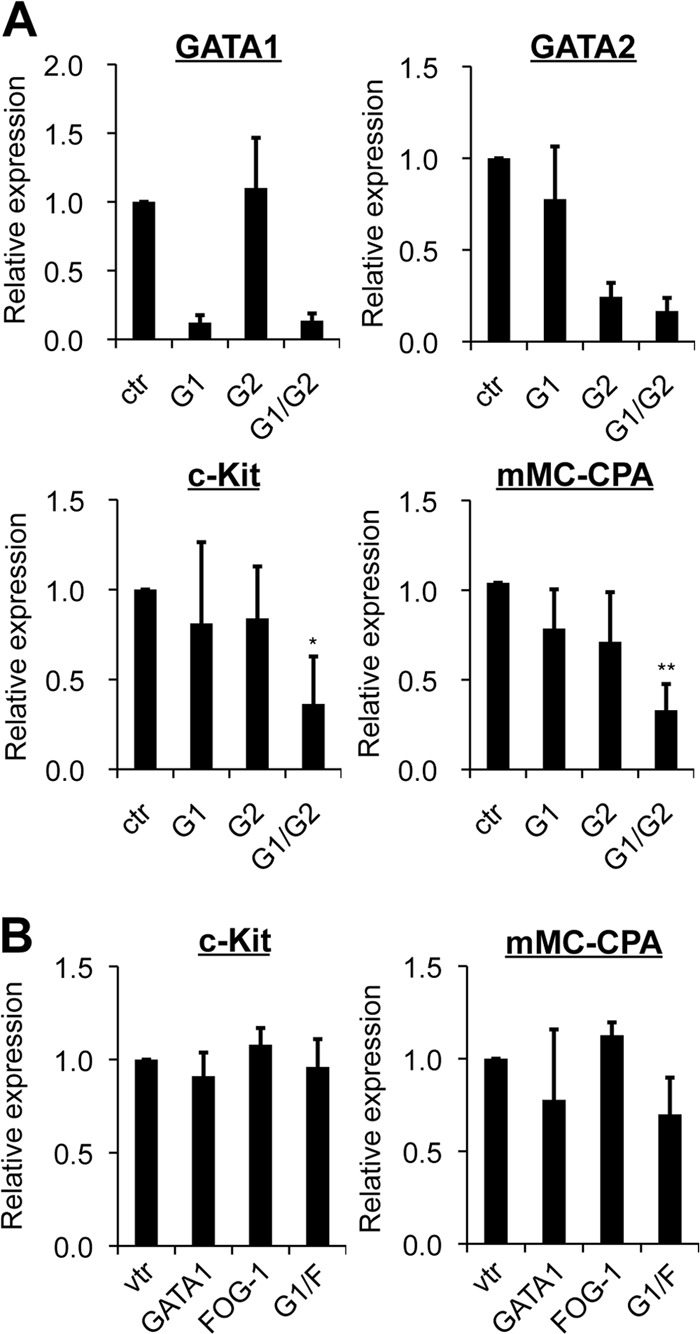
GATA1 and GATA2 play redundant roles in activating mast cell-specific genes in BMMCs. (A) mRNA levels of GATA1, GATA2, c-Kit, and mMC-CPA in BMMCs at days 30 to 40 of culturing were measured by qRT-PCR. The cells were transfected with GATA1 (G1) and/or GATA2 (G2) siRNA or control siRNA (ctr). *, P < 0.05, and **, P < 0.01 compared with the control. (B) mRNA levels of c-Kit and mMC-CPA were measured by qRT-PCR in BMMCs transfected with a control vector expressing GFP (vtr), GATA1, or FOG-1 or cotransfected with GATA1 and FOG-1 (G1/F). In both panels, data were obtained from 3 independent experiments. The value from cells transfected with control siRNA (A) or control vector (B) was set to 1.
GATA factor binding to the conserved GATA sites within the Gata1 and Gata2 loci in BMMCs.
Since the our experiments revealed no solid evidence of any cross-regulation of Gata1 and Gata2 gene expression in mast cells, we subsequently examined GATA factor binding to the conserved GATA sites within the Gata1 and Gata2 loci in BMMCs. Quantitative ChIP assays were performed across the Gata1 and Gata2 loci on days 14, 22, and 38 following BMMC differentiation (Fig. 6). GATA factor binding in MEL cells was simultaneously examined as an erythroid control. Interestingly, GATA1 occupancy was detected at the kb −3.9, −2.8, and +9.5 regions of the Gata2 locus in BMMCs, despite the failure of GATA1 to regulate Gata2 gene expression (Fig. 6B). Notably, this set of regions of the Gata2 locus was also occupied by GATA2 itself (Fig. 6B). Given that the GATA2 mRNA level was increased during BMMC differentiation (Fig. 2), a positive autoregulatory loop by GATA2 might play an important role in Gata2 gene regulation in BMMCs. When similar analyses were performed across the Gata1 locus, significant GATA1 binding was detected at the dblGATA and kb +3.5 regions of the Gata1 locus (Fig. 6C). The distal kb −25 region of the Gata1 gene has been described as a species-specific cis-acting element of the mouse Gata1 gene in erythroid cells (49). However, we did not observe any significant GATA1 binding to this region in BMMCs. To our surprise, neither GATA1 nor GATA2 binding was appreciably increased at the G1HE region of the Gata1 locus throughout the BMMC culture period (Fig. 6C). In contrast, GATA1 was highly enriched at the G1HE of the Gata1 locus in MEL cells (Fig. 6C). SCL associates with GATA factors and acts in a cooperative manner in erythroid and endothelial cells (40, 51, 54). ChIP assays with an antibody to SCL showed significant binding of this factor at the kb +9.5 region of the Gata2 gene in BMMCs and MEL cells (Fig. 6B). It is noteworthy that this region contains a critical GATA-E box composite motif that is required for Gata2 gene expression in vascular endothelial cells (18, 53). Another well-known GATA-E box motif resides within the G1HE region of the Gata1 gene. Consistent with previous reports (49, 50), we observed that the G1HE region was clearly bound by SCL in MEL cells (Fig. 6C). In contrast, the BMMCs barely showed SCL occupancy of this site (Fig. 6C). Given that this region is devoid of GATA factor binding in BMMCs, SCL recruitment to the G1HE region might be dependent on GATA factors. In summary, these data indicate that the binding profile of GATA1, GATA2, and SCL to the Gata1 and Gata2 chromatin regions in BMMCs is quite different from that in MEL cells. In particular, GATA1, GATA2, and SCL only weakly bound to the G1HE region in BMMCs, whereas the G1HE was robustly occupied by GATA1 and SCL in MEL cells. Considering that the G1HE region is essential for Gata1 gene expression in erythroid cells, these data further support the possibility that Gata1 expression in mast cells is controlled by a novel molecular mechanism, which is distinct from that in erythroid lineages.
Fig 6.

Neither GATA1 nor GATA2 binding was enriched in the G1HE chromatin region in BMMCs. (A) Configurations of the Gata1 and Gata2 loci. White ovals and black boxes depict the GATA sites and exons, respectively. (B and C) Binding of GATA1, GATA2, and SCL to the Gata2 locus (B) and the Gata1 locus (C) was examined by ChIP assays. Chromatin fragments were prepared from BMMCs on the specified culture days (days 14, 22, and 28 of culturing) and from MEL cells. ChIP assays were carried out using anti-GATA1, anti-GATA2, and anti-SCL antibodies. Control experiments were performed using rat IgG in place of anti-GATA1 antibody and rabbit IgG instead of anti-GATA2 or SCL antibody. The values of PCR amplicons using immunoprecipitated chromatin relative to those of input are shown. The results were obtained from 4 independent assays. The kb −27.7 (−27.7) and exon 6 (ex6) regions were amplified as negative controls for Gata2 (B) and Gata1 (C), respectively.
Histone modification at the Gata1 locus is different between erythroid and mast cell lineages.
Chromatin modification by epigenetic mechanisms is essential for establishing and maintaining cellular identity in adult organisms. It is well known that acetylation of histone H3 (AcH3) and histone H4 (AcH4) is commonly distributed around enhancers and locus control regions (11). In addition, trimethylation of histone H3 lysine 4 (H3K4me3) is associated with the transcription start sites of active loci (1, 15). Since the G1HE region was not occupied by either GATA factor, we hypothesized that the region might be epigenetically inactivated in BMMCs. To clarify this issue, we performed ChIP assays across the Gata1 locus with an antibody specific for either AcH3 or H3K4me3 in day 30 to 40 BMMCs (Fig. 7A). As expected, we found a much lower level of AcH3 in the G1HE region in BMMCs than in MEL cells. In contrast, a high level of AcH3 accumulation was observed at the dblGATA and kb +3.5 sites in both BMMCs and MEL cells (Fig. 7A, top). H3K4me3 was also detected at the dblGATA and kb +3.5 sites, but not in the G1HE region, in BMMCs (Fig. 7A, bottom). In MEL cells, H3K4me3 was predominantly discernible at the dblGATA and kb +3.5 sites, while AcH3 was distributed broadly across the Gata1 genomic region, including the G1HE, as previously reported (Fig. 7) (49). We next performed ChIP assays to assess histone acetylation in the G1HE and dblGATA regions on days 14, 22, and 28 of BMMC differentiation (Fig. 7B). Both AcH3 and AcH4 levels in the G1HE region remained at a lower level in BMMCs than in MEL cells. In contrast, these levels were increased in the dblGATA region during BMMC differentiation (Fig. 7B). Collectively, these results suggest that histone acetylation at the Gata1 locus is differentially regulated between erythroid and mast cell lineages.
Fig 7.

Chromatin modification profiles of the Gata1 locus in BMMCs. (A) A ChIP assay was performed across the Gata1 locus using normal rabbit IgG, anti-AcH3 antibody, or anti-trimethylated lysine 4 of histone H3 (K4me3) antibody in BMMCs at days 30 to 40 of culturing. Immunoprecipitated DNA was quantified by real-time PCR in duplicate using primers specific for the regions containing GATA sites of the Gata1 locus. The exon 6 (ex6) coding region was amplified as a negative control. Values were normalized to a control ChIP using anti-histone H3 antibody and are shown as averages and SD of data obtained from 4 independent experiments. (B) Chromatin fragments were prepared from BMMCs on the specified culture days (days 14, 22, and 28 of culturing) and from MEL cells. ChIP assays were carried out in the G1HE and dblGATA regions using normal rabbit IgG (IgG), anti-AcH3 antibody, anti-AcH4 antibody, and anti-histone H3 antibody. Immunoprecipitated DNA was quantified by real-time PCR in duplicate using primers specific for the G1HE and dblGATA. Values were normalized to a control ChIP using anti-histone H3 antibody. *, P < 0.05, and **, P < 0.01 compared with the data from day 14.
The G1HE region is dispensable for endogenous Gata1 expression in mast cells.
Given the absence of active histone modifications in the G1HE region, we surmised that the G1HE is dispensable for Gata1 gene regulation in BMMCs. To explore this hypothesis in vivo, we took advantage of transgenic-mouse lines expressing GFP under the control of a bacterial artificial chromosome (BAC) clone harboring 196 kb of the mouse Gata1 gene (G1B-GFP). The GFP reporter expression of this transgenic-mouse line faithfully recapitulates endogenous Gata1 gene expression, with the GFP intensity dependent on the transgene copy number (42). In this study, we additionally used two transgenic mouse lines bearing mutations in the G1B-GFP backbone. The G1B-GFP G1HEmut (G1HEmut-GFP) mouse line harbors a mutation in the GATA box in the G1HE region (TTATCT to TGGCTT) (42). The G1B-GFP delta-dblGATA (ΔdblGATA-GFP) mouse line harbors a reporter with a deleted dblGATA site in the G1B-GFP backbone (Fig. 8A) (T. Moriguchi, J. Takai, M. Suzuki, K. Ohneda, and M. Yamamoto, unpublished data). BMMCs were isolated from each of these transgenic-mouse lines, and the frequencies of GFP-positive cells in the c-Kit+/FcεRIα+ mast cell fractions were examined (Fig. 8B). As anticipated, almost all c-Kit/FcεRIα-double-positive mast cells were positive for GFP fluorescence in the G1B-GFP mice (Fig. 8B). In contrast, the frequency of GFP-positive cells was markedly reduced in the ΔdblGATA-GFP mast cell fraction (Fig. 8B). This indicates that the double GATA region is necessary for GFP reporter expression in BMMCs. Consistent with the ChIP data, the GATA box mutation in the G1HE region showed little influence on GFP expression in mast cells and actually resulted in more than 79.2% ± 3.5% of cells positive for GFP in the G1HEmut-GFP BMMCs. This observation conflicts strikingly with the GFP expression seen in erythroid lineage cells from the same reporter mice (42). The frequency of the GFP-positive fraction was reduced to 25% of control in the early erythroid progenitor fraction, termed “BREP” (burst-forming units erythroid cell-related erythroid progenitor) (42), in the G1HEmut-GFP mice. It appears, therefore, that the GATA site of the G1HE is dispensable for Gata1 expression in BMMCs. Next, we examined the GFP expression in peritoneal mast cells (PMC) from the three GFP reporter lines. Flow cytometric analysis revealed that, in all transgenic lines, approximately 3% of the peritoneal cells were mast cells as defined by the expression of c-Kit and FcεRIα (Fig. 8C). As observed in BMMCs, almost all control G1B-GFP (wild type) peritoneal mast cells were GFP positive (99.4% ± 0.7%) (Fig. 8D). The frequency of GFP-positive cells was lower in ΔdblGATA-GFP peritoneal mast cells (80.5% ± 10.1%) (Fig. 8D), although not by as much as in BMMCs. GFP expression in G1HEmut-GFP peritoneal mast cells was comparable to that in controls (96.4% ± 5.3%) (Fig. 8D). Hence, the dblGATA region appears to play an important role in Gata1 expression in both BMMCs and peritoneal mast cells. However, the remaining GFP expression in the ΔdblGATA-GFP peritoneal mast cells implies that other cis-acting regions might be able to partly compensate for the loss of this region, particularly in peritoneal mast cells.
Fig 8.

The G1HE region is dispensable for Gata1 expression in BMMCs. (A) Structures of the G1B-GFP transgenes, namely, G1B-GFP, G1HEmut-GFP, and ΔdblGATA-GFP. The black boxes depict Gata1 gene exons. IE denotes the erythroid cell-specific first exon. GFP cDNA and the conserved GATA sites G1HE and dblGATA are represented by gray boxes and ovals, respectively. (B and D) GFP expression in cultured BMMCs (B) and PMC (D) prepared from G1B-GFP transgenic mice (G1B, G1HEmut, and ΔdblGATA) and nontransgenic control mice (non Tg). The bar graphs show the percentages of cells that were GFP positive within the c-Kit/FcεRIα-double-positive fraction. The results are shown as averages and SD of data obtained from 3 independent experiments. (C) Flow cytometric analysis of peritoneal mast cells isolated from GFP reporter mice stained for c-Kit and FcεRIα expression. The numbers represent the average percentages and SD of c-Kit/FcεRIα-double-positive cells within the indicated gates. The data were obtained from 3 mice for each transgenic line.
The GdC minigene is sufficient to drive Gata1 expression in mast cells.
We moved on from focusing on the cis-acting regions containing the conserved GATA sites to exploring another possibility, that an undefined, novel cis-acting region mediates Gata1 gene expression in mast cells. To this end, we utilized two more different types of reporter mouse lines harboring a modified G1B-GFP transgene (Fig. 9A). One transgene carried a large deletion spanning the three critical cis-acting regions, i.e., G1HE, dblGATA, and CACCC, and the mice were designated ΔGdC-GFP reporter mice. The CACCC domain is a 300-bp region that resides in the 5′-flanking region of the IE promoter. The two CACCC boxes within this domain were shown to be essential for Gata1 expression in erythroid cells (30, 48). We previously constructed a 659-bp DNA fragment referred to as the “GdC minigene” by cis linking the three essential elements, G1HE, dblGATA, and CACCC, and demonstrated that the GdC minigene was sufficient to drive cell-type-specific reporter expression during primitive erythropoiesis (32). To further delineate the functional sufficiency of the GdC minigene fragment in the context of the G1B-GFP reporter, the GdC minigene fragment was inserted into the deletion site of the ΔGdC-GFP transgene to generate G1B-GFP GdC minigene (MG-GFP) reporter mice (J. Takai, T. Moriguchi, M. Suzuki, L. Yu, K. Ohneda, and M. Yamamoto, unpublished data). GFP reporter expression was examined in BMMCs and peritoneal mast cells prepared from the three transgenic lines (Fig. 9B and D). The frequencies of c-Kit+/FcεRIα+ cells in peritoneal cells were comparable among all transgenic lines (Fig. 9C). We found that almost all BMMCs from the ΔGdC-GFP reporter mice lacked GFP expression (Fig. 9B). Similarly, no GFP-positive cells were detected in c-Kit+/FcεRIα+ peritoneal mast cells from the ΔGdC-GFP mice (Fig. 9D). It is worth noting that the first intronic kb +3.5 region, which was occupied by both GATA factors (Fig. 6C) and was hyperacetylated (Fig. 7A), remained intact in the ΔGdC-GFP transgene. Thus, the kb +3.5 region per se is inadequate for promoting Gata1 gene expression in mast cells. Interestingly, insertion of the GdC minigene into the ΔGdC-GFP backbone successfully reinstated GFP expression in BMMCs (Fig. 9B). Likewise, GFP expression from the MG-GFP reporter was observed in the peritoneal cell population, specifically, in the c-Kit+/FcεRIα+ fraction (Fig. 9D). Consistent results were observed in independent transgenic lines, namely, two ΔGdC-GFP lines (lines 32 and 995) and three MG-GFP lines (lines 19, 20, and 33) (data not shown). Collectively, these findings indicate that the GdC minigene contains cis-acting regions that are sufficient for cell-type-specific Gata1 gene expression in mast cells.
Fig 9.

The GdC Gata1 minigene is sufficient to recover GFP expression in G1B-GFP ΔGdC transgenic mice. (A) Structures of the G1B-GFP transgenes, namely, G1B-GFP, ΔGdC-GFP, and MG-GFP. (B and D) Percentages of cells that were GFP positive within the c-Kit/FcεRIα-double-positive fraction in BMMCs (B) and PMC (D) prepared from G1B-GFP transgenic mice (G1B, ΔGdC, and MG) and nontransgenic control mice (non Tg). (C) Flow cytometric analysis of peritoneal mast cells isolated from GFP reporter mice stained for c-Kit and FcεRIα expression. The numbers represent the average percentages and SD of c-Kit/FcεRIα-double-positive cells within the indicated gates. The data were obtained from 3 mice for each transgenic line.
DISCUSSION
This study provides evidence that GATA factor expression is differentially regulated between the erythroid and mast cell lineages in two major respects. First, whereas GATA1 and GATA2 regulate each other's expression during erythroid cell differentiation, our data show no indication of such cross-regulation in BMMCs and RBL-2H3 cells. Second, our results show that the G1HE, a critical cis-acting region for Gata1 gene expression in erythroid cells, is epigenetically inactivated and inaccessible for transcription factors in BMMCs. Although the precise roles of GATA1 and GATA2 during mast cell development remain elusive, our data indicate that GATA1 and GATA2 are functionally redundant in their abilities to activate at least two mast cell-specific genes, c-Kit and mMC-CPA genes. This is in sharp contrast to the case of erythroid cells, in which GATA1 and GATA2 play unique roles. Taken together, these findings definitely further our understanding of the lineage-specific functions of the GATA factors during mast cell development.
A previous study showed that targeted disruption of the Gata2 gene resulted in the absence of mast cell precursors in in vitro-cultured embryonic stem cells, suggesting that GATA2 is required for the cell fate decision to develop into mast cells (46). In addition, we and other groups have shown that GATA1 functions in the later stage of mast cell maturation (14, 23, 25). These reports led us to anticipate that the expression profiles of GATA1 and GATA2 during mast cell development might mirror those in erythroid cells. However, contrary to our expectations, we found that GATA2 expression continued to increase, whereas GATA1 expression remained at a low level during BMMC culturing. Furthermore, we demonstrated that the forced expression or siRNA-mediated knockdown of GATA1 did not affect GATA2 expression in RBL-2H3 cells and BMMCs. Similarly, GATA1 expression was not affected by the GATA2 overexpression or repression in RBL-2H3 cells. Of particular note is the absence of GATA1-mediated Gata2 repression, which has been well documented in G1E-ER-GATA1 erythroblasts (9, 22, 34). Although GATA1 failed to repress Gata2 gene expression, our ChIP analyses showed that GATA1 bound to the “GATA switch” sites of the Gata2 locus in BMMCs (Fig. 6B). Since these regions were also occupied by GATA2 itself, even the forced expression of GATA1 might be insufficient to overcome the GATA2-mediated positive autoregulatory loop. Alternatively, GATA1 might fail to repress Gata2 gene transcription because of the absence of FOG-1, a key cofactor for GATA switching. Recent studies have revealed that FOG-1 plays critical roles in eliminating mast cell differentiation. Overexpression of FOG-1 in multipotential myeloid progenitors inhibits mast cell differentiation. Furthermore, the exogenous introduction of FOG-1 even redirects committed mast cells to other hematopoietic lineages (3, 41). Importantly, Cantor et al. reported that the retroviral expression of FOG-1 resulted in a reduced GATA2 transcript level at the granulocyte-monocyte progenitor (GMP) stage, as observed in erythroblasts (3). We observed that the simultaneous overexpression of both GATA1 and FOG-1 partially repressed Gata2 expression to 60% preferentially in day 30 to 40 BMMCs. This observation implies that a time window might exist for the FOG-1–GATA1 complex to repress GATA2 during mast cell development. Further analyses at each time point of BMMCs should be of particular importance in unveiling the molecular mechanism for the differentiation stage-specific function of FOG-1.
The present study demonstrates that the G1HE is dispensable for Gata1 gene expression in mast cells by three different approaches. First, in BMMCs, neither GATA1 nor GATA2 bound to the G1HE chromatin region. Second, in BMMCs, no active histone marks were detected in the G1HE region. Lastly, in the BMMCs and peritoneal mast cells of G1B-GFP transgenic mice, a mutation in the G1HE region had little influence on reporter expression. These findings contrast sharply with previous studies showing that the G1HE is required for Gata1 gene expression in erythroid cells and megakaryocytes. For instance, we and other groups showed that a mutation in the GATA site within the G1HE region almost completely eradicated reporter expression in the yolk sac and fetal liver of developing embryos (31, 50). Closer examination using G1B-GFP reporter mice revealed that the G1HE is essential for Gata1 gene expression in the BREP, which is close to the bipotential megakaryocyte-erythroid progenitors (MEP) (42). Targeted deletion of the Gata1 upstream region containing the G1HE abrogated Gata1 expression in the MEP and megakaryocytes, as well as erythroid cells, in mice (8, 39). Considering the redundancy of G1HE in mast cell differentiation, the G1HE might be activated predominantly at the MEP stage and function as megakaryocyte-erythroid lineage-specific enhancer. Interestingly, MEPs from Gata1low bone marrow were highly proliferative and aberrantly inclined to differentiate into the mast cell lineage (8, 25). These observations indicate that activation of the G1HE in MEP is critically required for normal erythromegakaryocytic differentiation, while the absence of G1HE leads to alteration in lineage specification.
Our data demonstrated that the proximal double GATA site is bound by both GATA factors and is required for G1B-GFP reporter expression in BMMCs and peritoneal mast cells. In contrast to the G1HE, which is activated specifically in megakaryocytes and erythroid cells, the proximal double GATA site seems to act as a general cis-regulatory region capable of functioning in different hematopoietic lineages. Targeted deletion of the double GATA site in mice resulted in the selective loss of the eosinophil lineage, which was a less dramatic phenotype than had been originally presumed (55). Interestingly, bone marrow cells from the eosinophil-ablated ΔdblGATA mice are still capable of differentiating into eosinophils when subjected to cytokine stimulation in vitro (4). The GATA1 mRNA level of the cultured ΔdblGATA eosinophils remained comparable to that of wild-type cells by virtue of the activated alternative first exon, called IEB, located in the first intron (4). Meanwhile, it was reported that mast cells developed normally from ΔdblGATA bone marrow when the cells were cultured in the presence of IL-3 and SCF (55). Given the essential requirement for the double GATA site in mast cells demonstrated here, it would be intriguing to examine the GATA1 expression level and to clarify the potential usage of the alternative first exon in mast cells of the ΔdblGATA mice. In addition to the double GATA site, our ChIP analyses showed that the kb +3.5 region was bound by both GATA factors in BMMCs. The kb +3.5 region contains multiple GATA-GACT repeats and was shown to be essential for definitive erythropoiesis in fetal liver (32). However, we found that GFP expression was completely abrogated in BMMCs from G1B-GFP ΔGdC reporter mice, even though the kb +3.5 site was retained in this reporter construct. It seems, then, that the kb +3.5 site by itself is insufficient to direct reporter expression in mast cells. Importantly, the reporter expression was restored by inserting the GdC minigene into the ΔGdC-G1B-GFP transgene backbone. Collectively, these results underscore that positive autoregulation through the double GATA site is a core mechanism for Gata1 expression in mast cells.
ACKNOWLEDGMENTS
We thank Tania Bezak for critical reading of the manuscript.
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Sports, Science and Technology (K.O.) and by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (K.O.).
Footnotes
Published ahead of print 17 September 2012
REFERENCES
- 1. Barski A, et al. 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837 [DOI] [PubMed] [Google Scholar]
- 2. Bresnick EH, Lee HY, Fujiwara T, Johnson KD, Keles S. 2010. GATA switches as developmental drivers. J. Biol. Chem. 285: 31087–31093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cantor AB, et al. 2008. Antagonism of FOG-1 and GATA factors in fate choice for the mast cell lineage. J. Exp. Med. 205: 611–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dyer KD, et al. 2007. Eosinophils from lineage-ablated Delta dblGATA bone marrow progenitors: the dblGATA enhancer in the promoter of GATA-1 is not essential for differentiation ex vivo. J. Immunol. 179: 1693–1699 [DOI] [PubMed] [Google Scholar]
- 5. Evans T, Felsenfeld G. 1989. The erythroid-specific transcription factor Eryf1: a new finger protein. Cell 58: 877–885 [DOI] [PubMed] [Google Scholar]
- 6. Ferreira R, Ohneda K, Yamamoto M, Philipsen S. 2005. GATA1 function, a paradigm for transcription factors in hematopoiesis. Mol. Cell. Biol. 25: 1215–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fujiwara Y, Browne CP, Cunniff K, Goff SC, Orkin SH. 1996. Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc. Natl. Acad. Sci. U. S. A. 93: 12355–12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ghinassi B, et al. 2007. The hypomorphic Gata1low mutation alters the proliferation/differentiation potential of the common megakaryocytic-erythroid progenitor. Blood 109: 1460–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grass JA, et al. 2003. GATA-1-dependent transcriptional repression of GATA-2 via disruption of positive autoregulation and domain-wide chromatin remodeling. Proc. Natl. Acad. Sci. U. S. A. 100: 8811–8816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grass JA, et al. 2006. Distinct functions of dispersed GATA factor complexes at an endogenous gene locus. Mol. Cell. Biol. 26: 7056–7067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grunstein M. 1997. Histone acetylation in chromatin structure and transcription. Nature 389: 349–352 [DOI] [PubMed] [Google Scholar]
- 12. Gutiérrez L, et al. 2007. Gata1 regulates dendritic cell development and survival. Blood 110: 1933–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gutiérrez L, et al. 2008. Ablation of Gata1 in adult mice results in aplastic crisis, revealing its essential role in steady-state and stress erythropoiesis. Blood 111: 4375–4385 [DOI] [PubMed] [Google Scholar]
- 14. Harigae H, et al. 1998. Differential roles of GATA-1 and GATA-2 in growth and differentiation of mast cells. Genes Cells 3: 39–50 [DOI] [PubMed] [Google Scholar]
- 15. Heintzman ND, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39: 311–318 [DOI] [PubMed] [Google Scholar]
- 16. Hong W, et al. 2005. FOG-1 recruits the NuRD repressor complex to mediate transcriptional repression by GATA-1. EMBO J. 24: 2367–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ishijima Y, Ohmori S, Uenishi A, Ohneda K. 2012. GATA transcription factors are involved in IgE-dependent mast cell degranulation by enhancing the expression of phospholipase C-gamma1. Genes Cells 17: 285–301 [DOI] [PubMed] [Google Scholar]
- 18. Khandekar M, et al. 2007. A Gata2 intronic enhancer confers its pan-endothelia-specific regulation. Development 134: 1703–1712 [DOI] [PubMed] [Google Scholar]
- 19. Kobayashi-Osaki M, et al. 2005. GATA motifs regulate early hematopoietic lineage-specific expression of the Gata2 gene. Mol. Cell. Biol. 25: 7005–7020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maeda K, Nishiyama C, Ogawa H, Okumura K. 2010. GATA2 and Sp1 positively regulate the c-kit promoter in mast cells. J. Immunol. 185: 4252–4260 [DOI] [PubMed] [Google Scholar]
- 21. Martin DI, Zon LI, Mutter G, Orkin SH. 1990. Expression of an erythroid transcription factor in megakaryocytic and mast cell lineages. Nature 344: 444–447 [DOI] [PubMed] [Google Scholar]
- 22. Martowicz ML, Grass JA, Boyer ME, Guend H, Bresnick EH. 2005. Dynamic GATA factor interplay at a multicomponent regulatory region of the GATA-2 locus. J. Biol. Chem. 280: 1724–1732 [DOI] [PubMed] [Google Scholar]
- 23. Masuda A, et al. 2007. Essential role of GATA transcriptional factors in the activation of mast cells. J. Immunol. 178: 360–368 [DOI] [PubMed] [Google Scholar]
- 24. McDevitt MA, Fujiwara Y, Shivdasani RA, Orkin SH. 1997. An upstream, DNase I hypersensitive region of the hematopoietic-expressed transcription factor GATA-1 gene confers developmental specificity in transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 94: 7976–7981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Migliaccio AR, et al. 2003. GATA-1 as a regulator of mast cell differentiation revealed by the phenotype of the GATA-1low mouse mutant. J. Exp. Med. 197: 281–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Minegishi N, et al. 1998. Alternative promoters regulate transcription of the mouse GATA-2 gene. J. Biol. Chem. 273: 3625–3634 [DOI] [PubMed] [Google Scholar]
- 27. Minegishi N, et al. 1999. The mouse GATA-2 gene is expressed in the para-aortic splanchnopleura and aorta-gonads and mesonephros region. Blood 93: 4196–4207 [PubMed] [Google Scholar]
- 28. Morii E, et al. 2004. Roles of MITF for development of mast cells in mice: effects on both precursors and tissue environments. Blood 104: 1656–1661 [DOI] [PubMed] [Google Scholar]
- 29. Nagai T, et al. 1994. Transcription factor GATA-2 is expressed in erythroid, early myeloid, and CD34+ human leukemia-derived cell lines. Blood 84: 1074–1084 [PubMed] [Google Scholar]
- 30. Nicolis S, et al. 1991. An erythroid specific enhancer upstream to the gene encoding the cell-type specific transcription factor GATA-1. Nucleic Acids Res. 19: 5285–5291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nishimura S, et al. 2000. A GATA box in the GATA-1 gene hematopoietic enhancer is a critical element in the network of GATA factors and sites that regulate this gene. Mol. Cell. Biol. 20: 713–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ohneda K, et al. 2002. A minigene containing four discrete cis elements recapitulates GATA-1 gene expression in vivo. Genes Cells 7: 1243–1254 [DOI] [PubMed] [Google Scholar]
- 33. Onodera K, et al. 1997. GATA-1 transcription is controlled by distinct regulatory mechanisms during primitive and definitive erythropoiesis. Proc. Natl. Acad. Sci. U. S. A. 94: 4487–4492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pal S, et al. 2004. Coregulator-dependent facilitation of chromatin occupancy by GATA-1. Proc. Natl. Acad. Sci. U. S. A. 101: 980–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Romeo PH, et al. 1990. Megakaryocytic and erythrocytic lineages share specific transcription factors. Nature 344: 447–449 [DOI] [PubMed] [Google Scholar]
- 36. Salmon JM, et al. 2007. Aberrant mast-cell differentiation in mice lacking the stem-cell leukemia gene. Blood 110: 3573–3581 [DOI] [PubMed] [Google Scholar]
- 37. Shimizu R, Ohneda K, Engel JD, Trainor CD, Yamamoto M. 2004. Transgenic rescue of GATA-1-deficient mice with GATA-1 lacking a FOG-1 association site phenocopies patients with X-linked thrombocytopenia. Blood 103: 2560–2567 [DOI] [PubMed] [Google Scholar]
- 38. Shimizu R, Takahashi S, Ohneda K, Engel JD, Yamamoto M. 2001. In vivo requirements for GATA-1 functional domains during primitive and definitive erythropoiesis. EMBO J. 20: 5250–5260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shivdasani RA, Fujiwara Y, McDevitt MA, Orkin SH. 1997. A lineage-selective knockout establishes the critical role of transcription factor GATA-1 in megakaryocyte growth and platelet development. EMBO J. 16: 3965–3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shivdasani RA, Mayer EL, Orkin SH. 1995. Absence of blood formation in mice lacking the T-cell leukaemia oncoprotein tal-1/SCL. Nature 373: 432–434 [DOI] [PubMed] [Google Scholar]
- 41. Sugiyama D, et al. 2008. Differential context-dependent effects of friend of GATA-1 (FOG-1) on mast-cell development and differentiation. Blood 111: 1924–1932 [DOI] [PubMed] [Google Scholar]
- 42. Suzuki M, Moriguchi T, Ohneda K, Yamamoto M. 2009. Differential contribution of the Gata1 gene hematopoietic enhancer to erythroid differentiation. Mol. Cell. Biol. 29: 1163–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takahashi S, et al. 1997. Arrest in primitive erythroid cell development caused by promoter-specific disruption of the GATA-1 gene. J. Biol. Chem. 272: 12611–12615 [DOI] [PubMed] [Google Scholar]
- 44. Tanabe O, et al. 2007. The TR2 and TR4 orphan nuclear receptors repress Gata1 transcription. Genes Dev. 21: 2832–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tsai FY, et al. 1994. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature 371: 221–226 [DOI] [PubMed] [Google Scholar]
- 46. Tsai FY, Orkin SH. 1997. Transcription factor GATA-2 is required for proliferation/survival of early hematopoietic cells and mast cell formation, but not for erythroid and myeloid terminal differentiation. Blood 89: 3636–3643 [PubMed] [Google Scholar]
- 47. Tsai SF, et al. 1989. Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through expression in mammalian cells. Nature 339: 446–451 [DOI] [PubMed] [Google Scholar]
- 48. Tsai SF, Strauss E, Orkin SH. 1991. Functional analysis and in vivo footprinting implicate the erythroid transcription factor GATA-1 as a positive regulator of its own promoter. Genes Dev. 5: 919–931 [DOI] [PubMed] [Google Scholar]
- 49. Valverde-Garduno V, et al. 2004. Differences in the chromatin structure and cis-element organization of the human and mouse GATA1 loci: implications for cis-element identification. Blood 104: 3106–3116 [DOI] [PubMed] [Google Scholar]
- 50. Vyas P, et al. 1999. Different sequence requirements for expression in erythroid and megakaryocytic cells within a regulatory element upstream of the GATA-1 gene. Development 126: 2799–2811 [DOI] [PubMed] [Google Scholar]
- 51. Wadman IA, et al. 1997. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J. 16: 3145–3157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Walsh JC, et al. 2002. Cooperative and antagonistic interplay between PU.1 and GATA-2 in the specification of myeloid cell fates. Immunity 17: 665–676 [DOI] [PubMed] [Google Scholar]
- 53. Wozniak RJ, Boyer ME, Grass JA, Lee Y, Bresnick EH. 2007. Context-dependent GATA factor function: combinatorial requirements for transcriptional control in hematopoietic and endothelial cells. J. Biol. Chem. 282: 14665–14674 [DOI] [PubMed] [Google Scholar]
- 54. Wozniak RJ, et al. 2008. Molecular hallmarks of endogenous chromatin complexes containing master regulators of hematopoiesis. Mol. Cell. Biol. 28: 6681–6694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu C, et al. 2002. Targeted deletion of a high-affinity GATA-binding site in the GATA-1 promoter leads to selective loss of the eosinophil lineage in vivo. J. Exp. Med. 195: 1387–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zon LI, et al. 1993. Expression of mRNA for the GATA-binding proteins in human eosinophils and basophils: potential role in gene transcription. Blood 81: 3234–3241 [PubMed] [Google Scholar]