

Abstract
Autophagy is an intracellular pathway that can contribute to innate antiviral immunity by delivering viruses to lysosomes for degradation or can be beneficial for viruses by providing specialized membranes for virus replication. Here, we show that the picornavirus foot-and-mouth disease virus (FMDV) induces the formation of autophagosomes. Induction was dependent on Atg5, involved processing of LC3 to LC3II, and led to a redistribution of LC3 from the cytosol to punctate vesicles indicative of authentic autophagosomes. Furthermore, FMDV yields were reduced in cells lacking Atg5, suggesting that autophagy may facilitate FMDV infection. However, induction of autophagosomes by FMDV appeared to differ from starvation, as the generation of LC3 punctae was not inhibited by wortmannin, implying that FMDV-induced autophagosome formation does not require the class III phosphatidylinositol 3-kinase (PI3-kinase) activity of vps34. Unlike other picornaviruses, for which there is strong evidence that autophagosome formation is linked to expression of viral nonstructural proteins, FMDV induced autophagosomes very early during infection. Furthermore, autophagosomes could be triggered by either UV-inactivated virus or empty FMDV capsids, suggesting that autophagosome formation was activated during cell entry. Unlike other picornaviruses, FMDV-induced autophagosomes did not colocalize with the viral 3A or 3D protein. In contrast, ∼50% of the autophagosomes induced by FMDV colocalized with VP1. LC3 and VP1 also colocalized with the cellular adaptor protein p62, which normally targets ubiquitinated proteins to autophagosomes. These results suggest that FMDV induces autophagosomes during cell entry to facilitate infection, but not to provide membranes for replication.
INTRODUCTION
Macroautophagy is a highly conserved degradation pathway that recycles cytoplasmic organelles and proteins in response to starvation (48). During macroautophagy (here referred to as autophagy), cup-shaped isolation membranes expand to form double-membrane autophagosomes that engulf cytosolic cargo and then fuse with late endosomes or lysosomes for degradation (49). Autophagosome formation is regulated by the mTORC1 (mammalian target of rapamycin complex 1) kinase, which is a negative regulator of autophagy. Autophagy can therefore be activated in the absence of starvation by mTORC1 inhibitors, such as rapamycin (46) or torin (43). mTORC1 regulates a multimeric complex (the ULK-1 complex) made up of ULK-1 (unc-51-like kinase), FIP200 (focal adhesion kinase family interacting protein of 200 kDa), Atg13, and Atg101 (4, 8–11, 13, 17, 26). Other key autophagy components, such as the phosphatidylinositol 3-kinase (PI3-kinase) complex (Atg6/beclin-Atg14-Vps15-Vps34) and the Atg12 (Atg12-Atg5-Atg16) and LC3 conjugation systems operate downstream of ULK-1 (13, 47). Increased PI3-kinase activity leads to recruitment of the Atg12-Atg5-Atg16 complex to isolation membranes and lipidation of LC3 to generate LC3II. Membrane association of LC3II facilitates expansion of the isolation membrane and formation of autophagosomes. Engulfment of the cytosol is generally believed to be nonspecific, but ubiquitinylated proteins can be directed to autophagosomes by the linker protein p62 (also known as sequestersome 1), which binds both polyubiquitinated proteins and membrane-associated LC3II (32).
Delivery of viruses to lysosomes by autophagy can play a protective role during infection when it leads to virus degradation and increased exposure of antigens to the innate and acquired immune systems. Sindbis virus, for example, activates autophagy in neurones (31), and overexpression of beclin/Atg6 in neurones protects against Sindbis virus infection (22). Interestingly, Sindbis virus capsids bind p62 in vitro, raising the possibility that p62 mediates direct delivery of viruses to autophagosomes (31). This pathway may work in vivo, because mice lacking neuronal expression of Atg5 suffer a fatal infection that correlates with slowed clearance of viral antigens from neurones. Similarly, herpesviruses are taken into autophagosomes (39). The neurovirulence seen during herpes simplex virus infection is determined by the viral protein ICP34.5, which binds beclin/Atg6 and prevents activation of autophagy. Viruses lacking ICP34.5 show reduced replication in the central nervous system and lowered neurovirulence and are cleared rapidly by the adaptive immune response (30). Analogous studies in Drosophila show that autophagy can protect against vesicular stomatitis virus infection (37).
Picornaviruses are a family of single-stranded positive-sense RNA viruses that includes many important pathogens of humans and animals, such as poliovirus, coxsackieviruses, human rhinoviruses (HRV), and foot-and-mouth disease virus (FMDV). Viruses of the genus Enterovirus, family Picornaviridae (e.g., poliovirus and coxsackieviruses), generate double-membrane vesicles resembling autophagosomes both in cell culture (6, 45) and in vivo (18), and the nonstructural proteins required for replication of human pathogens, such as coxsackievirus and poliovirus, associate with autophagosomes (12, 16, 45). Double-membrane vesicles can also be induced by coexpression of poliovirus nonstructural proteins 2BC and 3A (40), and when expressed alone, poliovirus 2BC can induce lipidation of LC3 (42). Studies with poliovirus suggest that autophagy may actually promote rather than protect against infection, as activation of autophagy appears to increase intracellular virus yields while inhibition of autophagy, through silencing essential atg genes, reduces virus yields (16, 41). Current models suggest that for poliovirus, autophagosomes are induced either to provide new membranes for assembly of the replication complex or to facilitate nonlytic virus release from cells (16). Similar studies with coxsackieviruses and enterovirus 71 show that autophagy is also activated during infection to enhance the efficiency of viral replication (12, 16, 50). Although the above-mentioned studies suggest that picornaviruses activate autophagy to promote their replication, studies investigating the role of autophagy in HRV infection provide a complex picture. HRV1 does not activate autophagy, and modulation of autophagy does not affect replication (20). Similar results have been reported for HRV2 (3), but other studies suggest that replication of HRV2 is facilitated by autophagy (16, 20).
FMDV is the type species within the genus Aphthovirus in the family Picornaviridae. O'Donnell et al. (29) reported that FMDV triggers autophagy and that autophagy may facilitate virus replication, as inhibitors of autophagy decreased viral replication. While the overall genome organization and the viral proteins of FMDV and enteroviruses share a number of conserved characteristics, there are important differences in the functions of the nonstructural proteins. This is particularly apparent for the 2B, 2BC, and 3A proteins, which have been implicated in the regulation of autophagy by enteroviruses (40, 42). The FMDV 2B and 3A reading frames are approximately twice the length of their enterovirus counterparts, and there are functional differences in the proteins. The poliovirus 3A protein blocks protein secretion between the endoplasmic reticulum (ER) and the Golgi apparatus (7), but the 3A proteins of other picornaviruses, such as FMDV and the human pathogens hepatitis A virus and human rhinovirus 14, are unable to block membrane traffic (5, 27), and for FMDV, protein secretion is blocked by 2BC (27) or coexpression of 2B and 2C (28). Given the functional differences between the nonstructural proteins of FMDV and the enteroviruses and the apparent differences in the abilities of enteroviruses to trigger autophagy during infection, we further investigated the effect of FMDV on autophagy.
MATERIALS AND METHODS
Cells and viruses.
Chinese hamster ovary cells expressing a green fluorescent protein (GFP)-tagged LC3 (CHO GFP-LC3) (38) were cultivated in Ham's F-12 nutrient medium (Sigma) supplemented with 10% fetal calf serum (FCS). Mouse embryonic fibroblasts (MEFs) and Atg5 knockout MEFs (atg5−/− MEFs) were cultivated in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and nonessential amino acids. IBRS-2 cells were cultivated in Glasgow's modified Eagle's medium supplemented with 10% adult bovine serum. All cell culture media were further supplemented with 20 mM glutamine, penicillin (100 SI units/ml), and streptomycin (100 μg/ml). Working stocks of FMDV O1BFS and O1Kcad2 were prepared using baby hamster kidney (BHK) 21 and primary bovine thyroid cells, respectively. FMDV type O empty capsids were synthesized using a vaccinia virus expression system and purified using sucrose gradients, as described previously (1).
Antibodies and reagents.
The monoclonal antibodies 2C2, 3B5, and D9, which recognize the FMDV 3A, 3D, and VP1 proteins (type O viruses), respectively (24), were gifts from Emiliana Brocchi (IZS, Brescia, Italy). The rabbit polyclonal antibody against LC3B and the anti-gamma-tubulin antibody used to detect the microtubule-organizing center (MTOC) were both obtained from Sigma. A guinea pig polyclonal antibody raised against the C-terminal region was used to detect p62 (Progen). Alexa Fluor-conjugated secondary antibodies were from Invitrogen. Stock solutions of nocodazole (Sigma), concanamycin A (Fluka), wortmannin (Sigma), and scriptaid (Calbiochem) were prepared in dimethyl sulfoxide (DMSO) at 40 mM, 10 mM, 20 μM, and100 mM, respectively. DMSO alone did not affect FMDV yields or the distribution of LC3 or the viral 3A protein.
UV inactivation of virus.
UV inactivation was used to cross-link the FMDV RNA genome and prevent replication while retaining the capsid structure. The protocol used was as described previously (34). Briefly, the virus was transferred to a tissue culture plate on ice and exposed to a UV light source (UVP, Upland, CA) at a wavelength of 256 nm for 12 min at a distance of 10 cm from the UV source. Loss of infectivity was confirmed by the absence of cytopathic effect (CPE) on prolonged exposure to BHK cells, in addition to the absence of labeling for FMDV nonstructural protein 3A under a confocal microscope following exposure to CHO cells expressing GFP-LC3 for 5 h.
Western blotting.
CHO cells expressing GFP-LC3 were lysed in 120 mM NaCl, 50 mM Tris, pH 7.5, 0.5% octylphenoxypolyethoxyethanol (IGEPAL), and 3× SDS-PAGE sample buffer (New England BioLabs) was added to the lysate. Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were probed with antibodies against LC3 or p62.
Drug treatment and infection of cells.
Inhibitors (nocodazole, scriptaid, or wortmannin) were added to cells for 0.5 h (nocodazole and scriptaid) or 1 h (wortmannin) prior to addition of virus and were present throughout infection. Concanamycin A was added to infected cells 1 h postinfection. The cells were inoculated with FMDV O1Kcad2 (multiplicity of infection [MOI] = 0.8) or O1BFS (MOI = 2), UV-inactivated FMDV type O1BFS (at a dilution equivalent to an MOI of 2), or empty FMDV type O capsids (50 μg/ml) diluted in cell culture medium containing 1% FCS for 1 h at 37°C. Unbound virus or capsid was removed by washing, and the cells were returned to 37°C in cell culture medium with reduced FCS for the remainder of the infection time. Mock treatments (including mock infections) were carried out as described above using cell culture medium containing 1% FCS.
Activation of autophagy by starvation.
To activate autophagy by starvation, the cells were washed with phosphate-buffered saline (PBS) and incubated in Earle's balanced salt solution (EBSS) (which lacks amino acids) for 2 h at 37°C. Control cells were incubated in cell culture medium.
Immunofluorescence microscopy.
Following fixation, cells on glass coverslips were incubated for 15 min with 0.1% Triton X-100 in PBS, washed, and incubated in block buffer (10 mM Tris, 150 mM NaCl, 1 mM CaCl2, 0.5 mM MgCl2, 10% normal goat serum, and 1% gelatin) for 0.5 h. Primary antibodies were added for 1 h in block buffer. The cells were washed and incubated with Alexa Fluor-conjugated secondary antibodies in block buffer for 45 min. After washing, the coverslips were mounted onto microscope slides using Vectashield mounting medium with DAPI (4′,6-diamidino-2-phenylindole) (Vector Laboratories, Peterborough, United Kingdom). All data were collected sequentially to eliminate cross talk of the fluorescent dyes using a Leica SP2 scanning laser confocal microscope. For pixel density analysis, images were analyzed using Imaris ×64 7.2.3 software (Bitplane Software Incorporated). Punctate structures within the images were identified using the “spots” function, and larger structures, such as nuclei, were identified using the “surfaces” function within the software. These object layers were used to create the rendered images and to assimilate data concerning vesicle diameter and number.
Growth curves.
MEFs or atg5−/− MEFs were incubated for 1 h at 37°C with FMDV O1BFS in reduced-serum (1%) cell culture medium (MOI = 5) to allow virus internalization. The cells were then washed using a low-pH buffer (0.1 M citric acid in 140 mM NaCl, pH 5.2) to inactivate extracellular virus. The wells were washed with reduced-serum cell culture medium to restore the pH to neutrality, and infection continued at 37°C. At the indicated times, supernatants were taken, and the cells were freeze-thawed three times to release intracellular virus. The extracellular and intracellular virus titers were determined by plaque assay on BHK cells.
RESULTS
FMDV infection causes redistribution and lipidation of LC3.
Activation of autophagy leads to the formation of autophagosomes containing LC3 that appear as LC3 punctae in fluorescence micrographs. Figure 1A shows the response to FMDV infection of CHO cells expressing LC3 as a fusion protein with GFP (CHO GFP-LC3). The top row (Fig 1A, i to iv) shows cells viewed at low magnification at increasing times postinfection, while the middle row (Fig 1A, v to viii) shows cells at high magnification counterlabeled for FMDV nonstructural protein 3A as a marker for virus replication. Punctate GFP-LC3 signals were absent at the time of infection (Fig 1A, i and v) but were present at 1 h after addition of virus (Fig 1A, ii and vi). In ∼30% of the cells, the punctae were dispersed throughout the cytosol, while in other cells, the signal was concentrated close to the nucleus. By 2 h postinfection, when 3A was first detected (Fig 1A, vii), the GFP-LC3 signal was concentrated close to the nucleus in >95% of the cells that showed GFP-LC3 punctae (Fig 1A, iii and vii). At 3 h postinfection, when the levels of 3A were high, the GFP-LC3 signal was greatly diminished and dispersed throughout the cytoplasm (Fig 1A, iv and viii). These results show that FMDV generates LC3 punctae that resemble autophagosomes very early during infection, prior to the detection of the viral 3A protein.
Fig 1.

Redistribution of LC3 and p62 and LC3 processing during FMDV infection. CHO GFP-LC3 cells were infected for increasing times with FMDV O1BFS (MOI = 2). (A) Autophagosome production was analyzed by following the redistribution of GFP-LC3 (green). Low-magnification (i to iv) and high-magnification (v to viii) images are shown. FMDV nonstructural protein 3A (red) was detected using antibody 2C2. Labeling for 3A is not shown for images i to iv so that the GFP-LC3 signal can be clearly seen. Labeling for p62 (red) is shown in images ix to xii. The signal for GFP-LC3 is not shown for images ix to xii so that p62 labeling can be clearly seen. Cell nuclei are blue. Scale bars = 30 μm (i to iv) or 10 μm (v to xii). (B) Infected cell lysates were separated by SDS-PAGE and analyzed by Western blotting using an antibody to LC3. The upper and lower blots show the conversion of GFP-tagged and endogenous LC3, respectively. (C) Infected cell lysates were separated by SDS-PAGE and analyzed by Western blotting using an antibody to p62.
Certain proteins are recognized for delivery to autophagosomes by an “autophagy receptor” protein called p62 that binds to both ubiquitinated proteins and LC3 in the autophagosome membrane (32). The distribution of p62 was followed during FMDV infection as a second marker for formation of autophagosomes. Figure 1A, ix to xii, shows that infection of CHO GFP-LC3 cells with FMDV generates punctate structures containing p62. These were first seen at 1 h, when they were dispersed through the cytoplasm (Fig 1A, x), and after 2 h, they clustered close to the nucleus (Fig 1A, xi) in a pattern that resembled that for LC3. Also, similar to LC3, the p62 signal diminished and dispersed at 3 h postinfection (Fig 1A, xii).
We also investigated whether FMDV induced lipidation of LC3, which is essential for translocation of LC3 from the cytosol to autophagosomes. The lipidated form of LC3 (LC3II) has increased mobility on SDS-PAGE relative to the unlipidated form. Figure 1B (top) shows a Western blot using an antibody to LC3 to detect GFP-LC3. The levels of LC3II clearly increased following infection, and consistent with the results described above, increased levels were first seen 30 min postinfection and peaked at 2 to 3 h. Figure 1B, bottom, shows that endogenous LC3 is also converted to the lipidated form in infected cells with kinetics similar to those of the GFP-tagged protein. Figure 1A shows that at 3 h postinfection, the signal for p62 is diminished. Figure 1C shows a Western blot for p62 and that the signal for p62 is also diminished at 3 h postinfection. Together, these observations suggest that a proportion of p62 is degraded during infection, which suggests that FMDV may trigger authentic autophagy.
Wortmannin does not inhibit induction of GFP-LC3 punctae by FMDV.
The class III PI3-kinase vps34 is required for the formation of isolation membranes in response to starvation (23) and is inhibited by the PI3-kinase inhibitor wortmannin. We compared the effects of wortmannin on the generation of GFP-LC3 punctae in CHO cells in response to starvation or incubation with FMDV (Fig. 2). Wortmannin prevented the formation of GFP-LC3 punctae when cells were starved (Fig. 2i and ii), but the drug was unable to prevent formation of GFP-LC3 punctae in response to FMDV (Fig. 2iii and iv). These data suggest that autophagosomes induced by FMDV do not require class III PI3-kinase activity, and therefore, their formation may be independent of the vps34-Atg14L-p150-Beclin1 complex.
Fig 2.
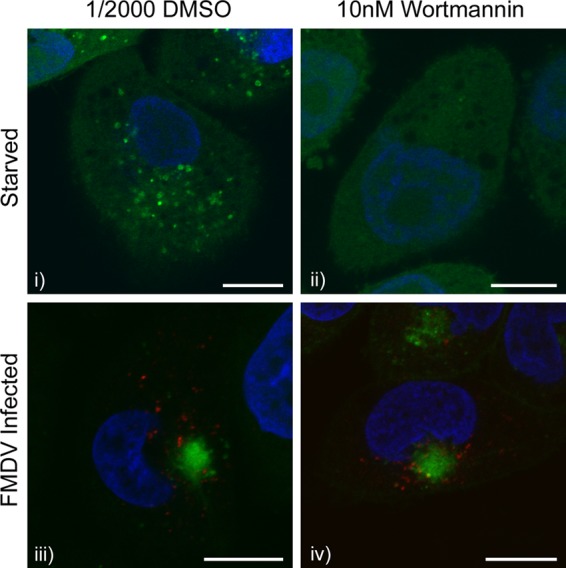
Redistribution of LC3 induced by FMDV is insensitive to wortmannin. CHO GFP-LC3 cells were pretreated with 1/2,000 DMSO (solvent control) (i and iii) or 10 nM wortmannin (ii and iv) for 1 h. The cells were then either starved for 2 h in EBSS (i and ii) or infected with FMDV O1BFS (MOI = 2) for 2 h (iii and iv) in the presence of wortmannin. Cells were fixed and processed for immunofluorescence microscopy. LC3 was visualized using the natural fluorescence of GFP (green). (iii and iv) FMDV nonstructural protein 3A (red) was detected by immunostaining using antibody 2C2. Cell nuclei are blue. Scale bars = 10 μm.
Redistribution of endogenous LC3 by FMDV requires the autophagy protein Atg5.
Generation of LC3 punctae does not necessarily indicate activation of autophagy. For example, severe acute respiratory syndrome (SARS) coronavirus induces the association of the nonlipidated form of LC3 with membranes rather than LC3II (33). These LC3-positive structures are known as edemosomes and transport proteins from the ER to endosomes. They form independently of autophagy and do not require the essential autophagy protein Atg5 (33). Given that FMDV nonstructural proteins locate to the ER and modulate ER structure and function (27, 28), it was possible that LC3 punctae induced by FMDV could represent edemosomes rather than autophagosomes. The effect of FMDV on LC3 distribution was therefore analyzed further by immunostaining for endogenous LC3 in MEFs that lack Atg5 (atg5−/− MEFs). Figure 3 shows that under resting conditions, levels of LC3 punctae are low in wild-type (wt) MEFs (Fig. 3i and iii) and increase markedly in response to starvation (Fig. 3ii) or FMDV infection (Fig. 3iv). When the experiment was repeated using atg5−/− cells, LC3 punctae were not generated following starvation (Fig. 3vi) or FMDV infection (Fig. 3viii). The LC3 punctae induced by FMDV were therefore dependent on Atg5, which supports the conclusion that the LC3 punctae are autophagosomes rather than edemosomes.
Fig 3.
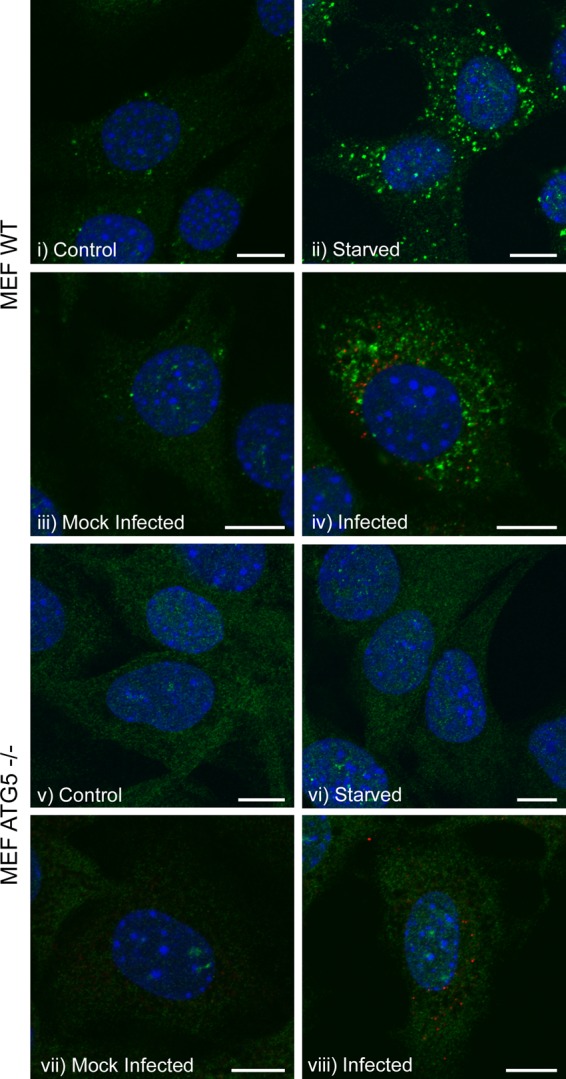
Redistribution of LC3 induced by FMDV is dependent on Atg5. MEFs or atg5−/− MEFs were either starved for 2 h in EBSS (ii and vi, respectively) or infected with FMDV O1BFS (MOI = 2) for 2 h (iv and viii, respectively). (i and v, and iii and vii) Control and mock-infected cells, respectively. Cells were fixed and processed for immunofluorescence microscopy. Autophagosome production was analyzed by immunostaining for endogenous LC3 (green), and FMDV nonstructural protein 3A (red) was detected using antibody 2C2. Cell nuclei are blue. Scale bars = 10 μm.
LC3 punctae are induced in the absence of virus replication.
The LC3-containing punctae induced by FMDV were detected very early during infection and before expression of 3A (Fig. 1). This led us to test whether autophagy was activated during cell entry rather than as a consequence of virus replication. FMDV was inactivated by UV irradiation, and loss of infectivity was confirmed by the absence of immunofluorescence signal for 3A (at 5 h postinfection) and loss of cytopathic effect following prolonged exposure to BHK cells (data not shown). Figure 4 shows that UV-inactivated FMDV induced punctate LC3 signals in CHO GFP-LC3 cells (Fig. 4ii) that were similar in distribution to those induced by live virus (Fig. 1). FMDV empty capsids that lacked a viral genome also induced LC3 punctae in CHO GFP-LC3 cells (Fig. 4iv). LC3 punctae were also induced by UV-inactivated FMDV in wild-type MEFs (Fig. 4vi), but not in atg5−/− MEFs (Fig. 4viii). These results confirm that formation of LC3 punctae is dependent on Atg5 and show that FMDV replication is not required to induce LC3 punctae.
Fig 4.
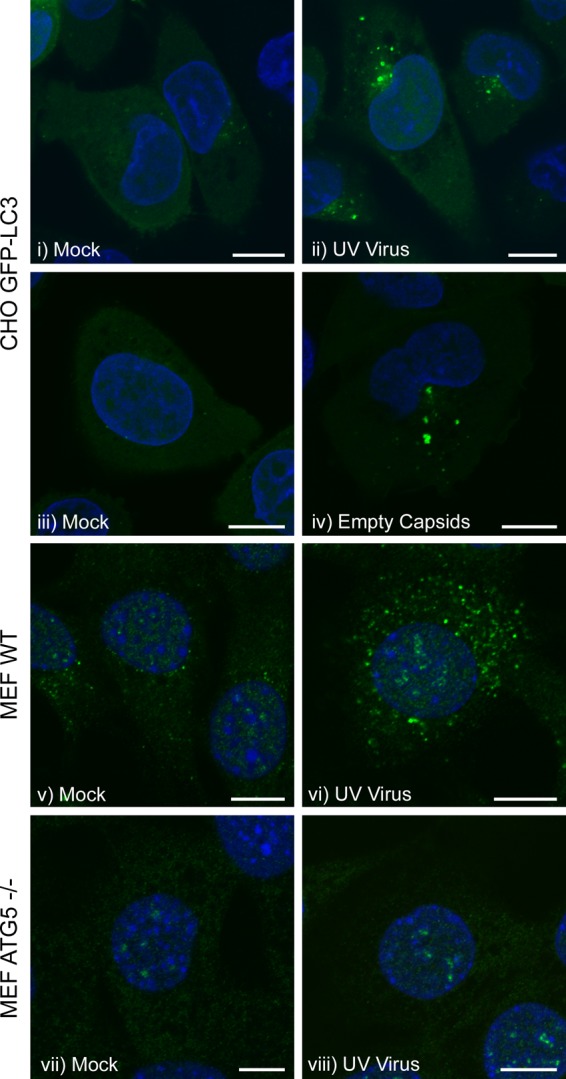
Effect of FMDV capsids and UV-inactivated FMDV on the distribution of LC3. (i to iv) CHO GFP-LC3 cells. (v and vi) MEFs. (vii and viii) atg5−/− MEFs. The cells were incubated for 2 h with UV-inactivated FMDV O1BFS (at a dilution equivalent to an MOI of 2) (ii, vi, and viii) or with 50 μg/ml FMDV O1-Manisa empty capsids (iv) in medium containing 1% FCS. (i, iii, v, and vii) Mock-infected cells. GFP-LC3 was visualized using the natural fluorescence of GFP. For MEFs and atg5−/− MEFs, autophagosome production was analyzed by immunostaining for endogenous LC3 (green). Cell nuclei are blue. Scale bars = 10 μm.
LC3 punctae induced by FMDV do not contain the viral nonstructural proteins but colocalize with VP1.
Previous work with poliovirus showed that coexpression of 2BC and 3A generates double-membrane vesicles that resemble autophagosomes and that during infection, 3A colocalizes with LC3 (16, 40). Similarly, a recent study reported that FMDV 2B, 2C, and 3A colocalize with LC3 in infected cells (29). We therefore analyzed the relationship between LC3 and FMDV nonstructural proteins in more detail. The locations of 3A at 2 h and 3D at 2.5 h postinfection of CHO GFP-LC3 cells are shown in Fig. 5A, ii (3A) and vi (3D). The GFP-LC3, 3A, and 3D signals were located close to the nucleus (Fig. 5A, i to iii and v to vii). The high-magnification images in Fig. 5A, iv and viii, show that the red and green signals were mostly separate and suggest that 3A and 3D do not colocalize with LC3.
Fig 5.

Localization of GFP-LC3 and nonstructural proteins 3A and 3D in cells infected with FMDV. CHO GFP-LC3 cells were infected with FMDV O1BFS (MOI = 2) for 2 h (A, i to iv) or 2.5 h (A, v to viii, and B, i to viii), fixed, and processed for immunofluorescence microscopy. (A) Analysis of GFP-LC3 and FMDV nonstructural proteins 3A (i to iv) and 3D (v to viii). LC3 was visualized using the natural fluorescence of GFP (green). FMDV 3A was detected by immunostaining using antibody 2C2 (red) and FMDV 3D using antibody 3B5 (red). The boxed regions of interest in the merged images (iii and vii) are shown at high magnification in images iv and viii. Cell nuclei are blue. Scale bars = 10 μm. (B) As for panel A, except that infection was carried out in the presence of 100 nM concanamycin A, which was added at 1 h postinfection.
Autophagosomes deliver their contents to lysososmes for degradation. Proteolysis of the GFP tag on LC3 or quenching of fluorescence at the low pH encountered in autophagosomes and lysosomes could account for the apparent lack of colocalization of 3A and 3D with GFP-LC3. To address this possibility, we treated cells with concanamycin A to inhibit the vacuolar ATPase, to raise the pH of endosomes and autophagosomes, and to inhibit proteolysis in lysosomes. The drug was added 1 h postinfection to avoid effects of the inhibitor on cell entry. Figure 5B shows that colocalization of 3A (Fig. 5B, i to iv) or 3D (Fig. 5B, v to viii) with GFP-LC3 was not observed in concanamycin A-treated cells, which supports our conclusion that these viral proteins do not associate with autophagosomes containing LC3. Interestingly, the patterns of GFP-LC3 were different in untreated and concanamycin A-treated cells (Fig. 5A, i, and B, i. This was due to the effects of concanamycin A, since DMSO alone (as a control for concanamycin A) did not affect the distribution of LC3 (data not shown).
Figure 6A, i to iv, shows the distribution of GFP-LC3 and the FMDV capsid protein VP1 (2 h postinfection) and that many punctae were positive for either VP1 or LC3, while others appeared to be positive for both proteins (identified as orange/yellow vesicles in the high-magnification images in Fig. 6A, iv), suggesting some degree of colocalization between the proteins. The results in Fig. 1 show that FMDV infection induced the formation of p62 punctae with a pattern of labeling similar to that of GFP-LC3. Therefore, we also investigated the relationship between VP1, p62, and LC3 in infected cells. Figure 6B, ii and iii, shows that the perinuclear structures containing LC3 were also positive for p62, suggesting that p62 may localize with FMDV-induced autophagosomes. Triple labeling of these cells (Fig. 6B, iv) showed that the LC3/p62-positive structures also contained VP1, adding support to the above conclusion that a proportion of the FMDV-induced autophagosomes contain VP1. However, the precise relationships between LC3, p62, and VP1 were difficult to resolve, as the fluorescence signals were concentrated close to the nucleus.
Fig 6.

Localization of GFP-LC3, p62, and VP1 capsid protein in cells infected with FMDV. CHO GFP-LC3 cells were infected with FMDV O1BFS (MOI = 2) for 2 h, fixed, and processed for immunofluorescence microscopy. Scale bars = 10 μm. (A) (i to iv) Analysis of GFP-LC3 and FMDV capsid protein VP1. LC3 was visualized using the natural fluorescence of GFP (green). FMDV capsid protein VP1 was detected by immunostaining using antibody D9 (red). The boxed region of interest in the merged image (iii) is shown at high magnification in image iv. Cell nuclei are blue. (B) Infected CHO GFP-LC3 cells were double labeled for FMDV capsid protein VP1 (i) (blue) and p62 (iii) (red). GFP-LC3 (ii) is shown in green. Cell nuclei were labeled with Topro-3 (pseudocolored gray). (iv) Merged image. (C) Infected CHO GFP-LC3 cells were labeled for the microtubule-organizing center (ii) (red) using an antibody against γ-tubulin. GFP-LC3 (i) is shown in green. (iii) Merge of images i and ii. Cell nuclei are blue. (D) (ii) CHO GFP-LC3 cells were infected with FMDV in the presence of 100 μM scriptaid and labeled for FMDV VP1 (red). (i) GFP-LC3 is shown in green. (iii) Merge of images i and ii. Cell nuclei are blue.
The distributions of LC3 induced by FMDV were noticeably different in MEFs and CHO GFP-LC3 cells (compare Fig. 1A, iii, and Fig. 3iv). In CHO cells, the signal resembled the large megaphagosomes formed in the pancreas during coxsackievirus infection (18), whereas in MEFs, the LC3 signal was dispersed throughout the cytoplasm as discrete punctae. A possible explanation for the pattern in CHO cells is that the perinuclear clustering of LC3 results from transport of autophagosomes along microtubules to the MTOC. Figure 6C, i to iii, shows that the perinuclear clustering of LC3 overlays the MTOC. Histone deacetylase 6 (HDAC6) is a cytosolic tubulin deacetylase that binds ubiquitin and the dynein microtubule motor protein. HDAC6 delivers ubiquitinated proteins to the MTOC and facilitates their incorporation into autophagosomes. These functions of HDAC6 are inhibited by scriptaid (14). Figure 6D, i, shows that scriptaid inhibited the perinuclear clustering of LC3, and punctae positive for LC3 were seen throughout the cytoplasm. Similarly, the perinuclear clustering of VP1 was also dispersed (Fig. 6D, ii), and many of the structures positive for VP1 were also positive for GFP-LC3, again indicating some colocalization (Fig. 6D, iii). No dispersal of LC3 or VP1 was seen in infected cells treated with DMSO alone (data not shown).
The above results suggest a role for microtubules in the location of LC3 in infected CHO GFP-LC3 cells. To confirm this, CHO GFP-LC3 cells were infected in the presence of nocodazole to depolymerize microtubules and immunostained for 3A, 3D, or VP1 (Fig. 7). The figure shows side-by-side comparisons of confocal images (Fig. 7i, iii, and v) and digital rendering of pixel densities in corresponding fluorescent punctae (Fig. 7ii, iv, and vi). As seen with scriptaid, in the presence of nocodazole, the GFP-LC3 signal was dispersed through the cytoplasm (Fig. 7i, iii, and v). Interestingly, disruption of microtubules also dispersed the signal for VP1 (Fig. 7i), 3A (Fig. 7iii), and 3D (Fig. 7v). No dispersal of LC3, VP1, 3A, or 3D was seen in infected cells treated with DMSO alone (data not shown). The dispersal of LC3 and viral proteins allowed us to calculate the degree of colocalization between LC3 and VP1, 3A, and 3D using rendered images (Fig. 7ii, iv, and vi show examples). We analyzed 837 fluorescent punctae in five infected CHO GFP-LC3 cells that were stained for VP1. Four hundred and forty-seven were positive for GFP-LC3, and of these, 224 were also positive for VP1. Thus, approximately half the autophagosomes induced by FMDV appear to be associated with capsid protein. A similar analysis was carried out for 3A and 3D, which showed that only 3.8% and 4.8% of LC3-positive punctae were positive for 3A or 3D, respectively. These results show that half of the LC3-positive autophagosomes formed during FMDV infection contain VP1, but virtually all are devoid of 3A and 3D.
Fig 7.

LC3 punctae generated during FMDV infection contain VP1, but not 3A or 3D. CHO GFP-LC3 cells were infected with FMDV O1BFS (MOI = 2) for 2.5 h in the presence of 10 μM nocodazole. The cells were fixed and immunostained for VP1 (i) (red), 3A (iii) (red), or 3D (v) (red). LC3 was visualized using the natural fluorescence of GFP (green). The panels show side-by-side comparisons of confocal images (i, iii, and v) and digital rendering of pixel densities in fluorescent punctae (ii, iv, and vi) for the same cells.
LC3 punctae induced by FMDV closely resemble autophagosomes generated by starvation.
Figure 7 shows that the large perinuclear GFP-LC3 signal induced in CHO cells by FMDV dispersed into small LC3 punctae that spread throughout the cytoplasm in response to nocodazole. This suggested that the perinuclear GFP-LC3 signal induced in CHO cells by FMDV represented an accumulation of LC3-positive autophagosomes rather than formation of large megaphagosomes (see above). Therefore, we compared the LC3 punctae induced by FMDV with those induced by starvation in several cell types, including porcine IBRS-2 cells, which are derived from a natural host of FMDV. Figure 8A, B, and C shows side-by-side comparisons of confocal images and digital rendering of pixel densities in fluorescent punctae for the same cells either in nutrient medium (control) or following starvation or infection. Figure 8A shows representative images from CHO GFP-LC3 cells, and average data from five cells analyzing the diameters of punctae are represented by blue bars in Fig. 8D. CHO cells in nutrient medium contained very few punctae, and starvation induced on average 28 punctae per cell, most with a diameter between 0.8 and 1.4 μm. MEFs (Fig. 8B) showed greater numbers of punctae (15 per cell) in nutrient medium, and starvation increased the numbers of punctae per cell to an average of 75, which had diameters similar (green bars in Fig. 8D) to those seen in CHO GFP-LC3 cells, while IBRS-2 cells (Fig. 8C) produced fewer punctae per cell (on average, 17) in response to starvation, which had slightly larger diameters (1.2 to 1.6um; red bars in Fig. 8D).
Fig 8.

LC3 punctae generated during FMDV infection closely resemble autophagosomes generated in response to starvation. CHO GFP-LC3 cells (A), MEFs (B), or IBRS-2 cells (C) were either mock treated, starved for 2 h, or infected with FMDV O1BFS (MOI = 2) for 2 h (MEFs) or 2.5 h (CHO GFP LC3 and IBRS-2 cells). CHO GFP-LC3 cells were infected in the presence of nocodazole. GFP-LC3 (green) was visualized using the natural fluorescence of GFP. For MEFs and IBRS-2 cells, autophagosome production was analyzed by immunostaining for endogenous LC3 (green). (A to C) Side-by-side comparisons of confocal images (i, iii, and v) and digital rendering of pixel densities in fluorescent punctae (ii, iv, and vi) for the same cells either in control nutrient medium (i and ii), following starvation (iii and iv), or following infection (v and vi). (D) Average numbers of punctae and their diameters calculated from analysis of at least 5 cells (green, MEFs; blue, CHO cells; red, IBRS-2 cells).
The LC3 punctae in starved cells were compared to those formed in cells following infection with FMDV (note that the LC3 punctae induced in CHO GFP-LC3 cells were dispersed by nocodazole prior to imaging). Similar to starvation, FMDV induced more punctae in CHO GFP-LC3 cells and MEFs than in IBRS-2 cells, and for each cell type, the diameters of the punctae generated by FMDV were indistinguishable from those generated by starvation (Fig. 8D). This provided further evidence that FMDV induces the formation of authentic autophagosomes and that the perinuclear structure seen in CHO GFP-LC3 cells represents an accumulation of autophagosomes rather than the formation of a structure that is unique to the cell line.
Induction of LC3 punctae by non-heparan sulfate binding FMDV.
The observation that autophagosomes are induced by empty capsids or UV-inactivated virus suggests that induction may be triggered by virus binding to cell surface receptors. Two classes of receptors can be used by FMDV to initiate infection: arginine-glycine-aspartic acid (RGD)-binding integrins are used by field strains and are believed to serve as receptors in the animal host, while heparan sulfate (HS) is used following adaptation of FMDV to cell culture (15). CHO cells lack any of the known integrin receptors of FMDV, and infection is mediated exclusively by HS (2). This raised the possibility that induction of autophagosomes was mediated via HS and that it may be a feature of adaptation to tissue culture. IBRS-2 cells express αvβ8 integrin that can bind FMDV, but we cannot rule out the possibility that binding of O1BFS to IBRS-2 cells is also predominantly through HS. To see if field strains of FMDV, which depend on the use of integrins for cell entry, can also induce autophagosomes, we infected IBRS-2 cells with FMDV O1Kcad2, which uses integrins as its sole receptor type. Figure 9A, i to iii, shows mock-infected cells, and Fig. 9A, iv to vi, shows cells infected with O1Kcad2. LC3 punctae were induced by O1Kcad2, showing that integrin-binding viruses can also induce autophagosome formation. The numbers of LC3 punctae in mock-infected and O1Kcad2-infected cells were determined and are shown in Fig. 9B. The numbers of LC3 punctae for starved and O1BFS-infected IBRS2 cells (calculated from the data shown in Fig. 8) were included for comparison. O1Kcad2 generated an average of 14 punctae per cell, similar to the 16 generated by starvation but less than the 22 generated by O1BFS (Fig. 9B). Taken together, our results show that LC3 punctae are induced when either integrins (IBRS-2 with O1Kcad2) or HS (CHO with O1BFS) serves as the major receptor and suggest that induction of LC3 punctae could be triggered by virus ligation of either receptor type. Alternatively, as FMDV must be delivered to acidic endosomes for infection, it is possible that induction of LC3 punctae is triggered by an as yet unidentified factor that is common to both integrin- and HS-mediated entry.
Fig 9.

Induction of LC3 punctae by non-heparan sulfate binding FMDV. (A) IBRS-2 cells were mock infected (i to iii) or infected with FMDV O1Kcad2 at an MOI of 0.8 (iv to vi) for 3 h, fixed, and processed for confocal microscopy. Autophagosome production was analyzed by immunostaining for endogenous LC3 (green), and FMDV 3A protein (red) was detected using monoclonal antibody 2C2. LC3 labeling is shown in confocal images alone (i and iv) or overlaid with the signal for FMDV 3A (ii and v) and also as a digital rendering of pixel densities in fluorescent LC3 punctae from the same cells (iii and vi). Cell nuclei are blue. (B) Average numbers of LC3 punctae per cell from uninfected and O1Kcad2-infected IBRS-2 cells calculated from immunofluorescence images using IMARIS software. The data for starved and O1BFS-infected IBRS-2 cells (calculated from the data in Fig. 8) are included for comparison. The means and standard errors of the mean for at least 8 cells are shown.
The role of autophagy in FMDV infection.
Reagents that activate or inhibit autophagy were used to test for the requirement for autophagy during FMDV replication and virus production. Activation of autophagy in CHO cells with rapamycin did not affect the virus yield (data not shown). Similarly, the autophagy inhibitor 3-methyladenine had no effect on the virus yield (data not shown). To clearly establish the role of autophagy in FMDV infection, we compared virus replication in wt and atg5−/− MEFs. When cells infected with FMDV (MOI = 0.5) were immunostained for the viral 3A protein, the proportions of infected cells were similar for wt and atg5−/− cells (data not shown), which suggests that autophagy was not required for cell entry. We also determined the intracellular and extracellular virus yields produced by wt and atg5−/− MEFs. Figure 10i and ii show a 90% reduction (at 8 h postinfection) in the extracellular virus yield in atg5−/− MEFs compared to wt MEFs. Fig. 10iii and iv show that yields of intracellular FMDV were reduced to similar extents (∼95%) in atg5−/− MEFs. Work on poliovirus (16) has shown that inhibition of autophagy produces its greatest effect by reducing levels of extracellular virus but does not appear to have a significant effect on intracellular virus yields, suggesting that autophagy may be required for nonlytic virus release from cells. The observation that the lowered yields of intracellular FMDV correlated with reduced extracellular virus suggests that decreased yields result from reduced replication rather than from a requirement for autophagy in release of virus from cells.
Fig 10.

Loss of Atg5 reduces FMDV yield. MEFs or atg5−/− MEFs were infected for 1 h with FMDV O1BFS (MOI = 5). The cells were washed in acid to inactivate extracellular virus, and infection continued in normal cell culture medium. (i and ii) Supernatants were taken at the indicated times, and PFU were titrated using BHK cells (extracellular titer). (iii and iv) The remaining cells were freeze-thawed, and PFU were titrated using BHK cells (intracellular titer). (i and iii) The results are shown as growth curves. The continuous lines represent MEFs, and the broken lines represent atg5−/− MEFs. (ii and iv) Virus yields normalized to MEFs at 8 h postinfection. The means and standard errors of the mean of three observations are shown for each data point. The experiment shown was repeated twice, giving similar results.
DISCUSSION
This study shows that FMDV infection causes a marked increase in the numbers of LC3 punctae in the cytoplasm. LC3 punctae were induced by FMDV in three different cell types and could be detected by immunostaining for endogenous LC3 or by following redistribution of GFP-LC3. This, coupled with the observations that induction of LC3 punctae required Atg5, a protein essential for autophagosome formation, and that LC3 was converted to LC3II, suggests that the LC3 punctae induced by FMDV were autophagosomes and not LC3-positive edemosomes induced during coronavirus infection (24). FMDV infection also increased the numbers of p62 punctae, and since they colocalized with LC3, we conclude that FMDV infection leads to the formation of authentic autophagosomes. However, induction of autophagosomes by FMDV appeared to differ from starvation in one important respect, since generation of LC3 punctae was not inhibited by wortmannin. This implies that autophagosome formation induced by FMDV does not require the class III PI3-kinase activity of vps34 in the Atg6/beclin-Atg14-Vps15-Vps34 complex. This observation could explain why FMDV replication is insensitive to the autophagy inhibitor 3-methyladenine, as it targets vps34 (35, 36). Nevertheless, the LC3 punctae do require the Atg5-Atg12-Atg16 complex that acts downstream of vps34. Noncanonical pathways of autophagy in which autophagosomes are formed independently of beclin-1 and vps34 have recently been described (35, 36). Like canonical autophagy, noncanonical pathways appear to depend on other key autophagy proteins, and autophagosomes appear to mature normally. Our data suggest that FMDV may trigger autophagosome formation using a noncanonical pathway, but further experiments will be needed to determine whether this is the case or if FMDV can trigger both canonical and noncanonical autophagy.
FMDV induced clusters of LC3 punctae next to the nuclei of CHO GFP-LC3 cells, but when CHO cells were starved or when MEFs or IBRS-2 cells were infected with FMDV, the LC3 punctae were dispersed through the cytoplasm. It was possible that the perinuclear GFP-LC3 signal may be unique to CHO cells, or it may be similar to the large LC3-positive megaphagosomes detected in pancreatic acinar cells infected with coxsackievirus B3 (18). Our results show that the perinuclear clustering of LC3 in infected CHO GFP-LC3 cells represented a microtubule-dependent accumulation of autophagosomes, as in cells treated with nocodazole, LC3 punctae were present throughout the cytosol. Cell treatment with nocodazole did not reduce the virus yield (data not shown), showing that clustering of LC3 was not required for infection. However, LC3 clustering was also inhibited by scriptaid, which reduced the virus yield by ∼90% at 6 h postinfection (data not shown). Currently, we do not know the mechanism by which scriptaid has an inhibitory effect on infection.
When dispersed using nocodazole, the LC3 punctae closely resembled those produced in starved cells or in MEFs and IBRS-2 cells infected with FMDV. The average diameters and numbers of punctae generated by FMDV were indistinguishable from those of punctae generated in the same cells by starvation. Also, the FMDV LC3 punctae induced by FMDV were much smaller than megaphagosomes (6.85 ± 0.38 μm in diameter) that form in pancreatic acinar cells infected with coxsackievirus B3 (18). Thus, our results support the conclusion that the clustering of LC3 in infected CHO cells results from an accumulation of autophagosomes at the MTOC rather than formation of megaphagosomes. The perinuclear concentration of autophagosomes in response to viral infection is not unique to CHO cells. Autophagosomes generated during infection of human rhabdosarcoma and neuroblastoma cells by enterovirus 71 also concentrate next to the nucleus (12). Our study also suggests that this perinuclear clustering of LC3 requires HDAC6, a tubulin deacetylase that links ubiquitinated proteins to dynein motors to facilitate delivery of proteins to the MTOC and autophagosomes (21). While this provides a mechanism for dynein-dependent delivery of autophagosomes to the MTOC of CHO cells, and possibly rhabdosarcoma and neuroblastoma cells infected by EV71, it does not occur in all cells, since it was not apparent in MEFs or IBRS-2 cells.
Induction of autophagosomes by FMDV occurred early after infection and did not appear to require virus replication, since UV-inactivated virus or empty capsids lacking viral RNA also induced autophagosomes. These observations are similar to those for vesicular stomatitis virus (37) and human cytomegalovirus (25), which activate autophagy independently of replication. FMDV, therefore, induces autophagosomes very early during infection, possibly during cell entry, which suggests that induction may be triggered by virus binding to cell surface receptors. However, our results show that autophagosomes are induced when either integrins (IBRS-2 with O1Kcad2) or HS (CHO with O1BFS) serves as the major receptor and suggest that autophagosome induction may result from virus ligation of either receptor type. Alternatively autophagosomes may be induced by a mechanism that is common to both integrin- and HS-mediated entry. Thus, FMDV appears to differ from poliovirus, where there is strong evidence that autophagosome formation is linked to replication and expression of viral nonstructural proteins (40, 42). However, although our study shows that autophagosomes are induced by empty FMDV capsids, it does not completely rule out the possibility that the viral RNA genome or nonstructural proteins can also activate autophagy during infection, as was seen for poliovirus (16, 42).
Yields of FMDV were reduced to 10% of control in cells lacking Atg5, suggesting that autophagy promotes FMDV replication and or assembly. This is in general agreement with previous studies on FMDV (29), where silencing of LC3 in MCF-10A cells produced a 75% decrease in yield while silencing Atg12 produced a 100-fold decrease in PFU. Several studies on picornavirus replication showed that inhibition of autophagy can produce large changes in replication, while the effects of rapamycin (which activates autophagy) are less pronounced. For example, rapamycin causes a 2- to 3-fold increase in poliovirus and HRV2 titer compared to a 10-fold decrease in the virus yield following inhibition of autophagy by 3-methyladenine. Similarly, for human enterovirus 71 (12), extracellular virus yields were reduced 1,000-fold by 3-methyladenine, but rapamycin and starvation caused a modest 1.5-fold increase in virus yields. The effects of rapamycin on FMDV can also vary between cell types. Rapamycin causes a 100-fold increase in FMDV yields from human mammary gland MCF-10A cells, but this falls to a 10-fold increase in bovine pharynx cells. However, we were unable to find definitive evidence for increased yields of FMDV when autophagy was activated in CHO cells by rapamycin (data not shown).
The autophagosomes induced by enteroviruses (such as poliovirus and coxsackievirus) are thought to facilitate virus replication by providing sites for assembly of the viral replication complex (19, 44). It was possible that autophagosomes induced by FMDV could also be used for assembly of a replication complex. Two observations argue against this. First, the LC3 punctae induced by FMDV were short-lived and fell in number as infection progressed. This decrease in LC3 punctae 2 h after infection was not restricted to CHO cells and was seen in MEFs and IBRS-2 cells. Second, we failed to find colocalization of LC3 with 3A or 3D, viral proteins that are believed to be involved in vRNA replication and therefore are likely to reside in the replication complex. Recent work by O'Donnell et al. (29) has shown colocalization of GFP-LC3 with FMDV 2B, 2C, and 3A in MCF-10A cells. It is therefore possible that LC3 punctae and the degree of colocalization can vary between cell types, making it important to quantify the levels of colocalization. Although we did not find colocalization with 3A or 3D, 50% of autophagosomes costained for VP1. This could indicate the use of autophagosomes for capsid assembly and/or degradation. However, we currently do not know why VP1 colocalizes with LC3, and further experiments will be required to understand if it is significant for virus replication.
In our study, punctae positive for GFP-LC3 and VP1 contained p62, and later during infection, the GFP-LC3 and p62 punctae dispersed and were reduced in number. p62 is an autophagy receptor protein involved in the delivery of ubiquitinated proteins to autophagosomes (32) and is degraded when autophagosomes fuse with lysosomes. The loss of LC3 and p62 punctae seen during infection may result from fusion of autophagosomes with lysosomes. The acute loss of signal may arise because LC3 and p62 proteins are not replenished once FMDV shuts down translation of host protein at the onset of replication. Unlike for Sindbis virus (31), we do not have direct evidence for binding of p62 to the FMDV capsid protein, so we are not able to confirm that p62 mediates virus delivery to autophagosomes. Interestingly, coxsackievirus B3 induces autophagy, but p62 is not degraded, suggesting that the virus may prevent autophagosome-lysosome fusion (45). This may explain the induction of large megaphagosomes seen in pancreatic acinar cells (18).
In addition to providing sites for replication of poliovirus, it has been proposed that the transit of poliovirus through autophagosomes may also promote “nonlytic” virus release (16, 41). Consistent with this model, electron micrographs show poliovirus (6, 16) and enterovirus 71 (12) in autophagosomes. Enteroviruses, which infect via the gastrointestinal tract, are relatively resistant to low pH and proteases and could survive in autophagosomes and lysosomes. In contrast, the FMDV capsid is highly sensitive to low pH, and transport to acidic autophagosomes and lysosomes would be expected to reduce virus yields. In this way, FMDV would be subject to removal by autophagy, as described for Sindbis virus (31) and herpes simplex virus (30). Similar to poliovirus, we also observed a reduction in the extracellular virus yield in autophagy-deficient cells; however, this was accompanied by a proportionate drop in intracellular virus, making it unlikely that autophagy facilitates nonlytic release of FMDV.
In conclusion, we have shown that FMDV induces autophagosomes at an early stage of the replication cycle, most likely during cell entry. This induction appears to be independent of the PI3-kinase activity of vps34, and in contrast to other picornaviruses, FMDV-induced autophagosomes did not colocalize with viral nonstructural proteins, making it unlikely that autophagosomes are used as sites of FMDV replication. Further, our results show that autophagy may be beneficial for FMDV, making it possible that autophagy removes factors from cells that would inhibit replication. Further analysis will be required to understand the mechanism of autophagosome induction during cell entry and the role this plays in subsequent virus replication.
ACKNOWLEDGMENTS
This work was supported by BBSRC grant BB/F012861/1 to T.W. and grant BB/F013949/1 to T.J. and in part by the Israeli Science Foundation (ISF) and the German Israeli Foundation (GIF) to Z.E.
We thank Noboru Mizushima of Tokyo Medical and Dental University for wild-type and atg5−/− MEFs and Emiliana Brocchi, IZS, Brescia, Italy, for 2C2, 3B5, and D9 antibodies.
Footnotes
Published ahead of print 19 September 2012
REFERENCES
- 1. Abrams CC, King AM, Belsham GJ. 1995. Assembly of foot-and-mouth disease virus empty capsids synthesized by a vaccinia virus expression system. J. Gen. Virol. 76:3089–3098 [DOI] [PubMed] [Google Scholar]
- 2. Berryman S, Clark S, Monaghan P, Jackson T. 2005. Early events in integrin alphavbeta6-mediated cell entry of foot-and-mouth disease virus. J. Virol. 79:8519–8534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brabec-Zaruba M, Berka U, Blaas D, Fuchs R. 2007. Induction of autophagy does not affect human rhinovirus type 2 production. J. Virol. 81:10815–10817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chan EY, Longatti A, McKnight NC, Tooze SA. 2009. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol. Cell. Biol. 29:157–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choe SS, Dodd DA, Kirkegaard K. 2005. Inhibition of cellular protein secretion by picornaviral 3A proteins. Virology 337:18–29 [DOI] [PubMed] [Google Scholar]
- 6. Dales S, Eggers HJ, Tamm I, Palade GE. 1965. Electron microscopic study of the formation of poliovirus. Virology 26:379–389 [DOI] [PubMed] [Google Scholar]
- 7. Doedens JR, Kirkegaard K. 1995. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 14:894–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ganley IG, et al. 2009. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284:12297–12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hara T, et al. 2008. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 181:497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hosokawa N, et al. 2009. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 20:1981–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hosokawa N, et al. 2009. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5:973–979 [DOI] [PubMed] [Google Scholar]
- 12. Huang SC, Chang CL, Wang PS, Tsai Y, Liu HS. 2009. Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J. Med. Virol. 81:1241–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Itakura E, Mizushima N. 2010. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6:764–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iwata A, Riley BE, Johnston JA, Kopito RR. 2005. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 280:40282–40292 [DOI] [PubMed] [Google Scholar]
- 15. Jackson T, King AM, Stuart DI, Fry E. 2003. Structure and receptor binding. Virus Res. 91:33–46 [DOI] [PubMed] [Google Scholar]
- 16. Jackson WT, et al. 2005. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156 doi:10.1371/journal.pbio.0030156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jung CH, et al. 2009. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20:1992–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kemball CC, et al. 2010. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J. Virol. 84:12110–12124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kirkegaard K, Taylor MP, Jackson WT. 2004. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat. Rev. Microbiol. 2:301–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klein KA, Jackson WT. 2011. Human rhinovirus 2 induces the autophagic pathway and replicates more efficiently in autophagic cells. J. Virol. 85:9651–9654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee JY, et al. 2010. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29:969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liang XH, et al. 1998. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 72:8586–8596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Longatti A, Tooze SA. 2009. Vesicular trafficking and autophagosome formation. Cell Death Differ. 16:956–965 [DOI] [PubMed] [Google Scholar]
- 24. McCahon D, et al. 1989. Evidence for at least four antigenic sites on type O foot-and-mouth disease virus involved in neutralization; identification by single and multiple site monoclonal antibody-resistant mutants. J. Gen. Virol. 70:639–645 [DOI] [PubMed] [Google Scholar]
- 25. McFarlane S, et al. 2011. Early induction of autophagy in human fibroblasts after infection with human cytomegalovirus or herpes simplex virus 1. J. Virol. 85:4212–4221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mercer CA, Kaliappan A, Dennis PB. 2009. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 5:649–662 [DOI] [PubMed] [Google Scholar]
- 27. Moffat K, et al. 2005. Effects of foot-and-mouth disease virus nonstructural proteins on the structure and function of the early secretory pathway: 2BC but not 3A blocks endoplasmic reticulum-to-Golgi transport. J. Virol. 79:4382–4395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moffat K, et al. 2007. Inhibition of the secretory pathway by foot-and-mouth disease virus 2BC protein is reproduced by coexpression of 2B with 2C, and the site of inhibition is determined by the subcellular location of 2C. J. Virol. 81:1129–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. O'Donnell V, et al. 2011. Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology 410:142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Orvedahl A, et al. 2007. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35 [DOI] [PubMed] [Google Scholar]
- 31. Orvedahl A, et al. 2010. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7:115–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pankiv S, et al. 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282:24131–24145 [DOI] [PubMed] [Google Scholar]
- 33. Reggiori F, et al. 2010. Coronaviruses hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 7:500–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Robinson L, et al. 2011. Foot-and-mouth disease virus exhibits an altered tropism in the presence of specific immunoglobulins, enabling productive infection and killing of dendritic cells. J. Virol. 85:2212–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R. 2008. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ. 15:1318–1329 [DOI] [PubMed] [Google Scholar]
- 36. Scarlatti F, Maffei R, Beau I, Ghidoni R, Codogno P. 2008. Non-canonical autophagy: an exception or an underestimated form of autophagy? Autophagy 4:1083–1085 [DOI] [PubMed] [Google Scholar]
- 37. Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. 2009. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 30:588–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shvets E, Fass E, Elazar Z. 2008. Utilizing flow cytometry to monitor autophagy in living mammalian cells. Autophagy 4:621–628 [DOI] [PubMed] [Google Scholar]
- 39. Smith JD, de Harven E. 1978. Herpes simplex virus and human cytomegalovirus replication in WI-38 cells. III. Cytochemical localization of lysosomal enzymes in infected cells. J. Virol. 26:102–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Suhy DA, Giddings TH, Jr, Kirkegaard K. 2000. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 74:8953–8965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taylor MP, Burgon TB, Kirkegaard K, Jackson WT. 2009. Role of microtubules in extracellular release of poliovirus. J. Virol. 83:6599–6609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taylor MP, Kirkegaard K. 2007. Modification of cellular autophagy protein LC3 by poliovirus. J. Virol. 81:12543–12553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thoreen CC, et al. 2009. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284:8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wileman T. 2006. Aggresomes and autophagy generate sites for virus replication. Science 312:875–878 [DOI] [PubMed] [Google Scholar]
- 45. Wong J, et al. 2008. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 82:9143–9153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wullschleger S, Loewith R, Hall MN. 2006. TOR signaling in growth and metabolism. Cell 124:471–484 [DOI] [PubMed] [Google Scholar]
- 47. Xie Z, Klionsky DJ. 2007. Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9:1102–1109 [DOI] [PubMed] [Google Scholar]
- 48. Yang Z, Klionsky DJ. 2010. Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12:814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang Z, Klionsky DJ. 2009. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 335:1–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yoon SY, et al. 2008. Coxsackievirus B4 uses autophagy for replication after calpain activation in rat primary neurons. J. Virol. 82:11976–11978 [DOI] [PMC free article] [PubMed] [Google Scholar]