

Abstract
Etravirine (ETR) is an expanded-spectrum nonnucleoside reverse transcriptase inhibitor (NNRTI) approved for use as an antiretroviral agent in treatment-experienced patients. Y181C and E138K in HIV-1 RT are among 20 different drug resistance mutations associated with ETR. However, E138K can be consistently selected by ETR when wild-type viruses but not viruses containing Y181C are grown in tissue culture. This study was carried out to evaluate any possible mechanisms that might explain antagonism between the Y181C and E138K mutations. Accordingly, we performed tissue culture studies to investigate the evolutionary dynamics of E138K in both a wild-type (WT) and a Y181C background. We also generated recombinant enzymes containing Y181C and E138K alone or in combination in order to study enzyme processivity, rates of processive DNA synthesis, enzyme kinetics, and susceptibility to ETR. We now show that the presence of the Y181C mutation prevented the emergence of E138K in cell culture and that the simultaneous presence of E138K and Y181C impaired each of enzyme activity, processivity, rate of processive DNA synthesis, and deoxynucleoside triphosphate (dNTP) affinity. The addition of E138K to Y181C also decreased the level of resistance to ETR compared to that obtained with Y181C alone.
INTRODUCTION
HIV-1 reverse transcriptase (RT) is a multifunctional enzyme that carries out RNA-dependent DNA polymerase (RDDP), DNA-dependent DNA polymerase (DDDP), and RNase H activities responsible for converting the single-stranded viral RNA genome into double-stranded DNA (dsDNA) that becomes integrated into host cell DNA (17). Due to its crucial role in viral replication, HIV-1 RT has been a major target for development of antiviral drugs. Currently, two classes of RT inhibitors, nucleoside and nonnucleoside, have been approved by multiple regulatory agencies for treatment of HIV-1 infection. The nucleoside reverse transcriptase inhibitors (NRTIs), which include zidovudine (AZT, ZDV), didanosine (ddI), stavudine (d4T), zalcitabine (ddC), lamivudine (3TC), emtricitabine (FTC), abacavir (ABC), and tenofovir disoproxil fumarate (TDF), compete with the natural deoxynucleoside triphosphate (dNTP) substrate and act as DNA chain terminators; the nonnucleoside reverse transcriptase inhibitors (NNRTIs), including the narrow-spectrum NNRTIs nevirapine (NVP), delavirdine (DLV), and efavirenz (EFV) and the expanded-spectrum NNRTIs etravirine (ETR, formerly known as TMC125) and rilpivirine (RPV, formerly known as TMC278), act allosterically by inducing conformational changes in the RT enzyme through binding to the NNRTI binding pocket (BP), located 10 Å from the polymerase active site, and by inhibiting the chemical step of polymerization (39). The conformational changes in the NNRTI BP induced by NNRTI binding distort the geometry of the DNA polymerase catalytic site (13), and in addition, NNRTI binding deforms the structural elements that comprise the primer grip which is involved in the positioning of a primer DNA strand into the polymerase active site (19). Either of these changes is able to inhibit the DNA polymerization reaction by preventing establishment of a catalytically competent RT-template/primer-dNTP ternary complex (38). Both NRTIs and NNRTIs are key components of highly active antiretroviral therapy (HAART), which has led to significant declines in HIV-associated morbidity and mortality, but both classes of drugs, like all other antiretrovirals, can be compromised by drug resistance.
NNRTI-based regimens, consisting of a dual NNRTI backbone plus an NNRTI (NVP or EFV), are common cost-effective potent regimens used in first-line HAART. However, one major limitation of narrow-spectrum NNRTIs is that they have a low genetic barrier for resistance, and a single mutation, such as K103N, is able to cause cross-resistance to all the drugs in this class (12, 18). Resistance to narrow-spectrum NNRTIs can develop quickly if low-grade HIV replication is permitted to occur, and this has spurred efforts to discover newer NNRTIs that have improved potency and a higher genetic barrier for resistance. The result has been the development of 2 expanded-spectrum NNRTI di-aryl-pyrimidine (DAPY) compounds, ETR and RPV (21), both of which have been approved for use in treatment-experienced patients and in drug-naive patients, respectively (10).
Y181C in HIV-1 RT is the second most common mutation, following only K103N in patients failing NNRTI-based therapy (45). In patients failing ETR-containing regimens, Y181C was shown to commonly occur (26, 42). HIV-1 containing Y181C alone showed moderate-level resistance to ETR (8, 41). The E138K mutation was shown to be the first mutation selected by ETR in cell culture passage experiments (5). Viruses containing E138K display moderate resistance to ETR (5, 41, 47), and E138K has been reported to emerge in HIV-1-infected patients who have failed an ETR-containing regimen (41, 42). Thus, E138K has the potential to be an important ETR resistance mutation, particularly in the case of wild-type (WT) viruses (6). At the same time, E138 and Y181C do not commonly occur together, as shown in several studies that have analyzed the resistance profiles of drug-experienced patients who received ETR as part of a second-line regimen (26, 41). The DUET studies showed that among the three patients who failed ETR and developed E138K (41), none of them either coselected Y181C or possessed Y181C at baseline. A separate study in NNRTI-experienced patients who failed ETR therapy showed that patients who harbor the Y181C at baseline selected mutations at position V179 and not E138K (26). Furthermore, selection studies performed in cell culture with wild-type and Y181C virus, with an NNRTI termed dapivirine (DAP) that is structurally related to ETR and RPV, also showed that the E138K mutation did not coemerge with Y181C (35).
We have hypothesized that antagonistic interactions may exist between E138K and Y181C in regard to RT function and ETR susceptibility. Previously, we demonstrated that RT containing the E138K mutation has low enzyme processivity and can be mutually compensated by the M184I/V mutations, leading to high-level resistance to both 3TC and FTC (47, 49). Here, we have performed tissue culture studies to investigate the evolutionary dynamics of E138K in a WT and Y181C background and used purified recombinant RT enzymes to study interactions between Y181C and E138K in regard to drug susceptibility, enzyme activity, enzyme processivity, dNTP affinity, and rates of processive DNA synthesis. We found that the presence of the Y181C mutation prevented the emergence of E138K in cell culture and that the addition of Y181C to E138K resulted in diminished levels of each of enzyme activity, processivity, and rate of DNA synthesis. Furthermore, steady-state kinetic analysis showed that the two mutations in tandem caused decreased dNTP binding and that the presence of E138K decreased levels of resistance conferred by Y181C against ETR.
MATERIALS AND METHODS
Chemicals, cells, viruses, and nucleic acids.
ETR was a gift of Janssen Pharmaceuticals (Titusville, NJ). The pRT6H-PROT plasmid DNA for expression and purification of HIV-1 heterodimeric reverse transcriptase was kindly provided by Stuart F. J. Le Grice, National Institutes of Health, Bethesda, MD.
Cord blood mononuclear cells (CBMCs) were obtained through the Department of Obstetrics, Jewish General Hospital, Montreal, Canada. HEK293T cells were obtained from the American Type Culture Collection (ATCC). The HIV-1 subtype B NL4-3 WT molecular cone and the TZM-bl cell line were obtained from the NIH AIDS Research and Reference Reagent Program. The NL4-3-derived viral clone containing the desired mutation Y181C was constructed using the QuikChange mutagenesis kit (Agilent Technologies Canada Inc., Mississauga, ON, Canada). NL4-3 WT and mutant viruses were generated by transfection of proviral plasmid DNAs into HEK293T cells as described previously using Lipofectamine 2000 (Invitrogen, Burlington, ON, Canada) (48). The primary HIV-1 WT and Y181C-containing subtype C isolates were obtained with informed consent from patients.
An HIV-1 RNA template ∼500 nucleotides (nt) in size, spanning the 5′ untranslated region (UTR) to the primer binding site (PBS), was transcribed in vitro from AccI-linearized pHIV-PBS DNA (4) by using an Ambion T7-MEGAshortscript kit (Invitrogen, Burlington, ON, Canada) as described previously (50). The oligonucleotides used in this study were synthesized by Integrated DNA Technologies Inc. (Coralville, IA) and purified by polyacrylamide-urea gel electrophoresis (47).
Selection of HIV-1 mutants in CBMCs under drug selection pressure.
CBMCs stimulated by phytohemagglutinin A (PHA) were cultured in 10% RPMI 1640 medium supplemented with 10% fetal calf serum (FCS), 20 U of human interleukin-2 (IL-2)/ml, 5 μg of hydrocortisone/ml, 2 mM l-glutamine/ml, 100 U of penicillin/ml, and 100 μg of streptomycin/ml. Cells in 24-well tissue culture plates were infected with recombinant viral clones at a similar multiplicity of infection (MOI). Selection for viral resistance mutations was performed using increasing concentrations of RT inhibitors at starting concentrations below the 50% effective concentration (EC50) as described previously (5, 30). As controls, all viruses were simultaneously passaged without drugs. Standard RT assays were performed weekly to monitor viral replication. Drug concentrations were increased at subsequent passages based on the ratio of the RT values of cell culture supernatant in control wells to those from test wells. Virus-containing culture media were harvested and kept at −80°C for subsequent standard genotypic analysis. Selections for resistance were performed over 25 weeks.
Phenotypic drug susceptibility assays.
Phenotypic susceptibility analysis of RT inhibitors was performed with recombinant HIV-1 NL4-3 clones in a TZM-bl cell-based in vitro assay as described previously (35). Briefly, ETR at variable concentrations was added to TZM-bl cells (104 cells/well) in 96-well plates grown in 100 μl Dulbecco's minimal essential medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (Gibco), 1% penicillin-streptomycin, and 1% l-glutamine (Invitrogen). Immediately after drug addition, cells were infected with WT or mutant viruses. At 24 to 48 h postinfection, cells were rinsed with 100 μl phosphate-buffered saline (PBS) and lysed with 50 μl/well cell lysis reagent (Promega). Cell lysates were then transferred to a white, opaque, 96-well plate (Corning). Luciferase assay reagent (Promega) was added to each well, and relative luminescence units (RLU)/well were measured by a luminometer (Microbeta2; Perkin Elmer). The EC50 (50% effective drug concentration) was calculated using the GraphPad Prism program (GraphPad Software, San Diego, CA).
Recombinant reverse transcriptase expression and purification.
Recombinant RTs in heterodimeric form were expressed from plasmid pRT6H-PROT and purified as described with minor modifications (24, 25). In brief, RT expression in Escherichia coli M15 (pREP4) (Qiagen, Mississauga, ON) was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at room temperature. The pelleted bacteria were lysed under native conditions with BugBuster protein extraction reagent containing Benzonase (Novagen, Madison, WI) according to the manufacturer's instructions. After clarification by high-speed centrifugation, the clear supernatant was subjected to the batch method of Ni-nitrilotriacetic acid (NTA) metal affinity chromatography (QIAexpressionist; Qiagen, Mississauga, ON, Canada). All buffers contained complete protease inhibitor cocktail (Roche, Mississauga, ON, Canada). Histidine-tagged RT was eluted using an imidazole gradient. RT-containing fractions were pooled, passed through DEAE-Sepharose (GE Healthcare, Mississauga, ON, Canada), and further purified using SP-Sepharose (GE Healthcare, Mississauga, ON, Canada). Fractions containing purified RT were pooled, dialyzed against storage buffer (50 mM Tris-HCl [pH 7.8], 50 mM NaCl, and 50% glycerol), and concentrated to 4 to 8 mg/ml with Centricon Plus-20 MWCO30 kDa (Millipore, Etobicoke, ON, Canada). Aliquots of proteins were stored at −80°C. Protein concentrations were measured by a Bradford protein assay kit (Bio-Rad Laboratories, Saint-Laurent, QC, Canada), and the purity of the recombinant RT preparations was verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The RNA-dependent DNA polymerase activity of each recombinant RT preparation was evaluated as described previously (33) using various concentrations of RT and a synthetic homopolymeric poly(rA)/p(dT)12-18 template/primer (T/P) (Midland Certified Reagent Company, Midland, TX).
RT inhibitor susceptibility assays.
Susceptibility to ETR was assayed using recombinant RT enzymes and a heteropolymeric HIV-1 PBS RNA template/primer (T/P) system as described previously (50). Briefly, RT reaction buffer containing 50 mM Tris-HCl (pH 7.8), 6 mM MgCl2, 60 mM KCl, dNTPs (5 μM each) with 2.5 μCi of [3H]-dTTP (70 to 80 mCi/mmol), 30 nM heterogeneous HIV-1 RNA template/primer, the same activity of RT enzymes, and variable amounts of RT inhibitors were included in 50-μl reaction volumes. In the reaction, the final concentrations of ETR were 0, 0.01, 0.03, 0.10, 0.30, 1.00, 3.00, and 10.00 μM. Reaction mixtures were incubated at 37°C for 30 min, terminated by adding 0.2 ml of 10% cold trichloroacetic acid (TCA) and 20 mM sodium pyrophosphate, and incubated for at least 30 min on ice. The precipitated products were filtered through a 96-well MultiScreen HTS FC filter plate (Millipore, Etobicoke, ON, Canada) and sequentially washed with 200 μl of 10% TCA and 150 μl of 95% ethanol. The radioactivity of incorporated products was analyzed by liquid scintillation spectrometry using a 1450 MicroBeta TriLux microplate scintillation and luminescence counter (Perkin Elmer, Woodbridge, ON, Canada). The 50% inhibitory concentration (IC50) of ETR was determined by nonlinear regression analysis using GraphPad Prism v5.01 (GraphPad Software, La Jolla, CA).
Processivity assays.
The processivity of recombinant RT enzymes was analyzed as described previously using a heteropolymeric HIV-1 PBS RNA template in the presence of a heparin enzyme trap to ensure a single processive cycle, i.e., a single round of binding and of primer extension and dissociation (47). The template/primer (T/P) was prepared by annealing the 497-nt HIV-1 PBS RNA with the [32P]-5′ end-labeled 25-nt DNA primer D25 at a molar ratio of 1:1, denatured at 85°C for 5 min, and then slowly cooled to 55°C for 8 min and 37°C for 5 min to allow for specific annealing of the primer to the template. RT enzymes with equal amounts of activity and 40 nM T/P were preincubated for 5 min at 37°C in a buffer containing 50 mM Tris-HCl (pH 7.8), 50 mM NaCl, and 6 mM MgCl2. Reactions were initiated by the addition of dNTPs at 0.5 μM or 200 μM and heparin trap (final concentration, 3.2 mg/ml) and incubated at 37°C for 30 min; then 2 volumes of stop solution (90% formamide, 10 mM EDTA, and 0.1% each of xylene cyanol and bromophenol blue) were added to stop the reaction. Reaction products were denatured by heating at 95°C and analyzed using 6% denaturing polyacrylamide gel electrophoresis and phosphorimaging. The effectiveness of the heparin trap was verified in control reactions in which the trap was preincubated with substrate before the addition of RT enzymes and dNTP.
Steady-state kinetics analysis.
Kinetics studies were carried out by a modification of a previously described method using homopolymeric poly(rA)/p(dT)12-18 and complementary dTTP as the nucleotide substrate (47). The reaction mixture (10 μl) contained 50 mM Tris-HCl (pH 7.8), 60 mM KCl, 6 mM MgCl2, 5 mM dithiothreitol (DTT), 0.5 U/ml poly(rA)/p(dT)12-18, RT enzymes, and variable concentrations of tracer [3H]-dTTP and cold dTTP (0.2 to 200 μM). Reactions were run at 37°C and quenched by adding 0.2 ml of 10% cold trichloroacetic acid and 20 mM sodium pyrophosphate; products were filtered onto Millipore 96-well MultiScreen HTS FC filter plates (MSFCN6B) and sequentially washed with 200 μl of 10% TCA and 150 μl of 95% ethanol. The radioactivity of incorporated products was analyzed by liquid scintillation spectrometry using a Perkin Elmer 1450 MicroBeta TriLux microplate scintillation and luminescence counter. The steady-state kinetic parameters, the maximum rate of metabolism (Vmax) and Km for nucleotide substrates, were determined by nonlinear regression analysis of the substrate concentration and initial velocity data using the Michaelis-Menten equation with the program GraphPad Prism v5.01 according to the manufacturer's instructions. The kcat was calculated using the equation kcat = Vmax/[E], where E stands for enzyme.
Analysis of the rate of processive DNA synthesis by gel-based RNA-dependent DNA polymerase assay.
The same 497-nt HIV-1 PBS RNA template and 5′ end [32P]-labeled D25 primers as those described previously (47) were used to assess the polymerization rate of processive DNA synthesis by recombinant RT enzymes in gel-based time course experiments. Final reaction mixtures contained 20 nM T/P, 400 nM RT enzyme, 50 mM Tris-HCl (pH 7.8), and 50 mM NaCl. Reactions were initiated by adding 6 mM MgCl2 and dNTPs at 200 μM, sampled at 40 s, 60 s, and 90 s, and mixed with 2 volumes of stop solution. Reaction products were separated by 6% denaturing polyacrylamide gel electrophoresis and analyzed as described previously (47).
RESULTS
Mutations in HIV-1 RT selected by ETR pressure in CBMC cell culture.
Previously, we showed that the most common NNRTI resistance-associated mutation selected in the presence of ETR in CBMCs was E138K, regardless of HIV-1 subtype (5). In the present study, we therefore compared WT and Y181C mutants of subtype B and C viruses in ETR cell culture selection experiments to investigate whether the Y181C mutation impacts the development of the E138K substitution. The results of Table 1 show that E138K consistently emerged from WT viruses of both subtypes; however, E138K did not emerge when Y181C was present at the beginning of the selection, indicating that Y181C prevents the emergence of E138K under ETR selection pressure.
Table 1.
Drug resistance mutations in HIV-1 RT selected by ETR in CBMCs
| Virus subtype | Baseline genotype | No. of wks of culture | Highest concn attained of ETR (μM) | Resistance mutation(s) genotyped |
|---|---|---|---|---|
| B | WT | 17 | 0.1 | L100I/L, E138K |
| WT | 25 | 0.25 | L100I, E138K, H221H/Y | |
| Y181C | 17 | 0.25 | Y181C | |
| Y181C | 25 | 0.5 | V179F, Y181C | |
| C | WT | 17 | 0.1 | E138K |
| WT | 25 | 0.25 | E138K, G190E/G | |
| Y181C | 17 | 0.25 | E138E/V, V179I, Y181C, Y188H/Y | |
| Y181C | 25 | 0.5 | E138V, V179I, Y181C, Y188H H208H/Y |
Purification and activity of recombinant HIV-1 RT enzymes.
Amino acid residue E138 in HIV-1 RT is part of the β7-β8 loop in the p51 subunit, located at the p66/p51 interface, which is a key structural element for RT dimerization, that involves the floor of the NNRTI binding pocket (14, 31, 32). It was previously shown that E138K did not interfere with either heterodimer formation or enzyme purification (2, 29, 47). Recombinant WT heterodimeric (p66/p51) RTs and RT enzymes containing each of the E138K, Y181C, and E138K/Y181C substitutions were purified to >95% homogeneity as demonstrated by Coomassie blue staining of SDS-PAGE gels (Fig. 1A). In these studies, the RT p66 and p51 subunits were processed to similar molar ratios. This verifies that the amino acid mutations E138K and Y181C and the double mutation E138K/Y181C did not affect protease cleavage, p66/p51 heterodimer formation, or RT purification.
Fig 1.
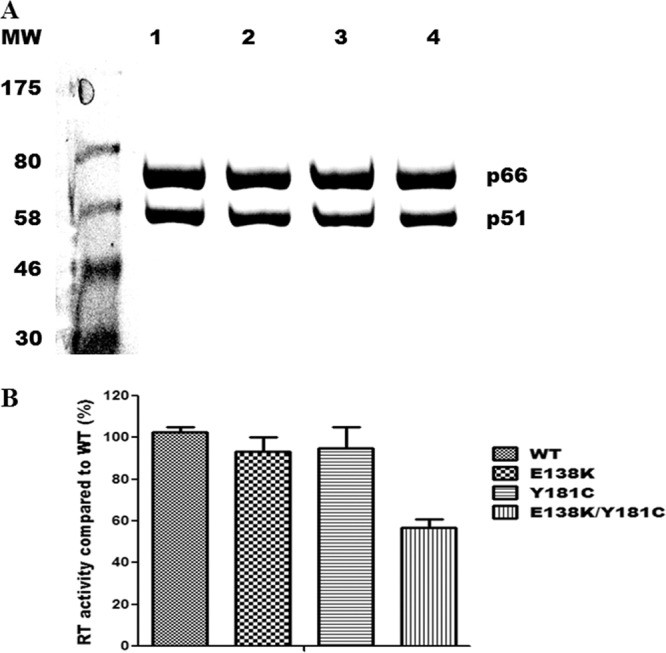
Purification of recombinant HIV-1 RTs. (A) Coomassie brilliant blue staining of purified heterodimer RTs following 8% SDS-PAGE. Purification of heterodimeric RT enzymes was achieved by attachment of a His6 tag at the C terminus of the p66 subunit through immobilized metal affinity chromatography (IMAC). MW (molecular weight) standards in kilodaltons are shown on the left. Lane 1, WT; lane 2, E138K; lane 3, Y181C; lane 4, E138K/Y181C. Positions of the purified recombinant RT heterodimers p66/p51 are indicated on the right. (B) Comparison of RNA-dependent DNA polymerase activities of recombinant subunit-selective mutant RT enzymes relative to those of the WT enzyme. DNA polymerase activity was assessed as described in Materials and Methods. Data from a representative experiment performed in triplicate are shown as means ± standard deviations.
To compare the activities of the recombinant enzyme preparations, RNA-dependent DNA polymerase activity was measured using synthetic poly(rA)/p(dT)12-18 template/primer. Recombinant RTs containing E138K or Y181C alone possessed activities similar to that of WT RT; the double mutant E138K/Y181C exhibited decreased activity (∼50%) relative to the WT. This result indicates that the copresence of E138K and Y181C impairs RT activity (Fig. 1B).
E138K decreases Y181C-based resistance to ETR.
Previous work showed that HIV-1 RT enzymes or viruses containing E138K or Y181C displayed modest phenotypic resistance to ETR (5, 46, 47). Now, we determined the impact of interactions between E138K and Y181C on susceptibility to ETR in an in vitro recombinant RT assay. The 50% inhibitory concentration (IC50) of ETR was determined by an in vitro RNA-dependent DNA polymerase assay using purified recombinant RT enzymes and a heteropolymeric HIV-1 PBS RNA template. The results in Table 2 show that E138K alone conferred modest resistance to ETR (2.3-fold change in IC50), consistent with previous reports (5, 47). The E138K/Y181C double mutant conferred a 2.4-fold change in IC50, compared to a 3.7-fold change for Y181C, indicating that the addition of E138K to Y181C did not enhance but rather diminished levels of resistance to ETR. A similar result was observed in the phenotyping assay with recombinant viruses (Table 3). The E138K/Y181C double mutant virus conferred a 5.1-fold change in EC50 against ETR, compared to an approximate 6-fold change for Y181C.
Table 2.
Susceptibility to ETR of recombinant and mutated RTs
| RT enzyme | IC50 (nM)a | FC resistanceb |
|---|---|---|
| WT | 97 ± 12 | |
| E138K | 223 ± 19 | 2.3 |
| Y181C | 359 ± 32 | 3.7 |
| E138K/Y181C | 233 ± 24 | 2.4 |
IC50s (50% drug inhibitory concentrations) were determined in recombinant RT assays using a heteropolymeric HIV-1 PBS RNA template. Data are the means and standard deviations (SD) of 3 to 6 independent experiments.
FC values are the fold changes in IC50 for mutated RTs compared to the IC50 of the WT RT.
Table 3.
Susceptibility to ETR of WT and mutated viruses
| Virus | EC50 (nM)a | FCb |
|---|---|---|
| WT | 1.65 ± 0.05 | |
| E138K | 5.4 ± 0.14 | 3.3 |
| Y181C | 9.95 ± 0.35 | 6.0 |
| E138K/Y181C | 8.45 ± 1.5 | 5.1 |
EC50s (50% effective drug concentrations) were determined in a phenotypic cell-based assay. Data are the means and standard deviations (SD) of 3 independent experiments.
FC values are the fold changes in EC50 for mutated viruses compared to the EC50 of the WT.
Y181C decreases the enzyme processivity of RT containing E138K.
Diminished HIV-1 RT processivity is a major determinant of impaired viral replication capacity, and impairments of processivity and viral replication can be influenced by dNTP concentration (7, 9, 28, 37). To investigate whether interactions between E138K and Y181C might impact enzyme processivity, we performed single-cycle processivity assays using recombinant WT RT enzyme or RT containing the E138K or Y181C mutation or both mutations at both low (0.5 μM) and high (200 μM) dNTP concentrations. Assays performed with 0.5 μM each dNTP showed that Y181C RT had similar processivity to wild-type RT and that the E138K-containing mutant enzyme that we tested had higher processivity than WT RT, consistent with previous results (47, 49). Although the processivity of the double E138K/Y181C mutant was severely impaired at low dNTP levels, all enzymes tested displayed similar processivity at high dNTP levels, based on production of full-length products (Fig. 2).
Fig 2.
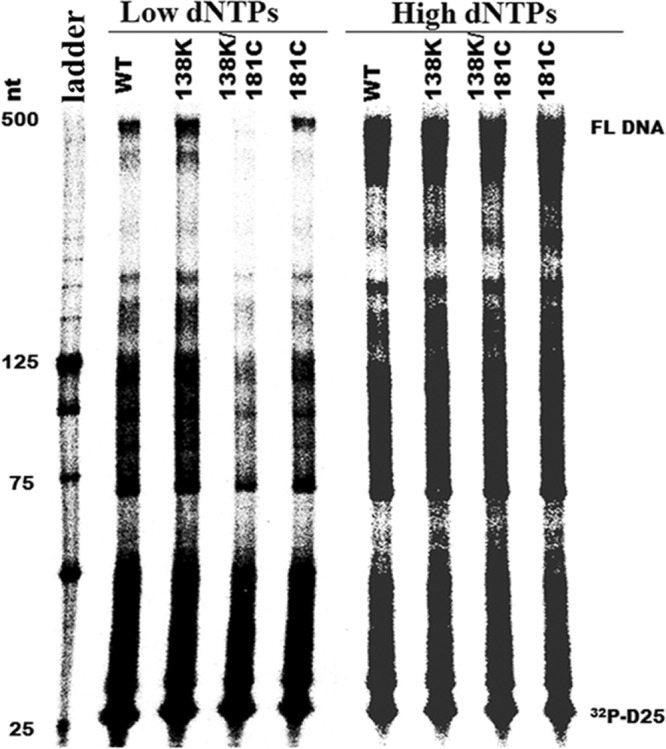
The Y181C mutation in HIV-1 RT diminishes enzyme processivity of E138K-containing enzymes at low dNTP concentrations. The processivity of purified recombinant RT enzymes was analyzed using 5′ end-labeled DNA primer (D25) annealed to an HIV-1 PBS RNA template as the substrate; the resulting full-length DNA (FL DNA) is 471 nt in size. Processivities were determined by the size distribution of DNA products in fixed-time experiments at two different concentrations of dNTPs in the presence of heparin trap. (A) Low dNTP concentration (0.5 μM). (B) High dNTP concentration (200 μM). Sizes of some fragments of the 32P-labeled 25-bp DNA ladder (Invitrogen, Burlington, ON, Canada) in nucleotide bases are indicated on the left side of the panel. All reaction products were resolved by denaturing 6% polyacrylamide gel electrophoresis and visualized by phosphorimaging. Positions of [32P]-labeled D25 primer (32P-D25) and the 471-nt full-length extension DNA (FL DNA) product are indicated on the right.
Y181C decreases the dNTP binding affinity of RT containing E138K.
Previous results of steady-state kinetic experiments showed that E138K RT has higher dNTP binding affinity (lower Km value) than WT and can restore the dNTP usage of RT containing the M184I/V mutations (47, 49). To assess the impact of the Y181C mutation on E138K RT dNTP binding affinity, steady-state kinetics studies were performed as described previously (47, 49). The results in Table 4 show that RT containing Y181C had a similar Km value to WT RT for dTTP, i.e., a difference of 1.1-fold. For E138K RT, the value was 0.42 of the WT, consistent with results showing that E138K has a lower Km than the WT (47, 49). For the double mutant E138K/Y181C, the fold change in Km was 2.2. Thus, Y181C seems to antagonize E138K in regard to dNTP binding affinity.
Table 4.
Kinetics parameters of recombinant RT enzymes as determined by steady-state kinetics analysisa
| Parameter | WT | E138K | Y181C | E138K/Y181C |
|---|---|---|---|---|
| kcat (min−1) | 15.7 ± 1.9 | 9.1 ± 0.9 | 16.8 ± 2.4 | 13.8 ± 1.4 |
| Km (μM) (FCb) | 5.2 ± 0.7 (1) | 2.2 ± 0.3 (0.42) | 5.7 ± 1.2 (1.1) | 11.5 ± 2.3 (2.2) |
The steady-state kinetics parameters kcat and Km for dTTP of WT HIV-1 RT and its mutant derivatives were determined using poly(rA)/p(dT)12-18 template/primers. The recombinant RT enzymes were purified in heterodimeric form, and mutations were introduced into specific subunits. Values are averages and SDs of representative experiments performed in triplicate.
Fold changes (FC) in Km for mutant RT variants compared to the Km of the WT.
Y181C does not restore the polymerization rate of E138K in processive DNA synthesis.
E138K caused a decrease in the polymerization rate at high but not low dNTP concentrations, and the addition of M184I/V restored the rate of processive DNA synthesis of RT E138K at high dNTP concentrations (47). To determine whether Y181C in RT can also compensate for a diminished polymerization rate, we performed RNA-dependent DNA polymerase reactions at high dNTP concentrations (200 μM) in time course experiments for 40 s, 60 s, and 90 s using the recombinant RT enzymes WT, E138K, Y181C, and E138K/Y181C. RT molecules were used at ∼20-fold excess over the substrate so that any RTs that dissociated from the primer terminus during synthesis would be rapidly replaced and the rate-limiting step would be nucleotide addition (16). The rate of polymerization was calculated as the number of nucleotide additions divided by the reaction time. The data show that the longest extension products were made after 60 s, and these were used to compare the polymerization rates of WT and mutant enzymes (Fig. 3). The WT RT enzyme showed a rate of processive DNA synthesis of ∼3.2 nt/s, consistent with previously reported maximum polymerization rates for WT HIV-1 RT (15, 16, 47). The Y181C RT variant had a rate of polymerization similar to that of WT RT, while the rate for E138K and the double mutant E138K/Y181C was ∼2.0 nt/s. These data confirm that E138K RT has an impaired rate of processive DNA synthesis and that the Y181C mutation cannot compensate for this deficit but rather exacerbates it.
Fig 3.
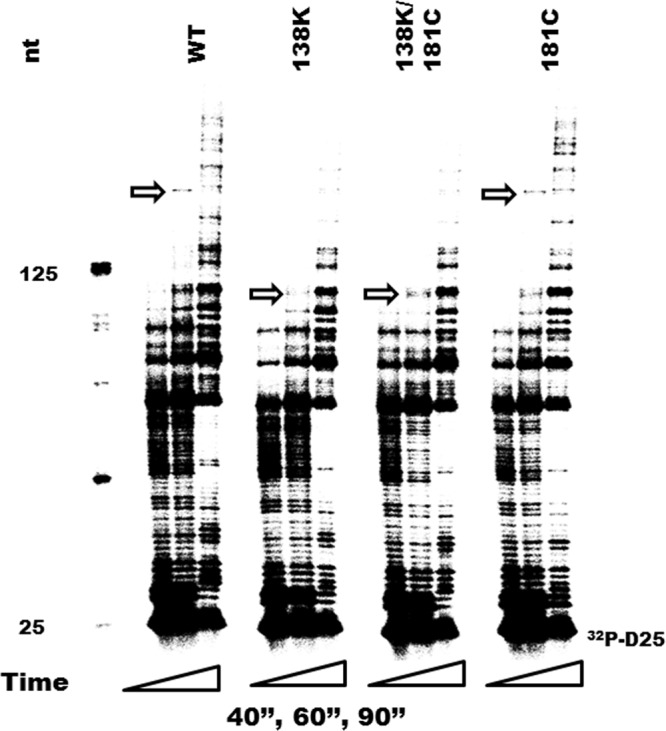
Time course experiments showing the effect of mutations Y181C, E138K, and E138K/Y181C in HIV-1 RT on rates of processive DNA polymerization. The [32P]-labeled D25 primer (32P-D25) was annealed to the 497-nt RNA template, and extension assays were performed at an excess of recombinant RT enzymes at dNTP concentrations of 200 μM. Reactions were stopped at 40 s (40”), 60 s (60′'), and 90 s (90′') (the first, second, and third columns for each mutation correspond to 40 s, 60 s, and 90 s, respectively). All reaction products were resolved by denaturing 6% polyacrylamide gel electrophoresis and visualized by phosphorimaging. Sizes of some fragments of the [32P]-labeled 25-bp DNA ladder (Invitrogen, Burlington, ON, Canada) in nucleotide bases are indicated on the left side of the panel. Positions of [32P]-labeled D25 primer (32P-D25) are indicated on the right. The longest extension products generated at 60 s are identified by arrows and indicate differences in polymerization rates.
DISCUSSION
HIV-1 that contains the Y181C mutation, associated with resistance to some NNRTIs, does not seem to be easily able to generate the E138K mutation following exposure to each of two DAPY next-generation NNRTIs, i.e., ETR and DAP; the latter is a compound that is being studied as an anti-HIV microbicide (35). Here, we have shown in cell culture selection experiments with ETR that Y181C prevents the emergence of E138K and have attempted to understand the mechanisms of antagonism between the Y181C and E138K mutations in HIV-1 RT. We have shown that Y181C antagonizes E138K by decreasing dNTP usage and impairing enzyme activity and processivity. E138K also diminishes levels of resistance conferred by Y181C to ETR.
In the phase III clinical trials (ECHO and THRIVE) that led to the approval of RPV, E138K was the most prevalent NNRTI mutation and was usually found together with M184I. As stated above, these mutations can mutually compensate each other in regard to RT polymerization rates at high dNTP concentrations (47). Here, we have shown that the interaction of Y181C and E138K is antagonistic, contrary to that seen between M184I and E138K. The Y181C mutation impairs enzyme processivity in the context of E138K by antagonizing dNTP usage. Coexistence of Y181C with E138K diminishes enzyme activity and the polymerization rate. These interactions render the doubly mutated Y181C RT impaired in regard to enzyme fitness and can explain the lower replication capacity associated with doubly mutated HIV-1, as also demonstrated here in cell culture experiments. It is noteworthy that Y181C was previously shown to be able to suppress the emergence of thymidine analogue mutations (TAMs) by antagonizing the ATP-mediated excision activity of RT containing TAMs (23, 36).
The presence of E138K alone and the combination of E138K plus Y181C yielded similar levels of resistance to ETR in our study, as determined in both phenotypic and biochemical assays. The fact that E138K plus Y181C does not enhance the resistance level compared to that with either E138K or Y181C alone demonstrates that the virus does not have a further advantage if it contains both mutations. This may be because this mutational combination results in impaired RT processivity and fitness, as shown here. Possibly, the two mutations together might improve the affinity of ETR to bind to RT compared to RT containing the single mutations alone, and further evaluation of this topic should be considered. At the same time, the differences between the EC50 and IC50 values for the double mutant and those for the Y181C mutant alone are not significant (results not shown). As ETR is a potent compound with high flexibility in structure, the fold decrease in resistance level observed, even a 2-fold change, might have clinical relevance. However, this is a case in which statistical analysis might be misleading, since the biochemical results do not seem to be significantly different. Two recent studies showed that the higher frequency of E138K and M184I among RPV plus FTC/TDF virologic failures in the ECHO and THRIVE studies is partly due to enhanced resistance to ETR/RPV for the double mutant E138K/M184I, albeit with less than a 2-fold change (20, 22). Thus, small differences in EC50 and/or IC50 that are not statistically significant can sometimes have clinical relevance, and it is probably best, therefore, to not emphasize this point.
We are currently studying how Y181C can decrease dNTP binding in a background of E138K. Y181 is located distant from the polymerase active site; the influence of Y181C on nucleotide binding might be due to long-range conformational changes at the RT active site. Pre-steady-state kinetic analyses showed that Y181C decreased ATP binding in a background of thymidine-associated mutations (TAMs) (36). Crystallography studies showed that TAMs, such as T215F/Y, can induce long-range conformational changes and that Y181C might disrupt these conformational changes (34). Previous steady-state kinetic analysis using a poly(rA)-oligo(dT) template showed that Y181C RT possessed an increased Km for dTTP (3) and only slightly decreased binding affinity (1.4-fold increase in Km) for the dGTP substrate using a poly(rC)-oligo(dG) template (11). Pre-steady-state kinetic analysis also showed that the Y181C mutation weakens nucleotide affinity by 2- to 3-fold compared to the WT (40). Detailed structural analysis of RTs containing the E138K/Y181C mutations, crystallized with dNTP and template/primer, may provide greater insight into the roles of these mutations in dNTP binding.
Neither K103N nor G190A has any impact on ETR (46, 48), and in the DUET studies, K103N had no effect on virological response to ETR (44). However, K103N was also shown in one study to confer increased susceptibility to ETR (27), an observation that was supported by docking analyses, which demonstrated increased affinity of ETR for RT containing K103N/S due to high conformational flexibility (1). Our study also showed that K103N can result in a lower IC50 for ETR relative to that for the WT.
Both ETR and DAP selection experiments in CBMCs using wild-type viruses showed that E138K is usually the first and the most prevalent mutation, irrespective of subtype (5, 35). E138K was also the most frequent NNRTI mutation to emerge in naïve patients failing RPV therapy in the ECHO and THRIVE studies. However, none of the other known NNTRI resistance mutations at codons 100, 101, 103, 106, 108, 190, etc., were observed with any regularity in either the cell culture selection studies or the clinical studies. Thus, it may be relevant to surmise that Y181C also prevents the emergence of these mutations under ETR pressure. A separate ETR resistance study (43) did not select E138K, but MT-2 rather than primary cells were used in that study. Our group has also shown enzymatically that the E138K mutation has a fitness advantage at low dNTP concentrations but is severely impaired in rates of DNA synthesis at high dNTP concentrations (47). Thus, dNTP pool size in the cells used in selection experiments can impact the selection of E138K, as has also recently been demonstrated in studies on both MT-2 cells and CBMCs, in which the latter generated E138K under ETR pressure while the former did not (49). It should be pointed out that CBMCs are more physiologically relevant than the MT-2 cell line. Although a different study (6) did show that E138K was selected in recombinant viruses containing both Y181C and V179I, it is possible that the presence of V179I might reverse the antagonism between E138K and Y181C, and this topic is worthy of investigation.
Emerging mutations in NNRTI-experienced patients failing ETR-containing regimens were V179I/F/L and Y181C (26). Y181C is usually found together with other resistance mutations, such as A98G, K101E/H, V108I, G190A/S, and H221Y. Tissue culture selection with ETR also showed that Y181C usually occurred with other resistance-associated mutations (TAMs), such as V179D/E/F. Although Y181C and V179F were also observed to coemerge in the DUET studies (42, 44), it is unclear whether other mutations that occur with Y181C can reverse the antagonism seen with E138K, and such studies are warranted.
In summary, we have shown that the Y181C mutation antagonizes E138K in regard to dNTP usage and enzyme processivity. The copresence of Y181C and E138K impairs enzyme fitness and also diminishes the level of resistance conferred by Y181C to ETR. To our knowledge, this is the first study to provide evidence that two ETR RAMs can have antagonistic interactions in terms of enzymatic function.
ACKNOWLEDGMENTS
We thank Stuart Le Grice for providing pRT6H-PROT DNA.
This work was supported by research grants from the Canadian Institutes of Health Research (CIHR).
We have no conflicts of interest to declare.
Footnotes
Published ahead of print 19 September 2012
REFERENCES
- 1. Alcaro S, et al. 2011. Docking analysis and resistance evaluation of clinically relevant mutations associated with the HIV-1 non-nucleoside reverse transcriptase inhibitors nevirapine, efavirenz and etravirine. ChemMedChem 6:2203–2213 [DOI] [PubMed] [Google Scholar]
- 2. Ambrose Z, et al. 2009. The human immunodeficiency virus type 1 nonnucleoside reverse transcriptase inhibitor resistance mutation I132M confers hypersensitivity to nucleoside analogs. J. Virol. 83:3826–3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Archer RH, et al. 2000. Mutants of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase resistant to nonnucleoside reverse transcriptase inhibitors demonstrate altered rates of RNase H cleavage that correlate with HIV-1 replication fitness in cell culture. J. Virol. 74:8390–8401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arts EJ, et al. 1994. Comparison of deoxyoligonucleotide and tRNA(Lys-3) as primers in an endogenous human immunodeficiency virus-1 in vitro reverse transcription/template-switching reaction. J. Biol. Chem. 269:14672–14680 [PubMed] [Google Scholar]
- 5. Asahchop EL, et al. 2011. Characterization of the E138K resistance mutation in HIV-1 reverse transcriptase conferring susceptibility to etravirine in B and non-B HIV-1 subtypes. Antimicrob. Agents Chemother. 55:600–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Azijn H, et al. 2010. TMC278, a next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI), active against wild-type and NNRTI-resistant HIV-1. Antimicrob. Agents Chemother. 54:718–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Back NK, et al. 1996. Reduced replication of 3TC-resistant HIV-1 variants in primary cells due to a processivity defect of the reverse transcriptase enzyme. EMBO J. 15:4040–4049 [PMC free article] [PubMed] [Google Scholar]
- 8. Benhamida J, Chappey C, Coakley E, Parkin NT. 2008. HIV-I genotype algorithms for prediction of etravirine susceptibility: novel mutations and weighting factors identified through correlations to phenotype. Antivir. Ther. 13:A142 [Google Scholar]
- 9. Caliendo AM, et al. 1996. Effects of zidovudine-selected human immunodeficiency virus type 1 reverse transcriptase amino acid substitutions on processive DNA synthesis and viral replication. J. Virol. 70:2146–2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Croxtall JD. 2012. Etravirine: a review of its use in the management of treatment-experienced patients with HIV-1 infection. Drugs 72:847–869 [DOI] [PubMed] [Google Scholar]
- 11. Debyser Z, et al. 1993. Kinetics of different human immunodeficiency virus type 1 reverse transcriptases resistant to human immunodeficiency virus type 1-specific reverse transcriptase inhibitors. Mol. Pharmacol. 43:521–526 [PubMed] [Google Scholar]
- 12. Delaugerre C, et al. 2001. Resistance profile and cross-resistance of HIV-1 among patients failing a non-nucleoside reverse transcriptase inhibitor-containing regimen. J. Med. Virol. 65:445–448 [PubMed] [Google Scholar]
- 13. Esnouf R, et al. 1995. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat. Struct. Biol. 2:303–308 [DOI] [PubMed] [Google Scholar]
- 14. Esnouf RM, et al. 1997. Unique features in the structure of the complex between HIV-1 reverse transcriptase and the bis(heteroaryl)piperazine (BHAP) U-90152 explain resistance mutations for this nonnucleoside inhibitor. Proc. Natl. Acad. Sci. U. S. A. 94:3984–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fuentes GM, Palaniappan C, Fay PJ, Bambara RA. 1996. Strand displacement synthesis in the central polypurine tract region of HIV-1 promotes DNA to DNA strand transfer recombination. J. Biol. Chem. 271:29605–29611 [DOI] [PubMed] [Google Scholar]
- 16. Gao L, et al. 2008. Apparent defects in processive DNA synthesis, strand transfer, and primer elongation of Met-184 mutants of HIV-1 reverse transcriptase derive solely from a dNTP utilization defect. J. Biol. Chem. 283:9196–9205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goff SP. 1990. Retroviral reverse transcriptase: synthesis, structure, and function. J. Acquir. Immune Defic. Syndr. 3:817–831 [PubMed] [Google Scholar]
- 18. Hirsch MS, et al. 2000. Antiretroviral drug resistance testing in adult HIV-1 infection: recommendations of an International AIDS Society-USA panel. JAMA 283:2417–2426 [DOI] [PubMed] [Google Scholar]
- 19. Hsiou Y, et al. 1996. Structure of unliganded HIV-1 reverse transcriptase at 2.7 A resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure 4:853–860 [DOI] [PubMed] [Google Scholar]
- 20. Hu Z, Kuritzkes DR. 2011. Interaction of reverse transcriptase (RT) mutations conferring resistance to lamivudine and etravirine: effects on fitness and RT activity of human immunodeficiency virus type 1. J. Virol. 85:11309–11314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Janssen PA, et al. 2005. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (R278474, rilpivirine). J. Med. Chem. 48:1901–1909 [DOI] [PubMed] [Google Scholar]
- 22. Kulkarni R, et al. 2012. The HIV-1 reverse transcriptase M184I mutation enhances the E138K-associated resistance to rilpivirine and decreases viral fitness. J. Acquir. Immune Defic. Syndr. 59:47–54 [DOI] [PubMed] [Google Scholar]
- 23. Larder BA. 1992. 3′-Azido-3′-deoxythymidine resistance suppressed by a mutation conferring human immunodeficiency virus type 1 resistance to nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 36:2664–2669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Le Grice SF, Cameron CE, Benkovic SJ. 1995. Purification and characterization of human immunodeficiency virus type 1 reverse transcriptase. Methods Enzymol. 262:130–144 [DOI] [PubMed] [Google Scholar]
- 25. Le Grice SF, Gruninger-Leitch F. 1990. Rapid purification of homodimer and heterodimer HIV-1 reverse transcriptase by metal chelate affinity chromatography. Eur. J. Biochem. 187:307–314 [DOI] [PubMed] [Google Scholar]
- 26. Marcelin AG, et al. 2012. Emerging mutations and associated factors in patients displaying treatment failure on an etravirine-containing regimen. Antivir. Ther. 17:119–123 [DOI] [PubMed] [Google Scholar]
- 27. Marcelin AG, et al. 2010. Factors associated with virological response to etravirine in nonnucleoside reverse transcriptase inhibitor-experienced HIV-1-infected patients. Antimicrob. Agents Chemother. 54:72–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Naeger LK, Margot NA, Miller MD. 2001. Increased drug susceptibility of HIV-1 reverse transcriptase mutants containing M184V and zidovudine-associated mutations: analysis of enzyme processivity, chain-terminator removal and viral replication. Antivir. Ther. 6:115–126 [PubMed] [Google Scholar]
- 29. Nissley DV, et al. 2007. Characterization of novel non-nucleoside reverse transcriptase (RT) inhibitor resistance mutations at residues 132 and 135 in the 51 kDa subunit of HIV-1 RT. Biochem. J. 404:151–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oliveira M, Brenner BG, Wainberg MA. 2009. Isolation of drug-resistant mutant HIV variants using tissue culture drug selection. Methods Mol. Biol. 485:427–433 [DOI] [PubMed] [Google Scholar]
- 31. Pandey PK, et al. 2002. Insertion of a small peptide of six amino acids into the β7-β8 loop of the p51 subunit of HIV-1 reverse transcriptase perturbs the heterodimer and affects its activities. BMC Biochem. 3:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pandey PK, Kaushik N, Talele TT, Yadav PN, Pandey VN. 2001. The β7-β8 loop of the p51 subunit in the heterodimeric (p66/p51) human immunodeficiency virus type 1 reverse transcriptase is essential for the catalytic function of the p66 subunit. Biochemistry 40:9505–9512 [DOI] [PubMed] [Google Scholar]
- 33. Quan Y, et al. 2003. Drug resistance profiles of recombinant reverse transcriptases from human immunodeficiency virus type 1 subtypes A/E, B, and C. AIDS Res. Hum. Retroviruses 19:743–753 [DOI] [PubMed] [Google Scholar]
- 34. Ren J, et al. 1998. Crystal structures of HIV-1 reverse transcriptase in complex with carboxanilide derivatives. Biochemistry 37:14394–14403 [DOI] [PubMed] [Google Scholar]
- 35. Schader SM, et al. 2012. In vitro resistance profile of the candidate HIV-1 microbicide drug dapivirine. Antimicrob. Agents Chemother. 56:751–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Selmi B, et al. 2003. The Y181C substitution in 3′-azido-3′-deoxythymidine-resistant human immunodeficiency virus, type 1, reverse transcriptase suppresses the ATP-mediated repair of the 3′-azido-3′-deoxythymidine 5′-monophosphate-terminated primer. J. Biol. Chem. 278:40464–40472 [DOI] [PubMed] [Google Scholar]
- 37. Sharma PL, Crumpacker CS. 1999. Decreased processivity of human immunodeficiency virus type 1 reverse transcriptase (RT) containing didanosine-selected mutation Leu74Val: a comparative analysis of RT variants Leu74Val and lamivudine-selected Met184Val. J. Virol. 73:8448–8456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sluis-Cremer N, Temiz NA, Bahar I. 2004. Conformational changes in HIV-1 reverse transcriptase induced by nonnucleoside reverse transcriptase inhibitor binding. Curr. HIV Res. 2:323–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smerdon SJ, et al. 1994. Structure of the binding site for nonnucleoside inhibitors of the reverse transcriptase of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U. S. A. 91:3911–3915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Spence RA, Anderson KS, Johnson KA. 1996. HIV-1 reverse transcriptase resistance to nonnucleoside inhibitors. Biochemistry 35:1054–1063 [DOI] [PubMed] [Google Scholar]
- 41. Tambuyzer L, Nijs S, Daems B, Picchio G, Vingerhoets J. 2011. Effect of mutations at position E138 in HIV-1 reverse transcriptase on phenotypic susceptibility and virologic response to etravirine. J. Acquir. Immune Defic. Syndr. 58:18–22 [DOI] [PubMed] [Google Scholar]
- 42. Tambuyzer L, et al. 2010. Characterization of genotypic and phenotypic changes in HIV-1-infected patients with virologic failure on an etravirine-containing regimen in the DUET-1 and DUET-2 clinical studies. AIDS Res. Hum. Retroviruses 26:1197–1205 [DOI] [PubMed] [Google Scholar]
- 43. Vingerhoets J, et al. 2005. TMC125 displays a high genetic barrier to the development of resistance: evidence from in vitro selection experiments. J. Virol. 79:12773–12782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vingerhoets J, et al. 2010. Resistance profile of etravirine: combined analysis of baseline genotypic and phenotypic data from the randomized, controlled phase III clinical studies. AIDS 24:503–514 [DOI] [PubMed] [Google Scholar]
- 45. Wainberg MA. 2003. HIV resistance to nevirapine and other non-nucleoside reverse transcriptase inhibitors. J. Acquir. Immune Defic. Syndr. 34(Suppl 1):S2–S7 [DOI] [PubMed] [Google Scholar]
- 46. Xu H, et al. 2009. Human immunodeficiency virus type 1 recombinant reverse transcriptase enzymes containing the G190A and Y181C resistance mutations remain sensitive to etravirine. Antimicrob. Agents Chemother. 53:4667–4672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu HT, et al. 2011. Compensation by the E138K mutation in HIV-1 reverse transcriptase for deficits in viral replication capacity and enzyme processivity associated with the M184I/V mutations. J. Virol. 85:11300–11308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu HT, Oliveira M, Quan Y, Bar-Magen T, Wainberg MA. 2010. Differential impact of the HIV-1 non-nucleoside reverse transcriptase inhibitor mutations K103N and M230L on viral replication and enzyme function. J. Antimicrob. Chemother. 65:2291–2299 [DOI] [PubMed] [Google Scholar]
- 49. Xu HT, et al. 2012. Subunit-selective mutational analysis and tissue culture evaluations of the interactions of the E138K and M184I mutations in HIV-1 reverse transcriptase. J. Virol. 86:8422–8431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu HT, et al. 2010. Comparative biochemical analysis of recombinant reverse transcriptase enzymes of HIV-1 subtype B and subtype C. Retrovirology 7:80. [DOI] [PMC free article] [PubMed] [Google Scholar]