

Abstract
Upon viral infection, pattern recognition receptors sense viral nucleic acids, leading to the production of type I interferons (IFNs), which initiate antiviral activities. Type I IFNs bind to their cognate receptor, IFNAR, resulting in the activation of signal-transducing activators of transcription 1 (STAT1). Thus, it has long been thought that double-stranded RNA (dsRNA)-induced STAT1 phosphorylation is mediated by the transactivation of type I IFN signaling. Foreign RNA, such as viral RNA, in cells is sensed by the cytoplasmic sensors retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA-5). In this study, we explored the molecular mechanism responsible for STAT1 phosphorylation in response to the sensing of dsRNA by cytosolic RNA sensors. Polyinosinic-poly(C) [poly(I:C)], a synthetic dsRNA that is sensed by both RIG-I and MDA-5, induces STAT1 phosphorylation. We found that the poly(I:C)-induced initial phosphorylation of STAT1 is dependent on the RIG-I pathway and that MDA-5 is not involved in STAT1 phosphorylation. Furthermore, pretreatment of the cells with neutralizing antibody targeting the IFN receptor suppressed the initial STAT1 phosphorylation in response to poly(I:C), suggesting that this initial phosphorylation event is predominantly type I IFN dependent. In contrast, neither the known RIG-I pathway nor type I IFN is involved in the late phosphorylation of STAT1. In addition, poly(I:C) stimulated STAT1 phosphorylation in type I IFN receptor-deficient U5A cells with delayed kinetics. Collectively, our study provides evidence of a comprehensive regulatory mechanism in which dsRNA induces STAT1 phosphorylation, indicating the importance of STAT1 in maintaining very tight regulation of the innate immune system.
INTRODUCTION
Pattern recognition receptors (PRRs) are receptors expressed by cells of the innate immune system and act as sensors to detect rapidly invading pathogens. PRRs recognize conserved pathogen-associated molecular patterns (PAMPs) and distinguish foreign organisms, such as bacteria, viruses, fungi, and parasites, from host cells. Subsets of PRRs trigger the activation of intracellular signaling pathways, leading to the activation of a series of innate antimicrobial immune responses (7, 19, 24). The repertoire of signal-transducing PRRs includes membrane-bound Toll-like receptors (TLRs) (23) and cytosolic receptors, such as RNA helicase retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs) (51) and Nod-like receptors (NLRs) (13).
In most types of cells, the RLR family members serve as cytoplasmic sensors for viral nucleic acids, whereas TLRs are the predominant receptors for viral nucleic acids in plasmacytoid dendritic cells, suggesting that PRRs trigger antiviral responses in a cell type-specific manner (16). The RLR family is composed of three members, RIG-I, melanoma differentiation-associated gene-5 (MDA-5), and laboratory of genetics and physiology 2 (LPG2). RIG-I and MDA-5 can be divided into three basic domains, the N-terminal tandem caspase activation and recruitment domain (CARD), the central helicase domain, and the C-terminal regulatory domain (33). RIG-I and MDA-5 both recognize viral RNA, which results in the exposure of the CARD. Downstream antiviral signaling is mediated by their downstream adaptor molecules. The signaling downstream of RIG-I and MDA5 is initiated by mitochondrial antiviral signaling protein (MAVS; also known as IPS-1, VISA, or Cardif) (20, 36, 45, 50) and triggers the activation of the transcription factors interferon-regulatory factor 3 (IRF3), IRF7, and NF-κB and the subsequent production of type I interferons (IFNs) (30).
Type I IFNs bind to a specific receptor, which is composed of two chains, designated the α-chain (IFNAR1) and the β-chain (IFNAR2). When an IFN interacts with its cognate receptor, a signal is rapidly transmitted within the cell. The primary signal transduction cascade promoted by type I IFNs is mediated by the Janus family of protein tyrosine kinase 1 (JAK1) signal transducers and activators of transcription (STAT) pathway (43). Receptor engagement subsequently leads to the activation of the IFN-stimulated regulatory factor 3 (ISGF3) transcription complex. ISGF3 is composed of STAT1 and STAT2, both of which are activated by JAK1, and IRF9 (also known as ISGF3γ or p48) (21). The activation of this transcriptional activator complex leads to the increased expression of IFN-induced genes, including (2′-5′) oligoadenylate synthetases, Mx proteins, and protein kinase R (PKR), inducing an antiviral state (43). In addition to the induction of ISGF3 complex formation, activated JAK1 also induces STAT1 homodimerization, allowing the dimer to bind to DNA elements called GASs (IFN-γ-activated sites) (40). IFN-γ also induces the expression of a variety of antiviral factors through the induction of STAT1 homodimerization (42). It is crucial to understand how STAT1, a key molecule in antiviral signaling, is regulated during innate immune responses. It is now evident that type I IFNs are produced during viral infection. In addition, type I IFN is known to activate STAT1. Therefore, the activation of STAT1 during viral infection was previously thought to be mediated simply by the transactivation of the type I IFN receptor. Bhattacharyya and coworkers have recently reported that polyinosinic-poly(C) [poly(I:C)], a synthetic double-stranded RNA, induces biphasic STAT1 phosphorylation in macrophages: no inhibitory effect of glucocorticoid (GC) on STAT1 phosphorylation was observed in the early phase, but significant inhibition of STAT1 phosphorylation by GC was observed in the later phase (2). In their study, they observed TLR3 signaling in response to poly(I:C) and found that, for TLR3 signaling, the suppression of STAT1 phosphorylation is due to both the induction of suppressor of cytokine signaling 1 (SOCS1) and the inhibition of type I IFN secretion. As described above, TLR3 predominantly responds to double-stranded RNA (dsRNA) in macrophages (1) and dendritic cells (16), whereas nonprofessional immune cells such as fibroblasts use RLRs in the recognition of dsRNA to produce type I IFN (16). Furthermore, extracellularly treated poly(I:C) is recognized by TLR3, whereas poly(I:C) introduced into cells is sensed by RLRs (2, 12, 51). These findings provided us with an idea about the involvement of intracellular RNA sensors, RLRs, in dsRNA-induced STAT1 activation. The overexpression of RIG-I has been shown to induce STAT1 phosphorylation, indicating that RIG-I is indeed associated with STAT1 signaling (14). However, no study demonstrating which RLRs mediate signaling associated with STAT1 phosphorylation in response to dsRNA has been published. In this study, we determined whether double-stranded RNA (dsRNA)-induced STAT1 activation is absolutely dependent on type I IFN. We observed that dsRNA-induced STAT1 phosphorylation is roughly divided into two phases: a predominantly type I IFN-dependent initial phosphorylation phase and a late phosphorylation phase that is independent of both RIG-MAVS and type I IFN. These results suggest that the regulation of STAT1 in the context of antiviral innate immunity is complex.
MATERIALS AND METHODS
Cell culture.
A549 cells were obtained from the American Type Culture Collection. Human IFNAR-deficient U5A cells and their parental 2fTGH cells were kindly provided by G. Stark (Cleveland Clinic Foundation Research Institute). These cells were grown in a 5% CO2 atmosphere at 37°C in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA). 293-flp cells (Invitrogen) were maintained in DMEM–10% FBS containing 100 μg/ml zeocin (Invitrogen).
Transfection.
Transient transfections of A549 cells were performed by plating cells at a density of 1.0 × 105 cells per 12-well culture plate 16 to 20 h prior to transfection. The cells were transfected with expression plasmids using Lipofectamine LTX (Invitrogen) or with poly(I:C) (Sigma-Aldrich) using Lipofectamine 2000 (Invitrogen) by following the manufacturer's instructions. The cells were incubated for the indicated times depending on the experiment and then further analyzed. RNA interference (RNAi) was performed by transfecting A549 cells with nonsilencing control small interfering RNA (siRNA) (Qiagen, Hilden, Germany) or siRNAs against RIG-I (SI03019646; Qiagen), MDA5 (SI03649037; Qiagen), and MAVS (SI04293471; Qiagen) using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol.
Plasmids.
MicroRNAs (miRNAs) targeting the 3′ untranslated region (UTR) of MAVS mRNA were designed with the BLOCK-iT RNAi Designer (Invitrogen). The oligonucleotide sequences were the following: miR-MAVS 2569 sense, 5′-TGCTGAAGACCAGAAGTCTGACATCTGTTTTGGCCACTGACTGACAGATGTCACTTCTGGTCTT-3′, and miR-MAVS 2569 antisense, 5′-CCTGAAGACCAGAAGTGACATCTGTCAGTCAGTGGCCAAAACAGATGTCAGACTTCTGGTCTTC-3′. The oligonucleotides used to create miRNA specific for β-galactosidase (LacZ), which served as a control, were supplied by Invitrogen. Each oligonucleotide pair was annealed and inserted into the pcDNA6.2-GW/EmGFP vector (Invitrogen). Following transfection of 293 cells with the plasmids, the miRNA levels were evaluated by observing the expression of emerald green fluorescent protein (EmGFP) using fluorescence microscopy; the knockdown efficiency for MAVS was confirmed by immunoblotting for MAVS. The EmGFP portion of the pcDNA6.2-GW/EmGFP-mir vectors was then removed by DraI digestion to increase the knockdown efficiency against MAVS. Subsequently, a BamHI-XhoI fragment from each plasmid containing specific miRNA sequences was transferred into the pcDNA5/FRT vector between BamHI and XhoI sites to generate the MAVS knockdown cells described below. The p3xFLAG-RIG-I-full-length (FLC), p3xFLAG-RIG-I-CARD, and p3xFLAG-RIG-I-ΔCARD vectors were constructed as previously reported (31).
Generation of stably MAVS-silenced cells.
The Flp-in system (Invitrogen) was used to generate stably MAVS-silenced cells according to the manufacturer's instructions. Site-specific DNA recombination was performed by the cotransfection of 293-flp cells with pcDNA5/FRT-mir MAVS or pcDNA5/FRT-mir LacZ and pOG44, which expresses Flp recombinase. MAVS-silenced cells were selected in hygromycin B-containing DMEM–10% FBS, and the knockdown efficiency was analyzed by immunoblotting for MAVS.
Quantitative RT-PCR.
Total RNA was extracted from the cells using an Illustra RNA isolation kit (GE Healthcare, Piscataway, NJ). One microgram of total RNA served as the template for first-strand cDNA synthesis in a reaction using an oligo(dT) primer and Moloney-murine leukemia virus (MMLV) reverse transcriptase (Invitrogen) under the conditions indicated by the manufacturer. A CFX96 PCR detection system (Bio-Rad, Hercules, CA) was used for the quantitative assessment of IFN-β and 18S rRNA. The sequences of the primers were the following: IFN-β-F (5′-CCTGTGGCAATTGAATGGGAGGC-3′), IFN-β-R (5′-CCTGTGGCAATTGAATGGGAGGC-3′), MX1-F (5′-GCCAGGACCAGGTATACAG-3′), MX1-R (5′-GCCTGCGTCAGCCGTGC-3′), G1P3-F (5′-GATTGCTTCTCTTCTCTCCTCCAAG-3′), G1P3-R (5′-TCGAGATACTTGTGGGTGGCGTAG-3′), OAS-F (5′-GAGCTCCTGACGGTCTATGC-3′), OAS-R (5′-CAAATTCACCGCATGTTCAC-3′), 18S rRNA-F (5′-ACTCAACACGGGAAACCTCA-3′), and 18S rRNA-R (5′-AACCAGACAAATCGCTCCAC-3′).
The amplification reactions were performed with SsoFast Evergreen DNA polymerase (Bio-Rad) according to the manufacturer's specifications. The amplification conditions were the following: 30 s at 95°C followed by 40 cycles of 5 s at 95°C and 10 s at 72°C. After the amplification was completed, melting curve analysis was performed by heating the reaction products slowly at 0.1°C per s to 95°C with continuous monitoring of the fluorescence. Melting curves were constructed and the data were quantitatively analyzed using a CFX manager.
ELISA for IFN-β.
A549 cells were transfected with poly(I:C), and then the IFN-β concentration in the medium was measured. Following incubation for up to 8 h, the conditioned medium was collected, the cell debris was pelleted by centrifugation at 12,000 rpm for 5 min at 4°C, and the supernatant was collected. The concentration of IFN-β in the conditioned medium was determined using a human IFN-β enzyme-linked immunosorbent assay (ELISA) kit (Kamakura Techno-Science, Japan).
Immunoblot analyses.
After a series of treatments, the cells were washed twice with phosphate-buffered saline (PBS) and lysed in hypotonic lysis buffer (10 mM Tris [pH 7.4], 100 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40). The lysate was cleared by centrifugation at 6,500 rpm for 15 min at 4°C. We subjected 5 to 10 μg of protein extracts to electrophoresis on 8 or 10% SDS-PAGE gels and then transferred the proteins to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA) that were blocked for 60 min at room temperature in 1× TBST buffer (50 mM Tris-HCl [pH 7.4], 250 mM NaCl, 0.1% Tween 20) containing 1% nonfat dry milk. The membranes were incubated overnight at 4°C with the primary antibody indicated in each case. The primary antibodies were anti-STAT1 and anti-phospho-STAT1 (Tyr 701) antibodies (Santa Cruz Biotechnology, Santa Cruz, CA), an anti-MDA-5 antibody (Immuno-Biological Laboratories, Japan), anti-RIG-I and anti-MAVS antibodies (Enzo Life Sciences, Miami, FL), an anti-DYKDDDDK tag antibody (Wako, Japan), and an anti-β-actin antibody (Sigma-Aldrich). After 5 washes with 1× TBST, the membranes were further incubated for 1 h at room temperature with the corresponding secondary antibody. Bovine anti-rabbit (Santa Cruz Biotechnology) or Zymax mouse IgG antibody (Invitrogen) coupled to horseradish peroxidase (HRP) at a 1:10,000 dilution in 1× TBST containing 5% nonfat dry milk was used. Thereafter, the membranes were washed 5 times with 1× TBST, followed by visualization of the immunoreactive bands using the Luminata Crescendo Western HRP substrate (Millipore). Quantification of the immunoreactivity was performed by densitometric analysis using NIH ImageJ software.
Immunofluorescence analyses.
Immunofluorescence staining was performed as reported previously (32). Briefly, A549 or U5A cells were grown on glass coverslips and then fixed with 4% formaldehyde for 20 min, permeabilized with 0.1% Triton X-100 for 10 min, and blocked with 3% bovine serum albumin for 1 h. The cells were then incubated for 1 h with mouse monoclonal anti-IFN-β (R&D systems Minneapolis, MN) (1:100) or rabbit polyclonal anti-STAT1 antibody (Santa Cruz Biotechnology) (1:200), respectively. After a washing step, the cells were incubated with Alexa 488-conjugated anti-mouse IgG or Alexa 555-conjugated anti-rabbit IgG (Invitrogen). The cells were mounted in Prolong gold antifade reagent (Invitrogen), and IFN-β and STAT1 were visualized by confocal laser scanning microscopy (C1si; Nikon, Japan).
RESULTS
Time-dependent STAT1 phosphorylation in response to poly(I:C) transfection in A549 cells.
A549 cells have been shown to express type I IFN in response to infection by Sendai virus and influenza A virus, both of which are RNA viruses and are sensed by RIG-I (29). These viral infections can induce the production of IFN-α and IFN-β in A549 cells. However, the kinetics of the expression of the different type I IFNs are different: the initial expression of IFN-β is observed 3 h after infection, and that of IFN-α is observed approximately 6 h after infection (29). The phosphorylation of STAT1 is also observed in response to viral infection for 3 h in A549 cells (26). Given that the virus-induced phosphorylation of STAT1 occurs in a type I IFN-dependent manner, STAT1 phosphorylation and type I IFN induction should proceed in parallel following viral infection. Therefore, we initially confirmed the kinetics of both responses after the introduction of a synthetic dsRNA, poly(I:C), into A549 cells. We examined the expression level of IFN-β as a model type I IFN because IFN-α does not appear to be involved in dsRNA-induced STAT1 phosphorylation, as it exhibits delayed kinetics compared to those of STAT1 phosphorylation (29). IFN-β mRNA was not detected in resting A549 cells. When poly(I:C) was introduced into the cells, the level of IFN-β mRNA increased rapidly starting 2 h after the introduction of poly(I:C) (Fig. 1A). A similar pattern was observed for the level of IFN-β protein; the protein could be detected beginning 4 h after transfection with poly(I:C) (Fig. 1B). A time course of poly(I:C)-induced STAT1 phosphorylation showed detectable phosphate incorporation after 3 h, reaching a maximal level at 4 to 6 h (Fig. 1C). Recombinant human IFN-β rapidly activated STAT1 within 5 min after treatment. Robust STAT1 phosphorylation was observed after 30 min of exposure to IFN-β, and then the level of phosphorylation gradually decreased (Fig. 1D).
Fig 1.

Kinetics of IFN-β production and STAT1 phosphorylation in response to poly(I:C) transfection. (A) A549 cells were transfected with poly(I:C) (200 ng), and the levels of IFN-β mRNA were determined by quantitative real-time PCR. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a normalization control for IFN-β. The data for IFN-β represent the averages from three determinations. The means (± standard deviations [SD]) from three experiments are shown. (B) A549 cells were transfected with poly(I:C) (200 ng) for up to 8 h, and then the levels of IFN-β protein in the conditioned medium were determined by ELISA. The means (± SD) from three experiments are shown. (C) A549 cells were transfected with poly(I:C) (200 ng) for 0 to 8 h. (D) A549 cells were treated with IFN-β (100 U/ml) for 0 to 8 h. The levels of phosphorylated STAT1 (pSTAT1) and STAT1 were analyzed by immunoblotting as described in Materials and Methods.
Influence of the IFN receptor (IFNAR) on poly(I:C)-induced STAT1 phosphorylation.
To evaluate the effect of transactivation by the type I IFN receptor on poly(I:C)-induced STAT1 phosphorylation, we added an anti-IFNAR neutralizing antibody to the culture medium to block type I IFN signaling, including the JAK1-STAT1 pathway. When the cells were preincubated with the neutralizing antibody, poly(I:C)-induced STAT1 phosphorylation was partially inhibited at 3 h (Fig. 2A). However, the inhibitory effect of this neutralizing antibody on poly(I:C)-induced phosphorylated STAT1 gradually decreased, and no effect was observed 6 h after its introduction. We note that the anti-IFNAR antibody still had neutralizing activity after 8 h of incubation in the culture medium; in cells preincubated with the neutralizing antibody for 8 h, STAT1 phosphorylation in response to a 15-min incubation with IFN-α2b was almost completely inhibited, even though IFN-α2b has a higher affinity for IFNAR than IFN-β does (Fig. 2A). We next asked whether the initial phosphorylation in response to poly(I:C) is indeed mediated by the production of IFN-β, because we were unable to detect IFN-β protein in conditioned medium from A549 cells transfected with poly(I:C) after 3 h by ELISA (Fig. 1B). We hypothesized that a small but sufficient amount of IFN-β must be produced to activate STAT1 after poly(I:C) transfection. To investigate this possibility, we performed immunofluorescence staining for IFN-β (Fig. 2B). In the resting state, little IFN-β immunoreactivity was observed. In contrast, polarized IFN-β was observed on certain areas of the cell surface 3 h after transfection with poly(I:C).
Fig 2.
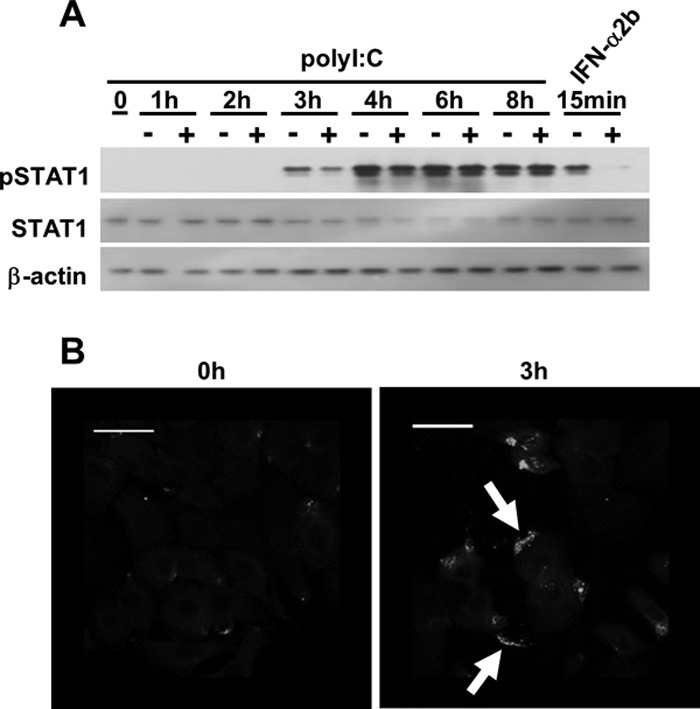
Influence of a neutralizing IFN receptor antibody on STAT1 phosphorylation in response to poly(I:C). (A) Following pretreatment with the anti-IFNAR neutralizing antibody (2.5 μg/well) for 1 h, A549 cells were transfected with poly(I:C) (200 ng) for up to 8 h. As a control, A549 cells were preincubated with the neutralizing antibody for 8 h, followed by treatment with IFN-α2b (200 ng/ml) for 15 min. Immunoblot analyses were performed using antibodies directed against pSTAT1, STAT1, and actin. The results are representative of two experiments conducted under similar conditions. (B) A549 cells were transfected with poly(I:C) for 3 h and then fixed with 4% paraformaldehyde and incubated with an anti-IFN-β antibody. IFN-β protein was detected with a secondary antibody conjugated to Alexa 488. White arrows indicate marginal IFN-β. The scale bar represents 100 μm.
Involvement of the RLR pathway in poly(I:C)-induced STAT1 phosphorylation.
Both RIG-I and MDA5 are intracellular receptors for dsRNA, and the recognition of dsRNA by these proteins is dependent on the length of the individual dsRNA (17). RLR-dependent antiviral signals are transduced by the adapter molecule MAVS (33). Therefore, we determined if the phosphorylation of STAT1 in response to poly(I:C) is mediated by the RLR/MAVS pathway using the RNAi technique. No suppressive effect of MDA-5 knockdown on poly(I:C)-induced STAT1 phosphorylation was observed (Fig. 3). The silencing of either RIG-I or MAVS significantly suppressed the initial phosphorylation of STAT1. In contrast, no inhibitory effect of RIG-I or MAVS silencing on STAT1 phosphorylation was observed when the cells were transfected with poly(I:C) for 8 h.
Fig 3.

Involvement of RIG-I, MDA-5, and MAVS in STAT1 phosphorylation in response to poly(I:C). A549 cells were transfected with control siRNA or gene-specific siRNAs against RIG-I, MDA-5, or MAVS for 48 h. After the incubation, the cells were transfected with poly(I:C) for up to 8 h. The levels of pSTAT1, STAT1, RIG-I, MDA-5, MAVS, and actin were analyzed by immunoblotting.
RIG-I-CARD is required for STAT1 phosphorylation.
We next determined if the RIG-I-dependent pathway is indeed involved in STAT1 phosphorylation. To demonstrate the direct involvement of RIG-I in initial STAT1 phosphorylation, we examined the relationship between overexpressed RIG-I proteins and STAT1 phosphorylation after the transfection, as shown in Fig. 4A. When the cells were transfected with an expression vector for the full-length form or the CARD domain of RIG-I, these proteins were produced 8 h after the transfections. At 8 h after transfection, the overexpression of RIG-I-CARD induced STAT1 phosphorylation, whereas the overexpression of the full-length RIG-I did not (Fig. 4A). We transfected the cells with vectors expressing RIG-I-full length, RIG-I-CARD, or RIG-IΔCARD, and after 8 h we confirmed that only RIG-I-CARD was able to activate STAT1 (Fig. 4B).
Fig 4.
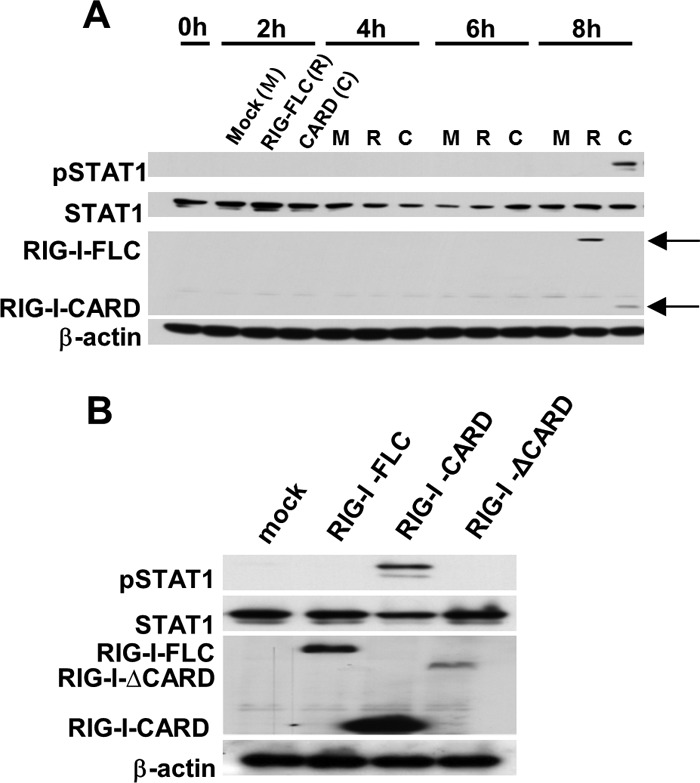
Time course of STAT1 phosphorylation following the transfection of A549 cells with vectors expressing RIG-I constructs. (A) A549 cells were transfected with FLC-RIG-I, RIG-I-CARD, or an empty control vector for up to 8 h. (B) A549 cells were transfected with RIG-I-FLC, RIG-I-CARD, RIG-I-ΔCARD, or an empty control vector for 8 h. Following the transfection, extracted samples were subjected to SDS-PAGE, and then the levels of pSTAT1, STAT1, exogenous RIG-I (FLAG tag), and actin were analyzed by immunoblotting.
RIG-I/MAVS-dependent and RIG-I/MAVS-independent signaling in STAT1 phosphorylation.
To examine the involvement of MAVS in RIG-I-mediated STAT1 phosphorylation, we attempted to cotransfect A549 cells with the RIG-I-CARD vector and siRNA against MAVS. However, we were unable to perform the experiments as desired, because the transient knockdown of MAVS unexpectedly suppressed RIG-CARD expression (data not shown). It is noteworthy that the other 3 siRNA that were designed to target MAVS showed similar effects, suggesting that the inhibition of RIG-I-CARD expression was not a nonspecific effect of the MAVS siRNA; in addition, no such effect was observed with control siRNA or siRNA targeting a gene unrelated to MAVS. To continue to investigate the involvement of MAVS, we generated stably MAVS-silenced cells using the 293-flp system, and we confirmed RIG-I-CARD was not inhibited in the MAVS knockdown cells. 293 cells, the parental cells of flp-in 293 cells, have been reported to show a level of type I IFN expression similar to that in A549 cells (15). When RIG-I-CARD was transfected into the control cells for 10 h, STAT1 was phosphorylated as expected. In the stably MAVS-silenced cells, phosphorylation was significantly inhibited. In contrast, 24 h after the transfection, RIG-I-CARD-mediated STAT1 phosphorylation was not inhibited in the MAVS-silenced cells (Fig. 5A). No significant production of IFN-β protein was observed in the conditioned medium from cells transfected with RIG-I-CARD after 10 h. Twenty-four hours after transfection with RIG-I-CARD, the level of IFN-β was elevated and that of IFN-β was significantly decreased in the conditioned medium from MAVS-silenced cells.
Fig 5.
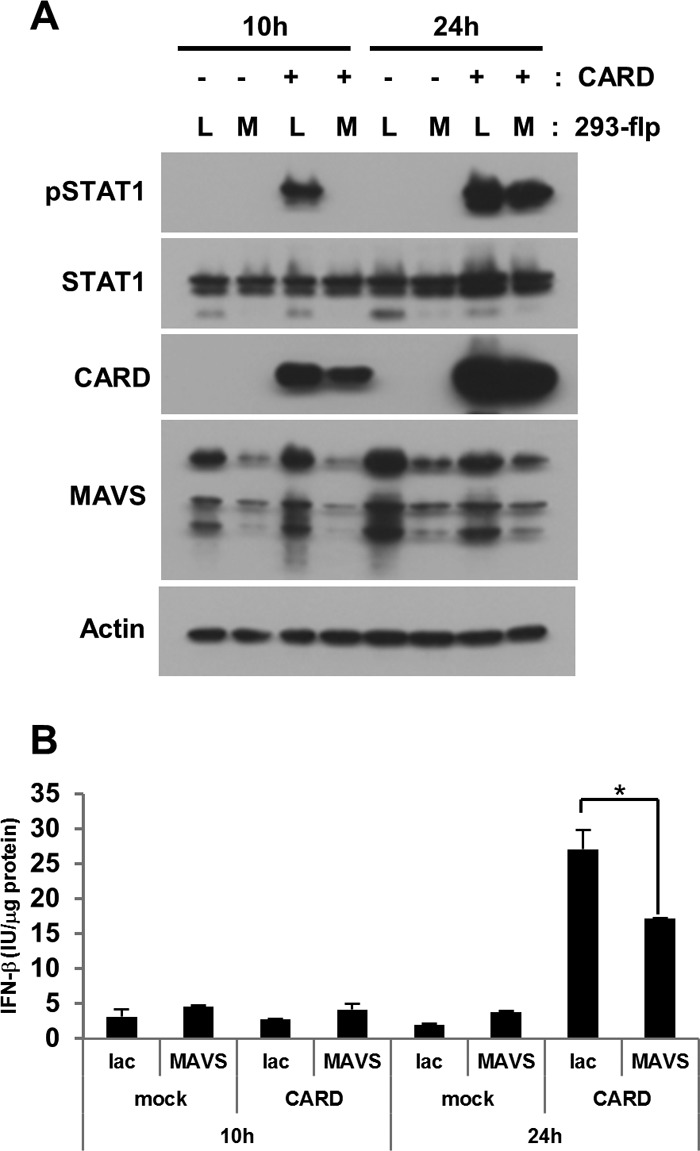
Effect of MAVS on RIG-I-CARD-induced STAT1 phosphorylation. Either stably MAVS-silenced 293-flp cells (MAVS) or a negative control, lacZ-silenced cells (Lac), were transfected with RIG-I-CARD (CARD) or an empty vector (mock) for the indicated periods. Following the transfections, cell lysates (A) or conditioned media (B) were collected. (A) The levels of the phosphorylated form of STAT1 (pSTAT1) and total STAT1 were analyzed by immunoblotting. (B) The levels of IFN-β protein were measured by ELISA. The means (± SD) from three experiments are shown. *, P < 0.01 by Student's t test.
When the type I IFN receptor was neutralized with a specific antibody, the initial phosphorylation of STAT1 induced by RIG-I-CARD was suppressed in control 293 cells (Fig. 6). In this experiment, a small amount of the phosphorylated form of STAT1 was detected in RIG-I-CARD-expressing MAVS-silenced 293 cells. This phosphorylation was almost completely inhibited in MAVS-silenced cells by blocking IFNAR. No significant effect caused by either an anti-IFNAR antibody or MAVS knockdown was observed on late STAT1 phosphorylation induced by RIG-I-CARD (Fig. 6).
Fig 6.

Effect of an anti-IFNAR neutralizing antibody on RIG-I-CARD-induced STAT1 phosphorylation. Either stably MAVS-silenced 293-flp cells (M) or lacZ-silenced cells (L) were transfected with FLAG-RIG-I-CARD or an empty vector (mock) for the indicated periods of time in the presence or absence of an anti-IFNAR neutralizing antibody (2.5 μg/well). As a control, we pretreated 293 cells with an anti-IFNAR antibody for 24 h and then incubated the cells with IFN-α2b for 15 min. Immunoblot analyses were performed using antibodies directed against pSTAT1, STAT1, MAVS, and FLAG (CARD).
IFNAR-deficient cells can induce STAT1 phosphorylation in response to poly(I:C).
To exclude the possibility that the IFNAR neutralizing antibody failed to block type I IFN signaling in response to poly(I:C), we utilized well-characterized IFNAR-deficient U5A cells (27). We initially characterized type II IFN, IFN-γ-stimulated STAT1 phosphorylation, showing that the JAK-STAT1 signaling pathway is intact in U5A cells as well as in A549 cells (Fig. 7A). In contrast, IFN-α2b failed to induce STAT1 phosphorylation in U5A cells, whereas it was able to activate STAT1 phosphorylation in A549 cells and parental cells of U5A, 2fTHG cells (Fig. 7A). These observations, combined with previous reports, indicated that in U5A cells, type I IFN fails to activate STAT1 phosphorylation due to IFNAR deficiency. We found that the introduction of poly(I:C) was able to induce STAT1 phosphorylation in U5A cells but with delayed kinetics relative to those in A549 cells (Fig. 7B). The overexpression of RIG-CARD in U5A cells for 24 h induced STAT1 phosphorylation (Fig. 8); however, the level of induction was much less than that in A549 and 293-flp cells.
Fig 7.

dsRNA can induce STAT1 phosphorylation in IFNAR-deficient cells. (A) Characterization of U5A cells. A549, 2fTGH, and U5A cells were treated with IFN-α2b (α2b; 200 pg/ml) or IFN-γ (γ; 5 ng/ml) for 30 min or left untreated (un). The graphs present densitometric band analysis normalized to total STAT1. (B) U5A cells were transfected with poly(I:C) (200 ng) for 0 to 8 h. Phosphorylated STAT1 (pSTAT1) and STAT1 were analyzed by immunoblotting. The graphs present densitometric band analysis normalized to total STAT1.
Fig 8.
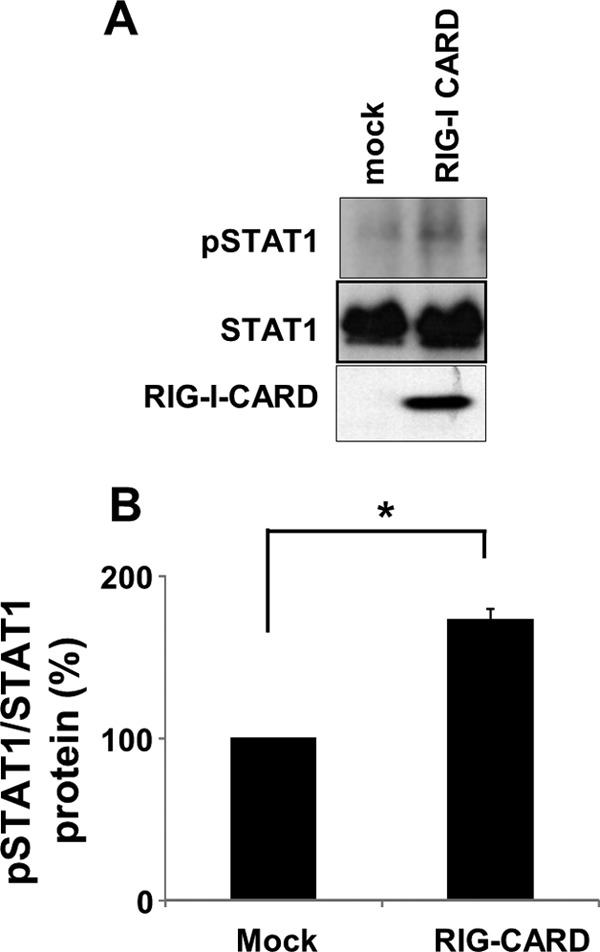
Effect of RIG-CARD on STAT1 phosphorylation in U5A cells. (A) The cells were transfected with either an empty control vector (mock) or a vector encoding the CARD of RIG-I for 24 h. The levels of pSTAT1, STAT1, and FLAG-RIG-CARD were analyzed by immunoblotting. The results are representative of three experiments conducted under similar conditions. (B) Densitometric analysis of pSTAT1 relative to total STAT1 based on the three independent experiments of panel A revealed a 173% increase in pSTAT1 in response to RIG-CARD in U5A cells. *, P < 0.001.
Type I IFN-independent STAT1 phosphorylation is unrelated to ISG induction.
STAT1-deficient mice have no innate responses to either viral or bacterial infection, because the first line of defense against potential pathogens requires the IFN response (35). Secreted IFN activates STAT1, and subsequently STAT1 accumulates in the nuclei, leading to the induction of IFN-stimulated genes (ISGs) (6). Therefore, we asked if IFN-independent STAT1 activation is associated with antiviral signaling. IFN-γ caused rapid accumulation of STAT1 in the nuclei of IFNAR-deficient cells (Fig. 9A). In contrast, no such nuclear accumulation was observed when IFNAR-deficient cells were transfected with poly(I:C) (Fig. 9A). This result allowed us to speculate on the different roles of IFN-independent STAT1 activation in response to poly(I:C). We then asked whether poly(I:C) can induce ISGs in IFN-deficient cells. We determined the expression levels of myxovirus resistance protein A (MxA) (37), interferon-inducible gene 6-16 (G1P3) (4), and oligoadenylate synthetase (OAS) (10), all of which are known ISGs, following transfection of the cells with poly(I:C). The transfection of both 2fTGH cells, the parental cells of U5A, and U5A cells with poly(I:C) induced the expression of IFN-β with similar expression patterns (Fig. 9B). Following transfection with poly(I:C), the mRNA levels of OAS, G1P3, and MxA were increased in 2fTGH cells in a time-dependent manner (Fig. 9C to E). However, no such mRNA enhancement was observed when poly(I:C) was introduced into IFNAR-deficient U5A cells (Fig. 9C to E). We confirmed that the expression of these ISGs was absolutely IFN dependent (Fig. 9F and G). Because ISGs are crucial molecules in antiviral actions (44), these data suggest that IFN-independent STAT1 activation is probably unrelated to innate antiviral immunity, because ISGs are essential to antiviral activities (25).
Fig 9.

Influence of the IFN receptor on ISG induction. (A) U5A cells were transfected with poly(I:C) (200 ng) for 8 h or treated with IFN-γ (5 ng/ml) for 30 min, followed by fixation with 4% paraformaldehyde. STAT1 (green) and DAPI (blue; nuclei) stains are shown. Both U5A cells and their parental 2fTGH cells were transfected with poly(I:C) (200 ng) (B to E) or treated with IFN-α2b (100 pg/ml) (F to H) for up to 8 h. The expression levels of IFN-β (B), OAS (C and F), G1P3 (D and G), and MxA (E and H) were quantified by real-time RT-PCR. The means (± SD) from three experiments are shown. *, P < 0.001.
DISCUSSION
Type I and type II IFNs activate STAT1 via specific receptors. In addition to the IFNs, IL-6 can cause STAT1 to be recruited to its signaling receptor, gp130 (41). The STAT1 protein is involved in a wide variety of intracellular responses, including cell differentiation and cell death, in addition to innate immunity (3, 49). A unique type II IFN, IFN-γ, attenuates cytokine-mediated cell growth by cross talk with antiapoptotic NF-κB signaling (46), most likely due to STAT1 acetylation, which allows the binding of STAT1 to NF-κB (22). Thus, the regulation and function of STAT1 are complicated and remain to be elucidated. Solid evidence has demonstrated the importance of STAT1 in antiviral innate immunity. The receptor for type I IFN activates JAK, leading to the phosphorylation of STAT1. In STAT1-impaired cells, little expression of IFN-inducible genes is detected in response to type I IFN; furthermore, the impairment of the STAT1-dependent response to type I IFN results in greater susceptibility to viral infection (8). These facts indicate that STAT1 is a cardinal molecule in innate immune responses to viral infection.
In 2000, Matikainen et al. reported that influenza A virus and Sendai virus activate STAT1 in macrophages (28). In that study, the phosphorylation of STAT1 was observed starting 3 h after viral infection, and the authors concluded that the phosphorylation was induced by the virus-induced type I IFN. Virus-induced STAT1 phosphorylation is transient compared to that induced by IFNs, indicating that viral proteins somehow interfere with STAT1 directly or with the production of type I IFN (28). Currently, little is known about the mechanism of STAT1 regulation in viral infection, especially following the sensing of viral RNA by RLRs. To exclude any effect of a viral protein on STAT1 activation and to observe the pure RLR-STAT1 axis, we used a synthetic dsRNA, poly(I:C). We found that poly(I:C) induces STAT1 phosphorylation in both a type I IFN-dependent manner and a type I IFN-independent manner depending on the period of time after the introduction of poly(I:C) into the cells. There is an early phase (<3 h) of STAT1 phosphorylation and a corresponding late phase (>8 h). In the initial phosphorylation phase, a neutralizing antibody targeting IFNAR markedly inhibited STAT1 phosphorylation. Competitive binding studies have demonstrated that IFN-β and IFN-α2b bind with similar affinities to the type I IFN receptor on human cells (39). As a positive control for STAT1 phosphorylation, we used 200 pg/ml of IFN-α2b, a concentration that is approximately equivalent to 130 IU/ml, because we found that the maximal amount of poly(I:C)-induced IFN-β production was approximately 120 IU/ml within 8 h (Fig. 1B). The blockade of the type I IFN receptor resulted in the complete suppression of IFN-α2b-mediated STAT1 phosphorylation, suggesting that the neutralizing antibody targeting IFNAR had sufficient activity to suppress type I IFN signaling. The silencing of either RIG-I or MAVS almost completely inhibited the phosphorylation of STAT1. These results suggest that dsRNA induces the initial STAT1 phosphorylation through a RIG-I-MAVS-dependent pathway; however, we could not exclude a type I IFN-independent pathway at this point, because the blockade of IFNAR incompletely suppressed the initial STAT1 phosphorylation. We observed that MDA-5, another sensor for poly(I:C) (18), is not involved in STAT1 phosphorylation, suggesting that among RLRs, RIG-I is the only molecule required for RLR/MAVS-mediated STAT1 phosphorylation. Neither immunofluorescence staining nor coimmunoprecipitation experiments showed a direct association between RIG-I or MAVS and STAT1 after transfection of cells with poly(I:C) (J. Dempoya and T. Matsumiya, unpublished observations), indicating that somehow RIG-I, which sensed the dsRNA, activates an intermediate signaling molecule that leads to the initial STAT1 phosphorylation in addition to the production of type I IFN.
IFNAR-deficient U5A cells can produce type I IFN in response to dsRNA or Sendai virus (34). In our experimental model, the transfection of cells with poly(I:C) also markedly increased the level of IFN-β expression in U5A cells. However, in the IFNAR-deficient cells, the level of STAT1 phosphorylation showed a different pattern than in A549 cells; contrary to our expectation, STAT1 was phosphorylated only from 8 h onward after the introduction of poly(I:C) into U5A cells. We also found that type I IFN signaling is associated with STAT1 accumulation and the induction of ISGs (Fig. 9). Taken together, these results suggest that only the initial STAT1 phosphorylation through type I IFN signaling is related to antiviral activity.
Fig 10.

Mechanism of dsRNA-mediated STAT1 phosphorylation. (A) Our results indicate that the initial phosphorylation of STAT1 is RIG-I-MAVS dependent. MDA-5 is not involved in the initial dsRNA-induced STAT1 phosphorylation. dsRNA-induced type I IFN is essential for the initial phosphorylation. In addition, there are presumably type I IFN-independent and RIG-MAVS-dependent pathways that activate STAT1. (B) Neither MAVS nor type I IFN is involved in late STAT1 phosphorylation. Late phosphorylation may be the result of the autophosphorylation of STAT1 or may be induced by an unknown molecule, which is represented by X, unrelated to those responsible for the initial antiviral signaling.
RIG-I has been shown to affect STAT1 phosphorylation, resulting in the inhibition of leukemia cell proliferation (14). Tetracycline-induced full-length RIG-I has been shown to be able to phosphorylate STAT1 at both Tyr701 and Ser727 in a U937-RIG-I tet-on system (14). These findings were useful for us to understand the relationship between RIG-I and STAT1 activation; however, the following two questions remained regarding our experimental model: how is dsRNA involved in this relationship, and what is the time dependence after the activation of RIG-I? As mentioned previously, following viral sensing, activated RIG-I exposes its N-terminal CARD (5). Indeed, only the CARD portion of RIG-I can activate antiviral signaling through the adaptor molecule MAVS (20, 51); moreover, the overexpression of full-length RIG-I alone does not affect antiviral signaling (51). Consistent with these reports, we observed that only RIG-I-CARD could induce STAT1 phosphorylation in A549 cells (Fig. 4), showing that the overexpression of RIG-I-CARD reflects the antiviral state more closely than the overexpression of full-length RIG-I. We next considered the kinetics of the expression of RIG-I-CARD after transfection, because we predicted that the induction of STAT1 phosphorylation should be followed by the expression of RIG-I-CARD as long as RIG-I-CARD reflects activated RIG-I. As expected, STAT1 phosphorylation was rapidly observed in RIG-I-CARD-transfected cells as soon as the protein level of RIG-I-CARD could be detected by immunoblotting, suggesting that RIG-I-CARD-mediated signaling is required for the initial STAT1 phosphorylation. Our findings also show that MAVS is essential in the CARD-mediated initial STAT1 phosphorylation. No detectable level of IFN-β was observed 8 to 10 h after RIG-I-CARD transfection in A549 cells (data not shown). The blockade of IFNAR suppressed the initial STAT1 phosphorylation in response to RIG-I-CARD. An amount of type I IFN undetectable by ELISA may participate in the initial phosphorylation of STAT1 by RIG-CARD, as we confirmed using immunofluorescence microscopy, the results of which are shown in Fig. 2B. However, some phosphorylated STAT1 remained even after blocking IFNAR. Taken together, our results indicate that the initial phase of STAT1 phosphorylation is regulated by the RIG-I-MAVS-dependent pathway. Type I IFN predominantly contributes to the initial STAT1 phosphorylation; however, another pathway may participate in this phosphorylation.
We also investigated the involvement of the type I IFN and RIG-MAVS pathways in late STAT1 phosphorylation in response to poly(I:C) introduction. Perry et al. reported that poly(I:C)-induced STAT1 phosphorylation could be detected in TANK-binding kinase-1 (TBK1)-deficient macrophages, albeit with delayed kinetics (38). In their study, poly(I:C) was added to the culture medium, where it is thought to be recognized by TLR3 (47). Consistent with the initial observation regarding the TLR3-mediated pathway, poly(I:C) induced late STAT1 phosphorylation in a type I IFN-independent fashion. One possibility explaining late STAT1 phosphorylation is that there is a positive feedback loop of STAT1 activation. It has been reported that overexpressed STAT1 can be phosphorylated in the absence of any stimulus (48); furthermore, both type I and type II IFNs are known to increase the level of STAT1 (11). Consequently, induced STAT1 might be autophosphorylated, a process through which it becomes independent of the type I IFN-independent pathway.
The silencing of MAVS did not affect the late STAT1 phosphorylation induced by the overexpression of RIG-I-CARD for 24 h, but it did produce IFN-β. Furthermore, RIG-CARD was able to induce STAT1 phosphorylation in IFNAR-deficient cells; therefore, another possibility is that the late STAT1 phosphorylation in response to dsRNA requires a CARD-containing RNA-binding molecule that is as yet unknown along with its adaptor in addition to RLRs and MAVS (Fig. 10).
Our findings underscore the importance of the RIG-I- and MAVS-dependent pathway for initial STAT1 phosphorylation following the recognition of dsRNA. Because the rapid induction of the innate antiviral response is critical for controlling viral replication (9), it is essential to understand how the signal from the cytosolic viral sensor RIG-I results in the expression of antiviral genes. We conclude that, in addition to transactivating type I IFN, the RIG-I-MAVS pathway might be able to directly activate STAT1 or at least trigger initial STAT1 phosphorylation. Although further studies are needed to elucidate the precise regulatory mechanisms governing the phosphorylation of STAT1 in response to dsRNA, we believe a signaling switch from an IFN-dependent pathway to an IFN-independent pathway in STAT1 activation is an important step in the innate immune response.
ACKNOWLEDGMENTS
This work was supported in part by a Grant-in-Aid for Scientific Research (KAKENHI, Japan; 23590560 to T.M.).
We thank George Stark for the 2fTGH and U5A cells and Janeth Tayone for technical assistance.
Footnotes
Published ahead of print 12 September 2012
REFERENCES
- 1. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738 [DOI] [PubMed] [Google Scholar]
- 2. Bhattacharyya S, Zhao Y, Kay TW, Muglia LJ. 2011. Glucocorticoids target suppressor of cytokine signaling 1 (SOCS1) and type 1 interferons to regulate Toll-like receptor-induced STAT1 activation. Proc. Natl. Acad. Sci. U. S. A. 108:9554–9559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bromberg JF, Fan Z, Brown C, Mendelsohn J, Darnell JE., Jr 1998. Epidermal growth factor-induced growth inhibition requires Stat1 activation. Cell Growth Differ. 9:505–512 [PubMed] [Google Scholar]
- 4. Cheriyath V, et al. 2007. G1P3, an IFN-induced survival factor, antagonizes TRAIL-induced apoptosis in human myeloma cells. J. Clin. Investig. 117:3107–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cui S, et al. 2008. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol. Cell 29:169–179 [DOI] [PubMed] [Google Scholar]
- 6. Darnell JE., Jr 1997. STATs and gene regulation. Science 277:1630–1635 [DOI] [PubMed] [Google Scholar]
- 7. Drummond RA, Saijo S, Iwakura Y, Brown GD. 2011. The role of Syk/CARD9 coupled C-type lectins in antifungal immunity. Eur. J. Immunol. 41:276–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dupuis S, et al. 2003. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat. Genet. 33:388–391 [DOI] [PubMed] [Google Scholar]
- 9. Fredericksen BL, Keller BC, Fornek J, Katze MG, Gale M., Jr 2008. Establishment and maintenance of the innate antiviral response to West Nile virus involves both RIG-I and MDA5 signaling through IPS-1. J. Virol. 82:609–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hovanessian AG, Justesen J. 2007. The human 2′-5′oligoadenylate synthetase family: unique interferon-inducible enzymes catalyzing 2′-5′ instead of 3′-5′ phosphodiester bond formation. Biochimie 89:779–788 [DOI] [PubMed] [Google Scholar]
- 11. Imaizumi T, et al. 2005. Involvement of retinoic acid-inducible gene-I in the IFN-γ/STAT1 signalling pathway in BEAS-2B cells. Eur. Respir. J. 25:1077–1083 [DOI] [PubMed] [Google Scholar]
- 12. Imaizumi T, et al. 2011. Polyinosinic-polycytidylic acid induces the expression of interferon-stimulated gene 20 in mesangial cells. Nephron Exp. Nephrol. 119:e40–e48 [DOI] [PubMed] [Google Scholar]
- 13. Inohara N, Nunez G. 2003. NODs: intracellular proteins involved in inflammation and apoptosis. Nat. Rev. Immunol. 3:371–382 [DOI] [PubMed] [Google Scholar]
- 14. Jiang LJ, et al. 2011. RA-inducible gene-I induction augments STAT1 activation to inhibit leukemia cell proliferation. Proc. Natl. Acad. Sci. U. S. A. 108:1897–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnsen IB, Nguyen TT, Bergstroem B, Fitzgerald KA, Anthonsen MW. 2009. The tyrosine kinase c-Src enhances RIG-I (retinoic acid-inducible gene I)-elicited antiviral signaling. J. Biol. Chem. 284:19122–19131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kato H, et al. 2005. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23:19–28 [DOI] [PubMed] [Google Scholar]
- 17. Kato H, et al. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205:1601–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kato H, et al. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105 [DOI] [PubMed] [Google Scholar]
- 19. Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131–137 [DOI] [PubMed] [Google Scholar]
- 20. Kawai T, et al. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988 [DOI] [PubMed] [Google Scholar]
- 21. Kotenko SV, et al. 2003. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 4:69–77 [DOI] [PubMed] [Google Scholar]
- 22. Kramer OH, et al. 2006. Acetylation of Stat1 modulates NF-kappaB activity. Genes Dev. 20:473–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kumar H, Kawai T, Akira S. 2009. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 388:621–625 [DOI] [PubMed] [Google Scholar]
- 24. Lee MS, Min YJ. 2007. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu. Rev. Biochem. 76:447–480 [DOI] [PubMed] [Google Scholar]
- 25. Liu SY, Sanchez DJ, Cheng G. 2011. New developments in the induction and antiviral effectors of type I interferon. Curr. Opin. Immunol. 23:57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu T, et al. 2004. Reactive oxygen species mediate virus-induced STAT activation: role of tyrosine phosphatases. J. Biol. Chem. 279:2461–2469 [DOI] [PubMed] [Google Scholar]
- 27. Lutfalla G, et al. 1995. Mutant U5A cells are complemented by an interferon-alpha beta receptor subunit generated by alternative processing of a new member of a cytokine receptor gene cluster. EMBO J. 14:5100–5108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matikainen S, et al. 2000. Influenza A and Sendai viruses induce differential chemokine gene expression and transcription factor activation in human macrophages. Virology 276:138–147 [DOI] [PubMed] [Google Scholar]
- 29. Matikainen S, et al. 2006. Tumor necrosis factor alpha enhances influenza A virus-induced expression of antiviral cytokines by activating RIG-I gene expression. J. Virol. 80:3515–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matsumiya T, Imaizumi T, Yoshida H, Satoh K. 2011. Antiviral signaling through retinoic acid-inducible gene-I-like receptors. Arch. Immunol. Ther. Exp. 59:41–48 [DOI] [PubMed] [Google Scholar]
- 31. Matsumiya T, et al. 2009. The levels of retinoic acid-inducible gene I are regulated by heat shock protein 90-alpha. J. Immunol. 182:2717–2725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matsumiya T, et al. 2010. Characterization of synergistic induction of CX3CL1/fractalkine by TNF-alpha and IFN-gamma in vascular endothelial cells: an essential role for TNF-alpha in post-transcriptional regulation of CX3CL1. J. Immunol. 184:4205–4214 [DOI] [PubMed] [Google Scholar]
- 33. Matsumiya T, Stafforini DM. 2010. Function and regulation of retinoic acid-inducible gene-I. Crit. Rev. Immunol. 30:489–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Melchjorsen J, et al. 2009. Differential regulation of the OASL and OAS1 genes in response to viral infections. J. Interferon Cytokine Res. 29:199–207 [DOI] [PubMed] [Google Scholar]
- 35. Meraz MA, et al. 1996. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 84:431–442 [DOI] [PubMed] [Google Scholar]
- 36. Meylan E, et al. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172 [DOI] [PubMed] [Google Scholar]
- 37. Pavlovic J, Schroder A, Blank A, Pitossi F, Staeheli P. 1993. Mx proteins: GTPases involved in the interferon-induced antiviral state. Ciba Found. Symp. 176:233–247 [DOI] [PubMed] [Google Scholar]
- 38. Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. 2004. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J. Exp. Med. 199:1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pfeffer LM, et al. 1998. Biological properties of recombinant alpha-interferons: 40th anniversary of the discovery of interferons. Cancer Res. 58:2489–2499 [PubMed] [Google Scholar]
- 40. Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5:375–386 [DOI] [PubMed] [Google Scholar]
- 41. Sanz E, Hofer MJ, Unzeta M, Campbell IL. 2008. Minimal role for STAT1 in interleukin-6 signaling and actions in the murine brain. Glia 56:190–199 [DOI] [PubMed] [Google Scholar]
- 42. Sartono E, et al. 1995. Elevated cellular immune responses and interferon-gamma release after long-term diethylcarbamazine treatment of patients with human lymphatic filariasis. J. Infect. Dis. 171:1683–1687 [DOI] [PubMed] [Google Scholar]
- 43. Schindler C, Darnell JE., Jr 1995. Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu. Rev. Biochem. 64:621–651 [DOI] [PubMed] [Google Scholar]
- 44. Sen GC, Ransohoff RM. 1993. Interferon-induced antiviral actions and their regulation. Adv. Virus Res. 42:57–102 [DOI] [PubMed] [Google Scholar]
- 45. Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682 [DOI] [PubMed] [Google Scholar]
- 46. Sironi JJ, Ouchi T. 2004. STAT1-induced apoptosis is mediated by caspases 2, 3, and 7. J. Biol. Chem. 279:4066–4074 [DOI] [PubMed] [Google Scholar]
- 47. Tanabe M, et al. 2003. Mechanism of up-regulation of human Toll-like receptor 3 secondary to infection of measles virus-attenuated strains. Biochem. Biophys. Res. Commun. 311:39–48 [DOI] [PubMed] [Google Scholar]
- 48. Tang L, Roberts PC, Kraniak JM, Li Q, Tainsky MA. 2006. Stat1 expression is not sufficient to regulate the interferon signaling pathway in cellular immortalization. J. Interferon Cytokine Res. 26:14–26 [DOI] [PubMed] [Google Scholar]
- 49. Wang H, et al. 2010. STAT1 activation regulates proliferation and differentiation of renal progenitors. Cell Signal. 22:1717–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu LG, et al. 2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19:727–740 [DOI] [PubMed] [Google Scholar]
- 51. Yoneyama M, et al. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730–737 [DOI] [PubMed] [Google Scholar]