

Abstract
Replication of plus-stranded RNA viruses takes place on membranous structures derived from various organelles in infected cells. Previous works with Tomato bushy stunt tombusvirus (TBSV) revealed the recruitment of either peroxisomal or endoplasmic reticulum (ER) membranes for replication. In case of Carnation Italian ringspot tombusvirus (CIRV), the mitochondrial membranes supported CIRV replication. In this study, we developed ER and mitochondrion-based in vitro tombusvirus replication assays. Using purified recombinant TBSV and CIRV replication proteins, we showed that TBSV could use the purified yeast ER and mitochondrial preparations for complete viral RNA replication, while CIRV preferentially replicated in the mitochondrial membranes. The viral RNA became partly RNase resistant after ∼40 to 60 min of incubation in the purified ER and mitochondrial preparations, suggesting that assembly of TBSV and CIRV replicases could take place in the purified ER and mitochondrial membranes in vitro. Using chimeric and heterologous combinations of replication proteins, we showed that multiple domains within the replication proteins are involved in determining the efficiency of tombusvirus replication in the two subcellular membranes. Altogether, we demonstrated that TBSV is less limited while CIRV is more restricted in utilizing various intracellular membranes for replication. Overall, the current work provides evidence that tombusvirus replication could occur in vitro in isolated subcellular membranes, suggesting that tombusviruses have the ability to utilize alternative organellar membranes during infection that could increase the chance of mixed virus replication and rapid evolution during coinfection.
INTRODUCTION
Replication of plus-strand RNA [(+)RNA] viruses takes place in membrane-bound viral replicase complexes (VRCs) in the cytoplasm of infected cells (9, 12, 29, 37, 39–41, 43, 67). Various (+)RNA viruses usurp different intracellular membranes, including endoplasmic reticulum (ER), mitochondrial, peroxisome, or endosomal membranes, to aid the replication process. Other viruses induce the formation of “viral replication organelles” or “membranous web” made from various intracellular membranes (4, 12, 14, 40, 67). The recruited membranes are thought to facilitate virus replication by (i) providing surfaces to assemble the VRCs, (ii) sequestering and concentrating viral and host components, (iii) protecting the viral RNA and proteins from nucleases and proteases (1), and (iv) facilitating regulated RNA synthesis by harboring the minus-strand RNA [(−)RNA] template for production of abundant (+)RNA progeny.
The emerging picture with several (+)RNA viruses is that their replication proteins bind to different lipids and recruit a number of host proteins, which are involved in lipid synthesis or modification, to the site of replication (14, 40, 62, 69). In addition, (+)RNA virus replication is also dependent on bending intracellular membranes that form characteristic viral structures, such as spherules (vesicles with narrow openings) or vesicles (9). Therefore, (+)RNA viruses likely recruit host proteins affecting membrane curvature, as shown for ESCRT (endosomal sorting complexes required for transport), reticulon, and amphiphysin proteins in the cases of tombusviruses, Brome mosaic virus, and Semliki Forest virus (1, 3, 10, 45). Lipids also affect membrane curvature and fluidity. Indeed, replication of several viruses has been shown to be affected by sterols, fatty acids, and phospholipids (6, 23, 27, 33, 74, 75).
Tomato bushy stunt virus (TBSV) is a small (+)RNA virus that has emerged as a model virus to study virus replication, recombination, and virus-host interactions due to the development of yeast (Saccharomyces cerevisiae) as a model host (40, 42, 48, 51, 84). Over 400 host genes/proteins that affected either TBSV replication or recombination have been identified via genome-wide screens of yeast genes or global proteomics approaches (21, 49, 71–73). The highly purified tombusvirus replicase complex (VRC) is known to contain the two viral replication proteins (i.e., p33 and p92pol) and 6 to 10 host proteins (28, 30, 70). These host proteins have different functions during TBSV replication. For example, heat shock protein 70 (Hsp70), eukaryotic elongation factor 1A (eEF1A) and the ESCRT family of host proteins are involved in the assembly of the VRC (1, 3, 29, 31, 55, 78, 79). In addition, eEF1A and eEF1Bγ facilitate minus-strand synthesis (31, 68), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Ded1 DEAD box helicase have been shown to promote viral (+)RNA synthesis (15, 25, 77), and the Pex19 shuttle protein is involved in targeting of the replication proteins to peroxisomes, the sites of replication (53).
The auxiliary p33 replication protein, which has RNA chaperone function, is an abundant protein and is essential for replication of TBSV in both yeast and plants (22, 47, 56, 76). The tombusvirus p33 is an integral membrane protein that has been shown to recruit the TBSV (+)RNA into replication. The current picture is that the p33 replication protein serves as the master regulator of TBSV replication by interacting with the viral RNA, p92pol, and numerous host proteins and host membranes. The host also targets p33 or the viral RNA via nucleolin, cyclophilins, or WW domain proteins to limit tombusvirus infections (2, 20, 32, 36, 57). On the other hand, the viral p92pol, which is a translational readthrough product containing the p33 sequence at its N terminus and a unique RNA-dependent RNA polymerase (RdRp) domain at the C terminus, is present in a smaller amount (84). Interaction between p92pol and p33 replication proteins is required for assembling the functional VRC (47, 50, 51, 54). Interestingly, the activation of the RdRp function of p92pol protein requires not only p33, cis-acting sequences present in the viral (+)RNA, and host factors but also host membranes (50–52, 56). This complex VRC assembly process and the many factors needed for the RdRp activation open the exciting questions of whether tombusviruses could utilize different cellular membranes or whether various heterologous combinations of tombusvirus replication proteins are functional.
Most tombusviruses, including TBSV, Cucumber necrosis virus (CNV), and Cymbidium ringspot virus (CymRSV), show preference for peroxisomal membranes (34, 44, 47). Interestingly, these viruses can also replicate efficiently on the ER membrane in the absence of peroxisomes, suggesting flexibility in intracellular membrane utilization (22, 53, 65). Another tombusvirus, Carnation Italian ringspot virus (CIRV), however, prefers to use mitochondrial membrane for replication (16, 81). Artificial retargeting of the CIRV replication proteins to the peroxisomes or of CymRSV to the mitochondria via chimeric constructs also supported CIRV and CymRSV replication (5), suggesting that these viruses could utilize more than one intracellular environment for their replication.
To analyze if tombusviruses are indeed capable of utilizing various intracellular membranes for their replication, we used in vitro approaches with recombinant viral proteins and isolated intracellular organelles/membranes. Interestingly, we found that TBSV, which originally uses the peroxisomal membrane, could also utilize ER and mitochondrial membranes for replication in vitro. On the other hand, CIRV, which originally utilizes the mitochondrial membranes, replicated on the isolated mitochondrial membranes, while it could use the ER membrane less efficiently in vitro. Using heterologous combinations of replication proteins and chimeric constructs, we identified that multiple domains in the replication proteins are determinants of membrane preference for tombusvirus replication. Altogether, the current study promotes the idea that TBSV is less restricted, while CIRV is more restricted, in utilizing various intracellular membranes for replication.
MATERIALS AND METHODS
Yeast strains and expression plasmids.
Saccharomyces cerevisiae strain BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) and pex3Δ single-gene deletion strains were obtained from Open Biosystems. Constructs pMAL-p33 and pMAL-p92, to express TBSV p33 (renamed here T33) and p92 (renamed T92) as fusion proteins to the C terminus of maltose binding protein (MBP) were described previously (55).
To generate the Escherichia coli expression constructs pMAL-p36, pMAL-p95, pMAL-C36-T92, pMAL-T33-C95, pMAL-T33c, pMAL-T92c, pMAL-T33tc, and pMAL-T92tc, we used the following approaches. The CIRV p36 sequence was amplified from CIRV full-length cDNA (obtained from A. White, York University, Canada) with primers 642 (5′-GTATTTGACACCGAGGG-3′) and 3230 (CCGCTCGAGCTATTTGACACCGAGGGATT). The CIRV p95 sequence was obtained by blunt-end ligation of the PCR product of C36 amplified by primers 642 and 643 (GGAGGCCTAGTGCGTCTAC) from CIRV cDNA, and the C95 C-terminal sequence was amplified by PCR using primers 644 (GGAGCTCGAGCTATTTGACACCCAGGGAC) and 970 (CCTAGGGAAAAACTGTCGGTA) and CIRV cDNA. C36-T92 chimeric sequence was obtained by blunt-end ligation of the PCR product of C36 sequence PCR amplified with primers 642 and 643 using CIRV full-length cDNA, and T92 C-terminal sequence was amplified by PCR with primers 6 (GGAGGCCTAGTACGTCTAC) and 826 (GATTACATTGTCCCTCTATCT) using TBSV full-length cDNA. T33-C95 chimeric sequence was obtained by blunt-end ligation of the PCR products of T33 (generated by PCR with primers 473 [GAGGAATTCGAGACCATCAAGAGAATG] and 3960 [GTATTTGACACCCAGGGAC]) and C-terminal sequence of C95 (generated by PCR with primers 644 and 970). The T33c sequence was obtained by blunt-end ligation of the PCR product obtained using primers 642 and 4102 amplified from CIRV cDNA and the PCR product obtained using primers 4099 and 810 amplified from TBSV cDNA. The T92c sequence was obtained by blunt-end ligation of T33c (generated by PCR with primers 642 and 3960) and T92 C-terminal sequence. The T33tc sequence was obtained by blunt-end ligation of the PCR product obtained using primers 642 and 4090 (ACGAGCCACACCCCGTTTAGC) and CIRV cDNA and the PCR product generated by using primers 4087 (GATTACATTGTCCCTCTATCT) and 810 (CCCGCTCGAGTCAAGCTACGGCGGAGTCGAGGA) and TBSV cDNA. The T92tc sequence was obtained by blunt-end ligation of T33tc (generated by PCR using primers 642 and 3960) and the PCR-amplified C-terminal sequence of T92. All the above PCR products were digested with EcoRI and XhoI restriction enzymes and inserted into pMAL-c2X (New England BioLabs).
To generate N-terminal glutathione S-transferase (GST)-His6 fusion proteins, the C-terminal sequence of TBSV p33 was PCR amplified using primers 633 (GGAGGAATTCATGGAGGGTTTGAAGGC) and 1593 (CGGCTCGAGCTATTTGACACCCAGGGACTCCTGT) and TBSV cDNA. The C-terminal sequence of CIRV p36 was PCR amplified using primers 633 (GGAGGAATTCATGGAGGGTTTGAAGGC) and 3230 (CCGCTCGAGCTATTTGACACCGAGGGATT) and CIRV cDNA. The PCR products were digested with BamHI and XhoI and cloned into BamHI/XhoI-digested pGEX-his (2).
To generate the pGD-L-T33, pGD-L-T92, pGD-L-C36, and pGD-L-95 constructs for agroinfiltration in plants, the PCR product of TBSV p33 sequence (using primers 788 [GGAGCTCGAGTCAAGCTACGGCGGAGTC]/810 [CCCGCTCGAGTCAAGCTACGGCGGAGTCGAGGA]) was digested with BamHI and XhoI, the TBSV p92 sequence (obtained using primers 4000 [CCAGAGATCTATGGAGACCATCAAGAGAATG]/826 [GATTACATTGTCCCTCTATCT]) was digested with BglII and XhoI, the PCR product of CIRV p36 sequence (obtained using primers 900 [ACGAGCCACACCCCGTTTAGC]/3230 [CCGCTCGAGCTATTTGACACCGAGGGATT]) was digested with BamHI and XhoI, and the PCR product of CIRV p95 sequence (obtained using primers 900 [ACGAGCCACACCCCGTTTAGC]/970 [CCTAGGGAAAAACTGTCGGTA]) was digested with BamHI and XhoI. The above PCR products were then separately inserted into pGD-L (1), which was digested with BamHI and XhoI, generating transient-expression vectors for agroinfiltration.
For the imaging experiments, we constructed the plasmids pESC-T33/DI72, pESC-C36/DI72, pYES-T92, and pYES-C95. For this, sequences of full-length TBSV p33 (primers 788/810), CIRV p36 (primers 900/3230), and p95 (primers 900/970) were PCR amplified and digested with BamHI and XhoI, while the PCR-amplified sequence of TBSV p92 (primers 4000/826) was digested with BglII and XhoI. Digested PCR products were then inserted into pESC-HisCNVp33-DI72 or pYES-CNVp92 digested with BamHI/XhoI.
To track viral protein localization in yeast cells, we cloned red-shifted green fluorescent protein (rsGFP) sequence upstream of the N termini of TBSV p33/p92 and CIRV p36/p95. The rsGFP sequence was PCR amplified with primers 1262 (CGGCGGATCCGGTAAAGGAGAAGAACTTTTCACT) and 1263 (CGGCGGATCCGAGTCCGGACTTGTATAGTTCA) using the pGDG vector as a template (provided by M. Goodin [7]), followed by digestion with BamHI, and inserted into pESC-C36/DI72 or pYES-C95 digested with BamHI, generating pESC-rsGFP-C36/DI72 and pYES-rsGFP/C95. The cDNA sequence of TBSV p33 was PCR amplified using primers 4000/810, while the cDNA sequence of TBSV p92 was PCR amplified with primers 4000/826. The obtained PCR products were digested with BglII, followed by ligation with the PCR-amplified rsGFP sequence digested with BamHI, generating the cDNAs of rsGFP-T33 and rsGFP-T92. The cDNAs of rsGFP-T33 and rsGFP-T92 were then digested with BamHI and XhoI and inserted into pESC-C36/DI72 or pYES-C95 digested with BamHI/XhoI, resulting in the pESC-rsGFP-T33/DI72 and pYES-rsGFP-T92 expression plasmids.
Agroinfiltration and RNA extraction.
Nicotiana benthamiana leaves were agroinfiltrated with A. tumefaciens cultures containing combinations of pGD-L-T33, pGD-L-T92, pGD-L-C36, or pGD-L-C95 as well as pGD-DI72sat and pGD-p19 as described previously (1). After 3.5 days postinfiltration, agroinfiltrated leaves were collected, and total RNA was extracted and subjected for Northern blot analysis as described previously (1).
Preparation of CFE and soluble fraction (S100).
The yeast cell extract (CFE) from yeast strain BY4741was prepared as described previously (54). For production of the S100 soluble fraction, yeast CFE was further centrifuged at 100,000 × g for 1 h, and the supernatant (S100) was carefully collected without disturbing the pellet and then stored at −80°C.
Purification of yeast microsomal membranes.
Yeast microsomes were prepared as previously described (80), except that yeast microsomes were washed in 30 mM HEPES-KOH (pH 7.4)–150 mM potassium acetate–2 mM magnesium acetate containing Complete Mini protease inhibitor cocktail (Roche Applied Science). The protein concentration of the obtained yeast microsomal membranes was 4 mg/ml.
Purification of intact yeast mitochondria.
Yeast intact mitochondria were purified as described previously (35). Briefly, yeast cells were made into spheroplasts by incubating with 5 mg/g (wet weight) Zymolyase 20T (Seikagaku), and then the spheroplasts were homogenized and lysed with a glass Dounce homogenizer in ice-cold homogenization buffer (0.6 M sorbitol, 10 mM Tris-HCl [pH 7.4], 1 mM phenylmethylsulfonyl fluoride [PMSF], 0.2% [wt/vol] bovine serum albumin [BSA]). The homogenized spheroplasts were then centrifuged at 3,000 × g for 5 min at 4°C, and the supernatant was subjected to additional centrifugation at 12,000 × g for 15 min to obtain the crude mitochondrial preparation. To further remove contaminating membranes, the crude mitochondrial preparation was subjected to two sequential centrifugations at 134,000 × g on a sucrose gradient (0.7 ml 60%, 1.5 ml 32%, 0.7 ml 23%, and 0.7 ml 15% [wt/vol] sucrose with 1 mM EDTA and 10 mM MOPS [morpholinepropanesulfonic acid]-KOH). The purified mitochondrial preparation was recovered between the 60%/32% sucrose gradient interface and stored in SEM buffer (250 mM sucrose, 1 mM EDTA, and 10 mM MOPS-KOH [pH 7.2], containing Complete Mini protease inhibitor cocktail [Roche Applied Science]) at −80°C. The protein concentration was about 3 mg/ml.
Isolation of oleate-induced peroxisomes using sucrose gradients.
The isolation of peroxisomes was as described previously (63). Yeast was grown in peroxisome induction medium containing 0.12% (wt/vol) oleic acid, 0.2% Tween 40, 0.5% Bacto peptone, and 0.3% yeast extract to an optical density at 600 nm (OD600) of 1.0. The cell wall was digested with Zymolyase to generate spheroplasts in MES (morpholineethanesulfonic acid) buffer (5 mM MES [pH 5.5], 1 mM EDTA, 1 mM KCl) with 1.2 M sorbitol and homogenized by gradually adding MES buffer until the sorbitol concentration reached to 0.65 M. Unlysed cells and cell debris were removed by centrifuging at 2,000 × g, and crude peroxisome preparations were collected via centrifugation at 20,000 × g. The crude peroxisome preparations were applied to a linear sucrose gradient (10% to 70%) using a Beckman VTi 50 rotor at 34,500 × g for 2.5 h. Fractions were collected and stored at −80°C. Before the in vitro replicase assembly assay, each membrane fraction (100 μl) was thawed on ice and diluted 5× with 30% sucrose–MES buffer, followed by centrifugation at 20,000 × g for 30 min. The pellets of each fraction were then carefully suspended into 10 μl of 30% sucrose–MES buffer. The protein density in peroxisome fractions 11 to 16 was about 1 mg/ml.
In vitro replication assay.
The yeast CFE-based replication assay was modified from that described previously (54) to study TBSV and CIRV replication using the isolated organelle preparations. The yeast cell extract (2 μl) or purified membrane fractions (1 μl) together with S100 soluble fraction (1 μl) was incubated at 25°C water bath for 1 h in 8 μl cell-free replication buffer A (containing 30 mM HEPES-KOH [pH 7.4], 150 mM potassium acetate, 5 mM magnesium acetate, and 0.6 M sorbitol) with 15 mM creatine phosphate, 1 mM ATP, CTP, and GTP, 0.025 mM UTP, 0.1 μl of [32P]UTP, 0.1 mg/ml creatine kinase, 0.1 μl of RNase inhibitor, 10 mM dithiothreitol, 0.5 μg DI-72 RNA transcript, and 0.5 μg recombinant MBP-fused viral proteins. The volume of the reaction mixture was then adjusted by adding 16 μl cell-free replication buffer B (containing 30 mM HEPES-KOH [pH 7.4], 150 mM potassium acetate, and 5 mM magnesium acetate) with 15 mM creatine phosphate, 1 mM ATP, CTP, and GTP, 0.025 mM UTP, 0.2 μl of [32P]UTP, 0.1 mg/ml creatine kinase, 0.2 μl of RNase inhibitor, 10 mM dithiothreitol, and 0.05 mg/ml actinomycin D. The reaction mixture was incubated at 25°C for 3 h. The reaction was terminated by adding 110 μl stop buffer (1% sodium dodecyl sulfate [SDS] and 0.05 M EDTA, pH 8.0), followed by phenol-chloroform extraction, isopropanol-ammonium acetate precipitation, and a washing step with 70% ethanol. RNA samples were electrophoresed in a denaturing gel (5% polyacrylamide gel containing 8 M urea) and analyzed by phosphorimaging (Typhoon; GE).
To test the effect of sorbitol on in vitro tombusvirus replication, the purified membrane preparations (1 μl) together with S100 fraction (1 μl) were incubated in a water bath at 25°C for 4 h in 24 μl of modified cell-free replication buffer A containing 0, 0.2, and 0.6 M sorbitol.
Micrococcal nuclease treatment of the in vitro replication assay mixture.
The replication assay was conducted as described above except that each sample was treated with 0.25 U/μl micrococcal nuclease and 1 mM CaCl2 at different time points after starting of the incubation in replication buffer A, as described previously (54). EGTA (2.5 mM) was added to each sample after 15 min of incubation of micrococcal nuclease to stop the nuclease digestion.
Replication assay to determine viral (+)RNA/(−)RNA ratio.
The replication assay to determine the viral (+)RNA/(−)RNA ratio was done as described in a previous publication (21). Briefly, 2 μg of in vitro transcripts of minus-strand and plus-strand DI-72 RNAs were separately dot blotted onto a Hybond XL membrane (Amersham), followed by UV cross-linking. In vitro replication products were hybridized to the blots in ULTRAhyb solution (Ambion) at 68°C and quantified after washing.
Western blotting.
Western blotting of yeast membrane proteins was done as described previously (79). The following antibodies were used: antiporin, anti-alkaline phosphatase (anti-ALP), anti-3-phosphoglycerate kinase (anti-PGK), and anti-dpm1 (purchased from Invitrogen, CA). Sec61p antibody was provided by Tom Rapoport, Harvard Medical School. Fox3p antibody was provided by Daniel J. Klionsky, University of Michigan.
In vitro membrane association assay.
[35S]methionine-labeled TBSV p33 and CIRV p36 were obtained in nuclease-treated rabbit reticulocyte lysates (Promega) in the presence of 400 μCi/ml [35S]methionine. One microliter of translation mixture was incubated at 25°C for 1 h with 2 μl purified yeast microsomes or purified yeast mitochondrial preparations and 2 μl S100 fraction in a 40-μl reaction mixture containing cell-free replication buffer A (30 mM HEPES-KOH [pH 7.4], 150 mM potassium acetate, 5 mM magnesium acetate, 0.6 M sorbitol) as well as 15 mM creatine phosphate, 1 mM ribonucleoside triphosphate (rNTP), 0.1 mg/ml creatine kinase, 0.1 μl of RNase inhibitor, 10 mM dithiothreitol, and 0.5 μg DI-72 RNA transcript. Each reaction mixture was diluted 3 times with cell-free reaction buffer A, followed by incubation on ice for 30 min. The samples were centrifuged at 100,000 × g for 2 h. The pellet was dissolved in SDS-PAGE sample buffer. Proteins from the supernatant fractions were precipitated in 10% trichloroacetic acid (TCA) and dissolved in SDS-PAGE sample buffer. Samples were separated by SDS-PAGE.
In vitro MBP pulldown assay.
MBP-tagged TBSV T33 (p33) and T92 (p92), CIRV C36 and C95, and GST-His6-tagged TBSV p33C and CIRV p36C were expressed in E. coli transformed with one of the following plasmids: pMAL-T33, pMAL-T92, pMAL-C36, pMAL-C95, pGEX-his-T33C, and pGEX-his-C36C. E. coli cultures were lysed by sonication, followed by affinity purification via amylose columns, and washed with cold column buffer with a high salt concentration (10 mM Tris-Cl [pH 7.4], 1 mM EDTA, 200 mM NaCl, 10 mM 2-mercaptoethanol) as described previously (60). The GST-His6-tagged p33C, GST-His6-p36C, or GST-His6 (negative control) was incubated with the MBP-tagged proteins for 1 h at 4°C and washed with column buffer with high salt. The bound proteins were eluted with column buffer with high salt supplemented with 10 mM maltose (60). The presence of GST-tagged proteins was analyzed by SDS-PAGE and Western blotting using an anti-GST antibody.
Confocal laser microscopy.
Visualization of Pho86-cyan fluorescent protein (CFP) ER marker protein and various combination of tagged viral proteins in live yeast cells was as described previously (22, 53). To visualize yeast mitochondrial distributions of various viral proteins, we transformed yeast with combinations of plasmids, i.e., pESC-GFP-T33/DI72, pESC-GFP-C36/DI72, pYES-GFP-T92, or pYES-GFP-C95 with pESC-T33/DI72, pESC-C36/DI72, pYES-T92 and pYES-C95, as described in the legend to Fig. 9. Transformed yeast was grown at 23°C in minimal medium supplemented with 2% galactose, and then we used rhodamine B (hexyl ester-perchlorate, a mitochondrion-specific dye used in yeast [catalog no. R-648MP; Invitrogen, CA]) to visualize yeast mitochondria (11, 46, 58) with an Olympus FV1000 microscope (Olympus America Inc., Melville, NY).
Fig 9.

Distribution of TBSV and CIRV replication proteins when expressed in heterologous combinations. (A) Confocal laser microscopy images show the colocalization of Pho86p-CFP (ER marker protein) with YFP-T92, YFP-T33, YFP-C95, or YFP-C36 expressed from the GAL1 promoter in a pex3Δ yeast strain. The description on the left shows the combination of replication proteins expressed in yeast. The merged images show the colocalization of Pho86p-CFP with YFP-tagged replication proteins. Differential interference contrast (DIC) images are shown on the right. (B) Localization of YFP-T92 or GFP-T92, YFP-T33 or GTP-T33, GFP-C95, or GFP-C36 expressed from the GAL1 promoter to the mitochondria in a pex3Δ yeast strain. We used rhodamine B (RhodB, red) fluorescent dye to visualize the mitochondria. See further details described for panel A. (C) Colocalization of YFP- or CFP-tagged TBSV and CIRV replication proteins in a pex3Δ yeast strain. See further details described for panel A. (D) ER localization of YFP-C95 or YFP-T33 in a pex3Δ yeast strain expressing heterologous combinations of tombusvirus replication proteins. See further details described for panel A. (E) Mitochondrial localization of GFP-C95 or GFP-T33 in a pex3Δ yeast strain expressing heterologous combinations of tombusvirus replication proteins. See further details described for panel A. (F) ER localization of YFP-T92 or YFP-C36 in a pex3Δ yeast strain expressing heterologous combinations of tombusvirus replication proteins. See further details described for panel A. (G) Mitochondrial localization of GFP-T92 or GFP-C36 in a pex3Δ yeast strain expressing heterologous combinations of tombusvirus replication proteins. See further details described for panel A. Yeast was grown under similar conditions and images were taken as described for panel A. Each experiment was repeated.
RESULTS
CIRV replication proteins support RNA replication in yeast cell-free preparations in vitro.
We have previously shown that TBSV can efficiently replicate in cell extracts (CFE) prepared from yeast (52, 54, 55). The CFE-based TBSV replication assay mixture contained purified recombinant TBSV p33 and p92pol replication proteins and T7 polymerase-made DI-72 plus-strand replicon RNA (repRNA) transcripts. The CFE supported one single cycle of replication, starting with (−)RNA synthesis on the added (+)repRNA transcripts, followed by robust synthesis of (+)repRNA progeny (52, 54, 55). The CFE-based assay recapitulated the known features of TBSV replication, including the requirement of cis-acting viral RNA elements, the dependence on viral and host factors, and the need for both membranous and soluble fractions of CFE. Other features of the assay included the following: asymmetrical replication, leading to 10- to 40-fold more (+)RNA than (−)RNA; association of the VRCs with membranes, which led to protection against RNases and proteases after the assembly of VRCs; and the release of (+)repRNA progeny to the soluble fraction during the reaction, while retaining the (−)repRNA in the VRCs (52, 54, 55).
To test whether CIRV replication proteins, which are originally associated with mitochondrial membranes (16, 81), could support repRNA replication in the CFE-based assay, we added purified recombinant p36 and p95pol (called C36 and C95 in this paper to discriminate them from the homologous TBSV p33 and p92pol, named T33 and T92) in combination with the TBSV-derived DI-72 (+)repRNA to yeast CFE. Interestingly, we observed the replication of repRNA in samples containing both C36 and C95 (Fig. 1A, lane 7), which reached about 10% of that supported by T33/T92 (lane 1). No replication was observed when C95 protein was omitted from the CFE-based assay mixture (Fig. 1A, lane 8), confirming that both CIRV replication proteins are required for repRNA replication. The supernatant or membrane fractions of CFE alone or the Triton X-100-treated CFE could not support repRNA replication in the presence of C36 and C95 (Fig. 1A, lanes 10 to 12), similar to what was observed with T33/T92 (lanes 4 to 6). Thus, these experiments showed that the CIRV C36/C95 replication proteins could support repRNA replication in CFE, albeit with reduced efficiency compared with the TBSV T33/T92 replication proteins.
Fig 1.

In vitro reconstitution of the CIRV replicase in yeast cell extract. (A) Purified recombinant p33 (named T33) and p92pol (named T92) replication proteins of TBSV or purified recombinant p36 (named C36) and p95pol (named C95) replication proteins of CIRV in combination with the TBSV-derived DI-72 (+)repRNA were added to the cell extract (lanes 1 and 7), to the membrane plus soluble fractions (lanes 3 and 9), to the soluble fraction (lanes 4 and 10), to the 1% Triton-treated membrane plus soluble fractions (lanes 5 and 11), and to the membrane fraction (lanes 6 and 12) of the yeast cell extract. The CFE-based replication assay mixture lacked T92 (lane 2) or C95 (lane 8) as a negative control. Denaturing PAGE analysis of the 32P-labeled repRNA products obtained is shown. The full-length repRNA is indicated by an arrowhead. The result of the CFE-based replication assay with T33 and T92 was chosen as 100% (lane 1). (B) The heterologous combinations of TBSV and CIRV replication proteins are functional in the CFE-based replication assay. The activity of the reconstituted tombusvirus replicases is estimated as for panel A. Denaturing PAGE analysis of the replicase products is as shown in panel A. (C) Detection of plus- and minus-stranded RNA products produced by the reconstituted TBSV and CIRV replicases in the CFE-based replication assay. The blot contains the same amounts of cold plus- and minus-strand DI-72 RNA, while the 32P-labeled repRNA probes were generated in the CFE-based replication assay. Note that we used 5 times more CIRV replication products than TBSV to increase the signal. The ratio of plus- and minus-strand RNA products was estimated. (D) The heterologous combinations of TBSV and CIRV replication proteins with the CIRV-derived DI-1 repRNA are functional in the CFE-based replication assay. See further details described for panel B. Each experiment was repeated.
To further test the CIRV replication process in the yeast CFE, we estimated the ratio of newly made plus-strand to minus-strand repRNA levels in the replication assay (Fig. 1C). This showed that, similar to the case for the TBSV replication proteins, the CIRV replication proteins also performed asymmetrical viral RNA synthesis by producing ∼10 times more new plus strands than − strands in the yeast CFE (Fig. 1C).
Since the repRNA was the TBSV-derived DI-72 in the above-described assays, it is possible that the reduced replication was due to the less efficient utilization of the heterologous repRNA by the CIRV replication proteins in comparison with repRNA replication supported by the homologous TBSV replication proteins. To test this possibility, we also used the CIRV-derived DI-1 repRNA (64) in the CFE-based assay. These experiments revealed that both repRNAs were used more efficiently by T33/T92 than by C36/C95 in vitro (Fig. 1B and D, lanes 1 and 2 versus 3 and 4). Thus, the viral replication proteins determine the efficiency of repRNA replication in this assay.
Heterologous combinations of replication proteins supports RNA replication in yeast cell-free preparations in vitro and in planta.
To test whether the heterologous combinations of tombusvirus replication proteins could support repRNA replication, we used the CFE-based assay with the purified recombinant proteins. The CFE-based assay revealed that the heterologous combinations of replication proteins did support repRNA replication (Fig. 1B and D, lanes 5 to 8), albeit less efficiently than T33/T92 (Fig. 1B and D, lanes 1 and 2). Moreover, we observed that the viral RdRp protein was the major factor controlling the efficiency of repRNA replication. Accordingly, the homologous combination of T33/T92 (Fig. 1B and D, lanes 1 and 2) or the heterologous combination of C36/T92 (lanes 5 and 6) supported more efficient replication than C36/C95 (lanes 3 and 4) or T33/C95 (lanes 7 and 8). Thus, it seems that T92 RdRp protein is ∼6- to 20-fold more active in the CFE-based assay than C95 RdRp. However, the small replication protein also affected the efficiency of replication, since the combinations of replication proteins that included T33 supported up to ∼3-fold more replication than C36 in a complex with the RdRp protein (compare T33/T92 and C36/T92; Fig. 1B and D, lanes 1 and 2 versus 5 and 6).
To test whether the tombusvirus replication proteins behave similarly in plant cells, we used an agroinfiltration-based approach to express the TBSV and CIRV replication proteins and DI-72 repRNA in Nicotiana benthamiana. These experiments also revealed that the T33/T92 combination supported repRNA replication more efficiently (Fig. 2, lanes 1 to 4) than C36/C95 (lanes 5 to 8). However, the use of heterologous combinations of tombusvirus replication proteins revealed that the T33 replication protein and not the T92 replication protein is responsible for the enhanced level of replication in plants (Fig. 2). This difference between in vitro and in planta data could be due to the ability of T33 or C36 to induce membrane proliferation in plant cells that promote more efficient replication (these features cannot manifest in the CFE). Nevertheless, the in planta experiments demonstrated that the heterologous combinations of tombusvirus replication proteins are functional in supporting repRNA replication.
Fig 2.

The heterologous combinations of TBSV and CIRV replication proteins are functional in N. benthamiana. The accumulation of DI-72 repRNA was measured by Northern blotting in N. benthamiana leaves. The expression of TBSV and CIRV replication proteins and the repRNA was launched from the 35S promoter in an Agrobacterium plasmid (introduced into the leaves via agroinfiltration). Samples were taken from the infiltrated leaves at 3.5 days after infiltration. Note that coagroinfiltration of single protein-expressing constructs with the repRNA-expressing construct did not result in repRNA accumulation (lanes 17 to 24). Each experiment was repeated. (B) Affinity binding (pulldown) assay to detect interaction between GST-His6-p33C (representing the C-terminal half of T33, involved in protein interaction) or GST-His6-p36C (representing the C-terminal half of C36) and the MBP-tagged TBSV and CIRV replication proteins. The MBP-tagged TBSV and CIRV replication proteins and the MBP produced in E. coli were immobilized on amylose-affinity columns. GST-His6-tagged p33C or GST-His6-p36C expressed in E. coli was then passed through the amylose affinity columns with immobilized MBP-tagged proteins. The affinity-bound proteins were specifically eluted with maltose from the columns. The eluted proteins were analyzed by Western blotting with anti-6×His or anti-GST antibody to detect the amount of GST-His6, GST-His6-p33C, or GST-His6-p36C specifically bound to MBP-tagged viral proteins. A similarly produced GST-His6 protein preparation was used as a negative control.
Tombusvirus replication depends on the interaction between the S1/S2 subdomains common in the T33/C36 RNA chaperone and T92/C95 RdRp proteins, which is needed for the assembly of the functional VRC (61). The above observations that the heterologous combinations of tombusvirus replication proteins support tombusvirus RNA replication in the CFE assay and in planta suggest that the heterologous replication proteins likely interact with one another. To test the heterologous interactions, we performed pulldown assays with immobilized MBP-tagged viral replication proteins and the GST-tagged T33C (the C-terminal half, p33C) or GST-C36C (the C-terminal half, p36C). This assay confirmed interaction between the heterologous replication proteins that was comparable to the interaction between the homologous replication proteins (Fig. 2B).
Tombusviruses can replicate in microsomal and mitochondrial preparations in vitro.
To better understand the roles of different subcellular membranes in tombusvirus replication and to test what subcellular membranes can be used for repRNA replication by TBSV and CIRV replication proteins in vitro, we isolated microsomal (representing the ER membrane), peroxisomal, and mitochondrial fractions from yeast, followed by in vitro replication assay with purified recombinant tombusvirus replication proteins (Fig. 3A). Interestingly, we found that the microsomal preparations, which lacked detectable peroxisomal and mitochondrial marker proteins (Fig. 3B, bottom panels), supported repRNA replication ∼16-fold more efficiently in the presence of T33/T92 than in the presence of C36/C95 and the S100 fraction of CFE (the membrane-free supernatant that provides essential soluble host proteins) (Fig. 3B, lane 1 versus 2). These data suggest that the isolated ER membrane can support the assembly of both TBSV and CIRV VRCs, although the CIRV replication proteins show poor activity in this environment.
Fig 3.

In vitro reconstitution of the TBSV and CIRV replicases in yeast microsome and mitochondrial preparations. (A) Scheme of the replication assays. The purified recombinant T33 and T92 as well as C36 and C95 replication proteins and the TBSV-derived (+)repRNA were used as described for Fig. 1. (B) Top panel, denaturing PAGE analysis of the 32P-labeled repRNA products obtained in the replication assays with the isolated yeast microsome preparation. The synthesized full-length repRNA is indicated by an arrowhead. The result from the replication assay with T33 and T92 was chosen as 100% (lane 1). Each experiment was repeated. Bottom panels, Western blot analysis of various marker proteins in the microsome preparation with the help of specific antibodies. The left lane represents the standard yeast proteins present in CFE as positive controls. (C) Detection of plus- and minus-strand RNA products produced by the reconstituted TBSV and CIRV replicases in the microsome-based replication assay. See further details described for Fig. 1C. (D) The TBSV and CIRV replication proteins are functional in the mitochondrion-based replication assay. Top panel, the activity of the reconstituted tombusvirus replicases is estimated as for panel B. Denaturing PAGE analysis of the replicase products is as shown for panel B. Note that we used the 2× purified mitochondria preparations for this assay. Bottom panels, Western blot analysis of various marker proteins in the microsome preparations with the help of specific antibodies. The left lane represents the standard yeast proteins from yeast induced with oleic acid as positive controls. The crude mitochondrial sample was prepared without sucrose density gradient centrifugation, while 1× and 2× indicate single and double sucrose density gradient-purified mitochondrial preparations, respectively. (E) Detection of plus- and minus-strand RNA products produced by the reconstituted TBSV and CIRV replicases in the mitochondrion-based replication assay. See further details described for Fig. 1C.
To test whether viral RNA synthesis includes the full cycle of replication in the microsomal preparations, we estimated the ratio of newly made plus-strand to minus-strand repRNA levels in the in vitro replication assay (Fig. 3C). Interestingly, both TBSV and CIRV replication proteins supported asymmetrical viral RNA synthesis by producing ∼11 to 14 times more new plus strands than minus strands in the microsomal preparations (Fig. 3C). Thus, even CIRV replication proteins are capable of supporting full replication, albeit less efficiently than the TBSV replication proteins, in the microsomal preparations.
Similar experiments with purified mitochondrial preparations revealed that both TBSV and CIRV replication proteins supported repRNA replication in vitro (Fig. 3D). Thus, unlike with the microsomal preparations, the CIRV replication proteins are fully active on the mitochondrial membrane, which is also used by CIRV in yeast and plants (16, 81). Both TBSV and CIRV replication proteins supported asymmetrical viral RNA synthesis by producing ∼9 to 10 times more new plus strands than minus strands in the mitochondrial preparations (Fig. 3E). Thus, these data indicate that the mitochondrial membrane can support full TBSV and CIRV replication in vitro.
Unfortunately, the isolated oleate-induced peroxisomal preparations did not support repRNA replication with TBSV and CIRV replication proteins (not shown). Therefore, we decided to test tombusvirus RNA replication using sucrose-gradient fractionated crude mitochondrial and peroxisomal preparations (Fig. 4). We found that fractions 12 to 16 of the crude peroxisomal preparation (obtained from yeast after induction with oleic acid), which had the highest concentration of Fox3 peroxisomal marker (Fig. 4A, lanes 12 to 16) while containing Sec61 ER marker or porin mitochondrial marker proteins in small amounts, did not support repRNA replication by C36/C95 and T33/T92. The only repRNA replication with C36/C95 and T33/T92 was seen with fractions 3 to 11, which contained the largest amount of contaminating ER and mitochondrial membranes (based on the presence of Sec61 ER and porin mitochondrial marker proteins in these fractions) (Fig. 4A, lanes 3 to 11). Based on these data, we conclude that the peroxisomal preparations obtained from yeast induced by oleic acid cannot support tombusvirus replication in vitro. This could be due to the fragile nature of the peroxisomes during the isolation procedure or to other, unknown factors.
Fig 4.

In vitro reconstitution of the TBSV and CIRV replicases in yeast membrane fractions. (A) Yeast was grown on oleate-rich medium to increase peroxisome numbers prior to isolation. The crude peroxisome sample was subjected to 10 to 70% sucrose density gradient centrifugation, and the fractions of the sucrose gradient were tested for the ability to support RNA replication by the CIRV or TBSV replicases assembled in vitro. The purified recombinant T33 and T92 as well as C36 and C95 replication proteins and the TBSV-derived (+)repRNA were used as described for Fig. 1. Top two panels, denaturing PAGE analysis of the 32P-labeled repRNA products obtained in the replication assays with various membrane fractions. The synthesized full-length repRNA is indicated by an arrow. The most active fraction in the replication assay was chosen as 100%. The fractions most enriched for peroxisome are boxed with dotted lines. The samples on the left represent the top of the gradient (10%), while the samples on the right are from the bottom of the gradient (70%). Bottom panels, Western blot analysis of various marker proteins in the membrane fractions with the help of specific antibodies. (B) The crude mitochondrial sample was subjected to 10 to 70% sucrose density gradient centrifugation, and the fractions of the sucrose gradient were tested for the ability to support RNA replication by the CIRV or TBSV replicases assembled in vitro. The most active fraction in the replication assay was chosen as 100%. The fractions most enriched for mitochondria are boxed with dotted lines. See further details described for panel A.
In contrast, fractions 17 and 18 of the crude mitochondrial preparation (after high-speed centrifugation in a sucrose gradient) supported repRNA replication by C36/C95 and T33/T92 at the highest efficiency (Fig. 4B, lanes 17 to 20). These fractions were enriched for mitochondria, since they had the porin mitochondrial marker protein in the highest concentration but contained Sec61 ER marker or peroxisomal, vacuolar, and cytosolic markers at low levels. These data confirmed that the enriched mitochondrial membrane could provide a suitable environment for tombusvirus VRC assembly and repRNA replication in vitro.
Since the ER membrane has been shown to support efficient TBSV replication in yeast (22, 53), we decided to use both the purified microsomal and mitochondrial preparations for the follow-up experiments. Accordingly, to further test whether the microsomal and mitochondrial preparations could assemble authentic tombusvirus VRCs, we performed time course experiments with micrococcal nuclease, which digests the unprotected viral RNA (Fig. 5A) (54, 55). Interestingly, similar to the case for CFE (54, 55), both microsomal and mitochondrial preparations with C36/C95 and T33/T92 replication proteins protected ∼15 to 24% of the newly made 32P-labeled repRNA (representing the minus- and plus-strand replication products) if added 60 min after the start of the assay (Fig. 5B and C, lanes 5 and 15). The addition of micrococcal nuclease during the first 15 min of the assay eliminated repRNA synthesis (Fig. 5B and C, lanes 2 and 12), likely due to the lack of (or incomplete) VRC assembly, which takes 30 to 60 min in vitro (54, 55). Altogether, we observed that the recruited repRNA becomes nuclease protected after 30 to 45 min in the microsome preparation in the presence of the viral replication proteins (Fig. 5B, lanes 3 and 13), while it takes 45 to 60 min in the mitochondrial preparation to assemble VRCs and protect the recruited repRNA from micrococcal nuclease (Fig. 5C, lanes 4 and 5 and lanes 14 and 15). These data suggest that both microsomal and mitochondrial preparations with C36/C95 and T33/T92 replication proteins could assemble authentic VRCs that protect the VRC-bound viral RNA from nucleases. In addition, it seems that the VRC assembly with C36/C95 and T33/T92 replication proteins is faster in the microsomal than in the mitochondrial preparations.
Fig 5.

The in vitro-assembled TBSV or CIRV replicases form an RNase-resistant structure in microsomal or mitochondrial preparations. (A) Scheme of the in vitro assay. The in vitro reconstitution of the TBSV or CIRV replicases is started by the addition of purified recombinant T33 and T92 as well as C36 and C95 replication proteins and the TBSV-derived (+)repRNA (zero time point) as described for Fig. 1. Note that we applied a 15-min treatment with micrococcal nuclease (which was inactivated by addition of EGTA at the end of the treatment) at various time points, followed by RNA synthesis up to 4 h (total length of incubation). (B and C) Denaturing PAGE analysis of the 32P-labeled repRNA products obtained. Note that only the VRC-bound (membrane-associated) repRNA is resistant to nuclease treatment and not the (+)repRNA released to the buffer from the VRCs. (B) Results with microsomal preparations; (C) results with mitochondrial preparations. (D) Intact organellar membranes are required for tombusvirus replication in vitro. Various amounts of sorbitol in the assay buffer were used to test TBSV and CIRV replication.
Osmotic pressure is important during the isolation of pure and intact mitochondria (35). To test whether the osmotic pressure is important during tombusvirus replication in vitro in microsomal or mitochondrial membranes, we compared the effects of different concentrations of sorbitol in the assay buffer. We found that in the absence of sorbitol in the assay buffer, neither TBSV nor CIRV could replicate in microsomal or mitochondrial preparations (Fig. 5D, lanes 3 and 7). These data suggest that intact microsomal or mitochondrial membranes should be maintained to support viral VRC assembly or replication. On the other hand, the purified tombusvirus replicase does not require sorbitol for RNA synthesis in vitro (50, 51), excluding that the replicase depends on sorbitol for function. We propose that the sorbitol is needed in the assay buffer to keep the organellar membranes intact and functional during the assay.
Multiple domains within the replication proteins are responsible for different level of tombusvirus replication in ER or mitochondrial membranes.
To test what domain of the tombusvirus replication proteins is responsible for the observed differences between the TBSV and CIRV in utilizing microsomal and mitochondrial membranes, first we used heterologous combinations of CIRV and TBSV replication proteins to support RNA replication in vitro based on microsomal and mitochondrial preparations. Interestingly, the heterologous combination of C36/T92 supported repRNA replication almost as efficiently as the homologous combination of T33/T92 (Fig. 6A, lanes 5 and 6 versus 1 and 2) in the microsomal preparation, while combinations of C36/C95 and T33/C95 replication proteins supported repRNA replication at only ∼10 to 15% of the level with T33/T92 (Fig. 6A, lanes 3 and 4 and lanes 7 and 8 versus lanes 1 and 2). Thus, we conclude that the T92 RdRp protein is far more active than the C95 RdRp protein and that T92 is the major determinant of the efficient use of the ER membrane for in vitro repRNA replication. In addition, we note that the T33 replication cofactor has a better stimulatory effect on the activity of the tombusvirus replicase than the C36 replication cofactor in case of the ER membrane.
Fig 6.

In vitro reconstitution of heterologous combinations of tombusvirus replicases in yeast microsome and mitochondrial preparations. (A) Denaturing PAGE analysis of the 32P-labeled repRNA products obtained in replication assays with the isolated yeast microsome preparation. The synthesized full-length repRNA is indicated by an arrow. The result from the replication assay with T33 and T92 was chosen as 100% (lanes 1 and 2). (B) Denaturing PAGE analysis of the 32P-labeled repRNA products obtained in the replication assays with the isolated yeast mitochondrial preparation. (C) SDS-PAGE analysis of microsome membrane association assay using [35S]methionine-labeled recombinant T33 or C36 and a microsome preparation in the presence of the soluble extract from yeast. The bottom panel (encircled) represents samples incubated in the absence of the microsome preparation. (D) Mitochondrial membrane association assay. [35S]methionine-labeled recombinant T33 or C36 was used with the mitochondrial preparation in the presence of the soluble extract from yeast. Asterisks represent 35S-labeled proteins. See further details described for panel C.
The picture was different with the mitochondrial preparation because of the improved stimulatory effect of the C36 replication cofactor on the activity of the tombusvirus replicase (Fig. 6B). Accordingly, a high replication level was supported by the C36/T92 and C36/C95 combinations (Fig. 6B). While the homologous combination of T33/T92 supported high-level replication (Fig. 6B, lanes 1 and 2), the heterologous combination of T33 and C95 supported the lowest level of repRNA replication (Fig. 6B, lanes 7 and 8), suggesting that the T33 protein is less efficient than C36 in the mitochondrial membrane. The observed differences between T33 and C36 are unlikely due to differences in membrane associations, since we found that both T33 and C36 replication proteins associated with microsomal and mitochondrial membranes efficiently in vitro (Fig. 6C and D).
Since the tombusvirus RdRp proteins have two major domains (Fig. 7A), we have made chimeric constructs between the TBSV T92 and CIRV C95 proteins, as shown in Fig. 7B. The N-terminal domain of the TBSV T92 and CIRV C95 RdRp proteins is identical with that of the T33 or C36 cofactor protein (Fig. 7A), while the C-terminal domain harbors the highly conserved RdRp functional motifs. Testing the chimeric RdRp proteins in microsomal preparations revealed that C36-T92 (in combination with C36 cofactor), carrying the RdRp domain of the TBSV T92 and the N-terminal C36 domain, supported repRNA replication at up to 40% of the level with the combination of T33 cofactor and T92 RdRp (Fig. 7C, lanes 7 and 8 versus 1 and 2). In contrast, the chimeric T33-C95 RdRp protein (in combination with T33 cofactor) supported repRNA replication poorly in microsomal preparations (lanes 5 and 6). Thus, these experiments indicate that the C-terminal RdRp domain in T92 is responsible for the efficient utilization of the ER membrane, while the homologous RdRp domain of C95 is less efficient in this environment. The data also support that the T33 overlapping domain within the RdRp protein is more active in the ER than the C36 overlapping domain.
Fig 7.
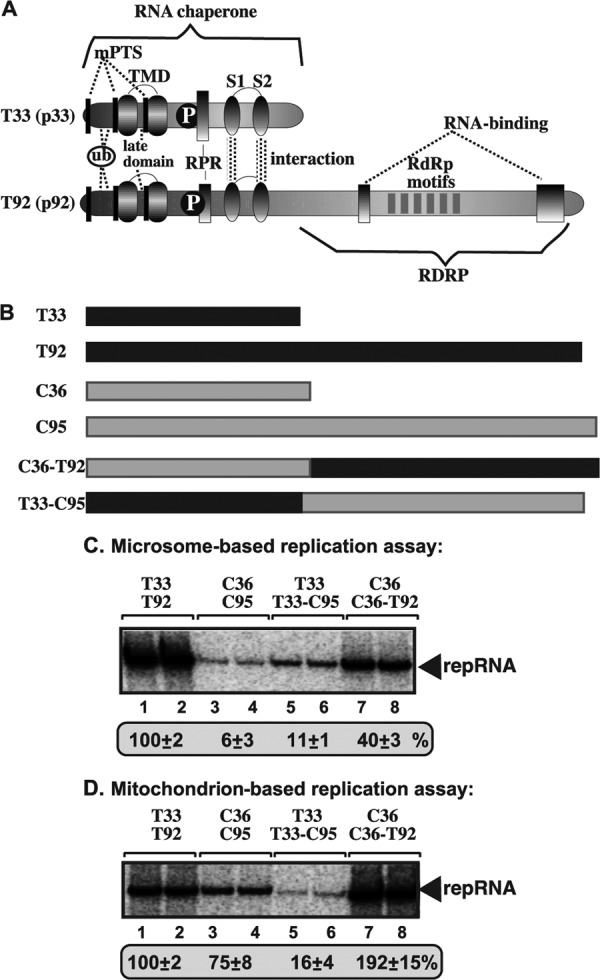
In vitro reconstitution of chimeric tombusvirus replicases in yeast microsome and mitochondrial preparations. (A) The known functional domains in the TBSV p33 RNA chaperone and the p92pol RdRp protein. The N-terminal segment in p92pol contains the same sequence as in p33 due to the strategy for overlapping expression of the TBSV genome, while the C-terminal region of p92pol carries the RdRp domain. mPTS, peroxisomal membrane targeting sequences; ub, monoubiquitinated region; TMD, transmembrane domains; late domain, sequence recognized by the ESCRT factors; P, phosphorylation sites; RPR, arginine-proline-rich RNA binding domain; S1 and S2, subdomains of the p33:p33/p92 interaction domain. (B) Schematic representation of the chimeric RdRp proteins made between the corresponding TBSV and CIRV replication proteins as shown. (C) Denaturing PAGE analysis of the 32P-labeled repRNA products obtained in replication assays using the chimeric RdRp proteins based on the isolated yeast microsome preparation. The synthesized full-length repRNA is indicated by an arrow. The result from the replication assay with T33 and T92 was chosen as 100% (lanes 1 and 2). (D) Denaturing PAGE analysis of the 32P-labeled repRNA products obtained in the replication assays with the chimeric RdRp proteins using the isolated yeast mitochondrial preparation. See further details described for panel C.
Similar experiments with mitochondrial preparations revealed that the RdRp domain of the TBSV T92 is still ∼3-fold more active than the corresponding domain of the CIRV C95 (compare the chimeric C36-T92 RdRp with C95 RdRp in combination with C36 cofactor) (Fig. 7D, lanes 7 and 8 versus 3 and 4). However, we also observed an ∼2-fold stimulatory effect of the C36 overlapping domain when present in the RdRp protein (compare the chimeric C36-T92 RdRp with T92 RdRp) (Fig. 7D, lanes 7 and 8 versus 1 and 2). In addition, the T33 cofactor and T33-C95 RdRp combination supported a low level of repRNA replication, demonstrating that the T33 overlapping domain is poorly adapted to the mitochondrial membrane, as noted above with the heterologous combination of replication proteins (Fig. 6B, lanes 7 and 8).
Since it seems that the T33 protein and the T33 overlapping domain when present in the RdRp protein are not well suited to support RNA replication in the mitochondrial membrane compared with the C36 cofactor, we made chimeras between T33 and C36 and their corresponding domains in the RdRp proteins, as shown in Fig. 8A and D. We have divided the T33 and C36 sequences into three subdomains: the N-terminal subdomain known to be involved in intracellular localization of T33 and C36 cofactors (34, 47, 81), the central subdomain that includes the two transmembrane sequences (TMD), and the C-terminal subdomain involved in protein-RNA and protein-protein interactions (59–61).
Fig 8.

In vitro reconstitution of additional chimeric tombusvirus replicases in yeast microsome and mitochondrial preparations. (A) Schematic representation of the chimeric replication proteins made between the corresponding TBSV and CIRV replication proteins as shown. The T33 and C36 sequences were divided into three segments based on the known functions/roles (see Fig. 7A). (B) Denaturing PAGE analysis of the 32P-labeled repRNA products obtained in replication assays using the chimeric tombusvirus replication proteins based on the isolated yeast microsome preparation. The synthesized full-length repRNA is indicated by an arrow. The result from the replication assay with T33 and T92 was chosen as 100% (lanes 1 and 2). (C) Denaturing PAGE analysis of the 32P-labeled repRNA products obtained in the replication assays with the chimeric tombusvirus replication proteins using the isolated yeast mitochondrial preparation. See further details described for panel B. (D) Coomassie blue-stained SDS-PAGE of the affinity-purified replication proteins expressed in E. coli as MBP fusion proteins.
Testing the chimeric constructs in the mitochondrial preparations revealed that replacing the N and TMD subdomains of T33 with those of C36 in the T33 cofactor and T92 RdRp resulted in a highly active chimera (T33c and T92c), which replicated the most efficiently (Fig. 8C, lanes 5 and 6 versus 1 and 2). This chimera, however, replicated the repRNA more efficiently even in the microsome preparations (Fig. 8B, lanes 5 and 6 versus 1 and 2), suggesting that chimeric tombusviruses might have increased potential to replicate in various intracellular membranes. These observations could be relevant for the evolution of tombusviruses (see Discussion).
Combinations of heterologous replication proteins show both ER and mitochondrial localization in yeast.
The in vitro data show that TBSV can efficiently replicate in both ER and mitochondrial membranes, while the CIRV replication proteins favor the mitochondrial membrane over the ER membrane to support viral repRNA replication. To test whether the tombusvirus replication proteins can indeed utilize these membranes in cells, we performed localization studies with GFP-, yellow fluorescent protein (YFP)-, and CFP-tagged proteins in yeast using homologous and heterologous combinations of the tombusvirus replication proteins.
As expected, the homologous combination of T33/T92 localized mostly in the ER membrane in yeast (in pex3Δ yeast to mimic the in vitro situation with isolated microsomal preparations by using yeast lacking peroxisomes) based on the Pho86 ER protein, while only a small fraction of C36 and C95 localized to the ER membrane, although these proteins were frequently located in the vicinity of the ER (Fig. 9A). In contrast, the homologous combination of C36 and C95 localized mostly in the mitochondrial membranes in yeast based on staining with rhodamine B (a mitochondrial dye used in yeast) (11, 46, 58), while only a small fraction of T33 and T92 localized in the mitochondrial membranes (Fig. 9B). Indeed, most of the T33 and T92 proteins did not colocalize with mitochondrial targeting sequence (MTS)-CFP mitochondrial marker protein (not shown) or with rhodamine B mitochondrial dye (Fig. 9B). Altogether, these experiments established that the homologous combination of T33/T92 is localized mostly in the ER (in pex3Δ yeast) with a fraction in the mitochondria, while C36/C95 is located in the mitochondrial membrane, as shown previously (22, 53, 81).
To test the membrane preference of the heterologous combinations of the tombusvirus replication proteins in pex3Δ yeast, first we performed colocalization studies. We found that the heterologous combinations of either T33/C95 or C36/T92 are colocalized in yeast cells (Fig. 9C). Second, we performed subcellular localization of YFP-C95 in pex3Δ yeast coexpressing T33, which showed partial colocalization with the CFP-Pho86 ER marker protein (Fig. 9D) and the mitochondrial dye (Fig. 9E). Similarly, YFP-T33 showed partial colocalization with the CFP-Pho86 ER marker protein (Fig. 9D) and the mitochondrial dye (Fig. 9E) in yeast coexpressing the heterologous C95. Thus, it seems that T33 and C95, although colocalized, are present in both the ER and mitochondria, with fractions of the T33 and C95 molecules divided between the two organelles.
Intriguingly, we observed a similar split/divided distribution of C36 and T92 between the ER and mitochondrial membranes (Fig. 9F and G) in heterologous coexpression studies with pex3Δ yeast. Therefore, we suggest that the tombusvirus replication proteins can be localized to both ER and mitochondrial membranes in yeast coexpressing the heterologous combinations (T33/C95 or C36/T92) of replication proteins. This distribution could be interesting during tombusvirus evolution by allowing less restricted use of subcellular membranes by putative interviral tombusvirus recombinants (see Discussion).
DISCUSSION
All known (+)RNA viruses of plants and animals depend on various subcellular membranes for their replication, yet we do not know why different (+)RNA viruses select different subcellular membranes/compartments for replication. Tombusviruses could be valuable for understanding the roles of various subcellular membranes in viral replication since they show different preferences. For example, TBSV, CNV, and CymRSV preferably utilize the peroxisomal membranes or, in the absence of peroxisomes, the ER membranes (22, 34, 44, 47, 53). On the other hand, CIRV replicates in the mitochondrial membranes (16, 81). At a late stage of tombusvirus replication in plants, however, large multivesicular bodies form that frequently contain ER membranes and mitochondria as well (66). These observations suggest complex interactions between subcellular membranes and tombusvirus replication proteins.
In this study, we have developed in vitro tombusvirus replication assays with isolated organelles (Fig. 3) or enriched organellar preparations (Fig. 4) to directly address the roles of the various subcellular membranes in tombusvirus replication. We have shown the following: (i) both plus- and minus-strand RNA syntheses occur in these assays; (ii) the process is asymmetrical, leading to an excess amount of plus over minus strands; (iii) a membrane-bound replicase complex forms; (iv) there is a requirement of cellular factors; (v) there is requirement of both p33 and p92 replication proteins for replication; (vi) the newly made (+)RNAs are released to the solution; and (vii) the (−)RNA is kept protected in the replicase bound to the membrane. All these pieces of evidence support that the tombusvirus replication in the isolated organelles is a complete cycle of an authentic replication process, similar to that developed using the whole CFE (54, 55).
Interestingly, we found that TBSV replication proteins utilized the isolated ER membrane efficiently for repRNA replication, while the CIRV replication proteins did not (Fig. 3 and 4). These data are in agreement with the in vivo observations that TBSV uses the ER membranes (in the absence of the peroxisomes), while CIRV favors the mitochondria for replication in yeast and plant cells (16, 22, 44, 47, 53, 81). Surprisingly, however, TBSV was also able to utilize the isolated mitochondria for replication (Fig. 3 and 4), suggesting that this tombusvirus could be less restricted in its ability to utilize subcellular membranes. Indeed, we did see some colocalization of TBSV T33 and T92 replication proteins with mitochondrial markers (based on both MTS-CFP (not shown) and a mitochondrial dye) (Fig. 9B) in pex3Δ yeast, suggesting that mitochondria are likely used for TBSV replication at some point, possibly at the late stage of replication when peroxisomal or ER membranes have already been fully exploited. Similarly, we observed some colocalization of CIRV C36 and C95 replication proteins with the ER marker protein (Fig. 9A), supporting that ER membranes might be targeted for CIRV replication. However, the activity of the CIRV replicases in the ER membranes is likely less robust than that in the mitochondrial membranes, based on the in vitro experiments with the isolated microsomes (Fig. 3). Altogether, the in vitro and in vivo experiments suggest that TBSV shows rather high flexibility in membrane utilization for replication, while CIRV is somewhat more restricted, at least in vitro.
Unfortunately, we failed to obtain peroxisomal preparations supporting either TBSV or CIRV replication from yeast cultured in oleic acid medium to induce peroxisome formation (Fig. 4). It is possible that peroxisomes are too fragile and damaged during the isolation procedure. It is also possible that the oleic acid induced peroxisomes are not suitable to support TBSV replication. Indeed, addition of oleic acid to the culture medium did not increase TBSV replication in yeast (T. Panavas and P. D. Nagy, unpublished data). Therefore, it is highly likely that the CFEs obtained from yeast support TBSV replication occurring mainly in the ER-derived membranes (Fig. 1). Because the ER is as suitable to support TBSV replication as the peroxisomes in yeast (22, 53), the obtained in vitro data are likely valuable in dissecting TBSV replication in vitro. We also propose that the CFE likely supports weak CIRV replication (compared with TBSV) due to the limiting amount of mitochondria present in the CFE prepared from yeast cultured under the standard conditions. Indeed, comparison of CFE (Fig. 1) and microsomal and mitochondrial preparations (Fig. 3 and 4) revealed similarity between CFE and microsomal preparations, suggesting that most of the in vitro repRNA replication in the CFE is likely supported by the ER membrane. This could be due to the growth conditions for yeast, which favor the presence of low numbers of mitochondria and peroxisomes but abundant ER membranes (13). The isolated mitochondrial preparation, however, supported CIRV-based repRNA replication efficiently, making this approach suitable for future mechanistic studies.
Combinations of heterologous replication proteins reveal remarkable flexibility of membrane usage by tombusviruses.
One of the surprising discoveries from the in vitro tombusvirus replication assays with the combinations of heterologous replication proteins is the extended ability of tombusviruses to utilize subcellular membranes more efficiently than some homologous combinations. For example, CIRV C36 and C95 colocalized more efficiently with the ER membranes when present in heterologous than when present in homologous combinations (Fig. 9). Moreover, the CIRV C36 protein became part of a more efficient replicase in the ER membranes when associated with T92 RdRp protein than in homologous combination with C95 (Fig. 6A) without becoming less efficient in the mitochondrial membrane (Fig. 6B). Also, the TBSV T92 RdRp showed increased activity in the mitochondrial membrane when combined with C36 cofactor than in combination with TBSV T33 (Fig. 6B). This suggests that tombusviruses might be able to utilize various subcellular membranes more efficiently during some coinfections with other tombusviruses than during single infections.
Even more interesting is the possibility of generation of chimeric tombusviruses due to RNA recombination between tombusviruses. RNA recombination is well documented for tombusviruses in vitro, in yeast, and in planta (8, 17–19, 38, 71, 72, 82, 83). The formation of chimeric tombusviruses could expand the efficiency of using various subcellular compartments by the tombusvirus replicase, based on the chimeric constructs tested for Fig. 7 and 8. Indeed, particular chimeric constructs replicated efficiently in both ER and mitochondrial preparations (e.g., T33c/T92c [Fig. 8]). We propose that the extra flexibility in membrane usage by these chimeric tombusviruses could be useful for tombusviruses when infecting some plant species, thus expanding the wide range of plants supporting tombusvirus replication. Accordingly, recombinant CIRV strains that had N-terminal sequences similar to that of the TBSV p33 replication protein and targeted the peroxisome for replication were recently isolated (24, 26). Thus, recombination involving the p33/p36 open reading frame (ORF) can occur in nature, creating new variants or strains.
Adaptation to subcellular membranes for robust tombusvirus replication depends on multiple domains within the replication proteins.
The heterologous combinations of tombusvirus replication proteins revealed that T92 and C95 are major determinants of repRNA replication in particular subcellular membranes. For example, the C95 RdRp was mostly functional in the mitochondrial preparations, while the T92 RdRp was active in both microsomal and mitochondrial preparations (Fig. 6C and D). Furthermore, the results with chimeric proteins suggest that the RdRp domain in T92 is very active in both microsomal and mitochondrial preparations (Fig. 7C and D), while the origin of the N-terminal, overlapping domain in the tombusvirus RdRp was also important for the activity of VRCs during replication. Therefore, we suggest that multiple domains within the replication proteins affect the ability and efficiency of the tombusvirus VRCs to support repRNA replication in particular subcellular membranes. It is also possible that C36 forms a single “domain” that favors mitochondrial targeting as a whole and that swapping its C-terminal and/or transmembrane region with corresponding regions of T33 disrupts the structure necessary for specific targeting to mitochondria.
Altogether, the developed in vitro tombusvirus replication assays based on CFE, isolated microsomes, and mitochondrial preparations will be powerful to gain mechanistic insights into the roles of membranes in (+)RNA virus replication, virus-host interactions, and possibly viral evolution.
ACKNOWLEDGMENTS
We thank Judit Pogany and Daniel Barajas for critical reading of the manuscript and for very helpful suggestions. We thank Daniel Barajas for providing the pGEX-his-T33C plasmid and Michael Goodin for sharing the pGDG vector. Sec61p and Fox3p antibodies were provided by Tom Rapoport from Harvard Medical School and by Daniel J. Klionsky from University of Michigan, respectively.
This work was supported by NIH, NIAID (grant 5R21AI079457-02), and by the University of Kentucky.
Footnotes
Published ahead of print 12 September 2012
REFERENCES
- 1. Barajas D, Jiang Y, Nagy PD. 2009. A unique role for the host ESCRT proteins in replication of Tomato bushy stunt virus. PLoS Pathog. 5:e1000705 doi:10.1371/journal.ppat.1000705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barajas D, Li Z, Nagy PD. 2009. The Nedd4-type Rsp5p ubiquitin ligase inhibits tombusvirus replication by regulating degradation of the p92 replication protein and decreasing the activity of the tombusvirus replicase. J. Virol. 83:11751–11764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barajas D, Nagy PD. 2010. Ubiquitination of tombusvirus p33 replication protein plays a role in virus replication and binding to the host Vps23p ESCRT protein. Virology 397:358–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bartenschlager R, Cosset FL, Lohmann V. 2010. Hepatitis C virus replication cycle. J. Hepatology. 53:583–585 [DOI] [PubMed] [Google Scholar]
- 5. Burgyan J, Rubino L, Russo M. 1996. The 5′-terminal region of a tombusvirus genome determines the origin of multivesicular bodies. J. Gen. Virol. 77:1967–1974 [DOI] [PubMed] [Google Scholar]
- 6. Castorena KM, Stapleford KA, Miller DJ. 2010. Complementary transcriptomic, lipidomic, and targeted functional genetic analyses in cultured Drosophila cells highlight the role of glycerophospholipid metabolism in Flock House virus RNA replication. BMC Genomics 11:183 doi:10.1186/1471-2164-11-183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chakrabarty R, et al. 2007. PSITE vectors for stable integration or transient expression of autofluorescent protein fusions in plants: probing Nicotiana benthamiana-virus interactions. Mol. Plant Microbe Interact. 20:740–750 [DOI] [PubMed] [Google Scholar]
- 8. Cheng CP, Nagy PD. 2003. Mechanism of RNA recombination in carmo- and tombusviruses: evidence for template switching by the RNA-dependent RNA polymerase in vitro. J. Virol. 77:12033–12047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. den Boon JA, Diaz A, Ahlquist P. 2010. Cytoplasmic viral replication complexes. Cell Host Microbe 8:77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Diaz A, Wang X, Ahlquist P. 2010. Membrane-shaping host reticulon proteins play crucial roles in viral RNA replication compartment formation and function. Proc. Natl. Acad. Sci. U. S. A. 107:16291–16296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Durr M, et al. 2006. Nonredundant roles of mitochondria-associated F-box proteins Mfb1 and Mdm30 in maintenance of mitochondrial morphology in yeast. Mol. Biol. Cell 17:3745–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fernandez-Garcia MD, Mazzon M, Jacobs M, Amara A. 2009. Pathogenesis of flavivirus infections: using and abusing the host cell. Cell Host Microbe 5:318–328 [DOI] [PubMed] [Google Scholar]
- 13. Hofmann L, et al. 2009. A nonproteolytic proteasome activity controls organelle fission in yeast. J. Cell Sci. 122:3673–3683 [DOI] [PubMed] [Google Scholar]
- 14. Hsu NY, et al. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141:799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang TS, Nagy PD. 2011. Direct inhibition of tombusvirus plus-strand RNA synthesis by a dominant negative mutant of a host metabolic enzyme, glyceraldehyde-3-phosphate dehydrogenase, in yeast and plants. J. Virol. 85:9090–9102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hwang YT, McCartney AW, Gidda SK, Mullen RT. 2008. Localization of the Carnation Italian ringspot virus replication protein p36 to the mitochondrial outer membrane is mediated by an internal targeting signal and the TOM complex. BMC Cell Biol. 9:54 doi:10.1186/1471-2121-9-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jaag HM, Lu Q, Schmitt ME, Nagy PD. 2011. Role of RNase MRP in viral RNA degradation and RNA recombination. J. Virol. 85:243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jaag HM, Nagy PD. 2010. The combined effect of environmental and host factors on the emergence of viral RNA recombinants. PLoS Pathog. 6:e1001156 doi:10.1371/journal.ppat.1001156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jaag HM, Pogany J, Nagy PD. 2010. A host Ca2+/Mn2+ ion pump is a factor in the emergence of viral RNA recombinants. Cell Host Microbe 7:74–81 [DOI] [PubMed] [Google Scholar]
- 20. Jiang Y, Li Z, Nagy PD. 2010. Nucleolin/Nsr1p binds to the 3′ noncoding region of the tombusvirus RNA and inhibits replication. Virology 396:10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang Y, Serviene E, Gal J, Panavas T, Nagy PD. 2006. Identification of essential host factors affecting tombusvirus RNA replication based on the yeast Tet promoters Hughes Collection. J. Virol. 80:7394–7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jonczyk M, Pathak KB, Sharma M, Nagy PD. 2007. Exploiting alternative subcellular location for replication: tombusvirus replication switches to the endoplasmic reticulum in the absence of peroxisomes. Virology 362:320–330 [DOI] [PubMed] [Google Scholar]
- 23. Kapadia SB, Chisari FV. 2005. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. U. S. A. 102:2561–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koenig R, Lesemann DE, Pfeilstetter E. 2009. New isolates of carnation Italian ringspot virus differ from the original one by having replication-associated proteins with a typical tombusvirus-like N-terminus and by inducing peroxisome- rather than mitochondrion-derived multivesicular bodies. Arch. Virol. 154:1695–1698 [DOI] [PubMed] [Google Scholar]
- 25. Kovalev N, Pogany J, Nagy PD. 2012. A co-opted DEAD-box RNA helicase enhances tombusvirus plus-strand synthesis. PLoS Pathog. 8:e1002537 doi:10.1371/journal.ppat.1002537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Laliberte JF, Sanfacon H. 2010. Cellular remodeling during plant virus infection. Annu. Rev. Phytopathol. 48:69–91 [DOI] [PubMed] [Google Scholar]
- 27. Lee WM, Ahlquist P. 2003. Membrane synthesis, specific lipid requirements, and localized lipid composition changes associated with a positive-strand RNA virus RNA replication protein. J. Virol. 77:12819–12828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li Z, Barajas D, Panavas T, Herbst DA, Nagy PD. 2008. Cdc34p ubiquitin-conjugating enzyme is a component of the tombusvirus replicase complex and ubiquitinates p33 replication protein. J. Virol. 82:6911–6926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li Z, Nagy PD. 2011. Diverse roles of host RNA binding proteins in RNA virus replication. RNA Biol. 8:305–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Z, et al. 2009. Translation elongation factor 1A is a component of the tombusvirus replicase complex and affects the stability of the p33 replication co-factor. Virology 385:245–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Z, et al. 2010. Translation elongation factor 1A facilitates the assembly of the tombusvirus replicase and stimulates minus-strand synthesis. PLoS Pathog. 6:e1001175 doi:10.1371/journal.ppat.1001175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lin JY, Mendu V, Pogany J, Qin J, Nagy PD. 2012. The TPR domain in the host Cyp40-like cyclophilin binds to the viral replication protein and inhibits the assembly of the tombusviral replicase. PLoS Pathog. 8:e1002491 doi:10.1371/journal.ppat.1002491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mackenzie JM, Khromykh AA, Parton RG. 2007. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe 2:229–239 [DOI] [PubMed] [Google Scholar]
- 34. McCartney AW, Greenwood JS, Fabian MR, White KA, Mullen RT. 2005. Localization of the tomato bushy stunt virus replication protein p33 reveals a peroxisome-to-endoplasmic reticulum sorting pathway. Plant Cell 17:3513–3531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meisinger C, Sommer T, Pfanner N. 2000. Purification of Saccharomcyes cerevisiae mitochondria devoid of microsomal and cytosolic contaminations. Anal. Biochem. 287:339–342 [DOI] [PubMed] [Google Scholar]
- 36. Mendu V, Chiu M, Barajas D, Li Z, Nagy PD. 2010. Cpr1 cyclophilin and Ess1 parvulin prolyl isomerases interact with the tombusvirus replication protein and inhibit viral replication in yeast model host. Virology 406:342–351 [DOI] [PubMed] [Google Scholar]
- 37. Miller S, Krijnse-Locker J. 2008. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 6:363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nagy PD. 2011. The roles of host factors in tombusvirus RNA recombination. Adv. Virus Res. 81:63–84 [DOI] [PubMed] [Google Scholar]
- 39. Nagy PD. 2008. Yeast as a model host to explore plant virus-host interactions. Annu. Rev. Phytopathol. 46:217–242 [DOI] [PubMed] [Google Scholar]
- 40. Nagy PD, Pogany J. 2012. The dependence of viral RNA replication on co-opted host factors. Nat. Rev. Microbiol. 10:137–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nagy PD, Pogany J. 2008. Multiple roles of viral replication proteins in plant RNA virus replication. Methods Mol. Biol. 451:55–68 [DOI] [PubMed] [Google Scholar]
- 42. Nagy PD, Pogany J. 2006. Yeast as a model host to dissect functions of viral and host factors in tombusvirus replication. Virology 344:211–220 [DOI] [PubMed] [Google Scholar]
- 43. Nagy PD, Wang RY, Pogany J, Hafren A, Makinen K. 2011. Emerging picture of host chaperone and cyclophilin roles in RNA virus replication. Virology 411:374–382 [DOI] [PubMed] [Google Scholar]
- 44. Navarro B, Russo M, Pantaleo V, Rubino L. 2006. Cytological analysis of Saccharomyces cerevisiae cells supporting cymbidium ringspot virus defective interfering RNA replication. J. Gen. Virol. 87:705–714 [DOI] [PubMed] [Google Scholar]
- 45. Neuvonen M, et al. 2011. SH3 domain-mediated recruitment of host cell amphiphysins by alphavirus nsP3 promotes viral RNA replication. PLoS Pathog. 7:e1002383 doi:10.1371/journal.ppat.1002383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nowikovsky K, et al. 2004. The LETM1/YOL027 gene family encodes a factor of the mitochondrial K+ homeostasis with a potential role in the Wolf-Hirschhorn syndrome. J. Biol. Chem. 279:30307–30315 [DOI] [PubMed] [Google Scholar]
- 47. Panavas T, Hawkins CM, Panaviene Z, Nagy PD. 2005. The role of the p33:p33/p92 interaction domain in RNA replication and intracellular localization of p33 and p92 proteins of Cucumber necrosis tombusvirus. Virology 338:81–95 [DOI] [PubMed] [Google Scholar]
- 48. Panavas T, Nagy PD. 2003. Yeast as a model host to study replication and recombination of defective interfering RNA of Tomato bushy stunt virus. Virology 314:315–325 [DOI] [PubMed] [Google Scholar]
- 49. Panavas T, Serviene E, Brasher J, Nagy PD. 2005. Yeast genome-wide screen reveals dissimilar sets of host genes affecting replication of RNA viruses. Proc. Natl. Acad. Sci. U. S. A. 102:7326–7331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Panaviene Z, Panavas T, Nagy PD. 2005. Role of an internal and two 3′-terminal RNA elements in assembly of tombusvirus replicase. J. Virol. 79:10608–10618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Panaviene Z, Panavas T, Serva S, Nagy PD. 2004. Purification of the cucumber necrosis virus replicase from yeast cells: role of coexpressed viral RNA in stimulation of replicase activity. J. Virol. 78:8254–8263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pathak KB, Pogany J, Xu K, White KA, Nagy PD. 2012. Defining the roles of cis-acting RNA elements in tombusvirus replicase assembly in vitro. J. Virol. 86:156–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pathak KB, Sasvari Z, Nagy PD. 2008. The host Pex19p plays a role in peroxisomal localization of tombusvirus replication proteins. Virology 379:294–305 [DOI] [PubMed] [Google Scholar]
- 54. Pogany J, Nagy PD. 2008. Authentic replication and recombination of Tomato bushy stunt virus RNA in a cell-free extract from yeast. J. Virol. 82:5967–5980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pogany J, Stork J, Li Z, Nagy PD. 2008. In vitro assembly of the Tomato bushy stunt virus replicase requires the host heat shock protein 70. Proc. Natl. Acad. Sci. U. S. A. 105:19956–19961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pogany J, White KA, Nagy PD. 2005. Specific binding of tombusvirus replication protein p33 to an internal replication element in the viral RNA is essential for replication. J. Virol. 79:4859–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Qin J, Barajas D, Nagy PD. 2012. An inhibitory function of WW domain-containing host proteins in RNA virus replication. Virology 426:106–119 [DOI] [PubMed] [Google Scholar]
- 58. Quesada I, Verdugo P. 2005. InsP3 signaling induces pulse-modulated Ca2+ signals in the nucleus of airway epithelial ciliated cells. Biophys. J. 88:3946–3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rajendran KS, Nagy PD. 2003. Characterization of the RNA-binding domains in the replicase proteins of tomato bushy stunt virus. J. Virol. 77:9244–9258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rajendran KS, Nagy PD. 2004. Interaction between the replicase proteins of Tomato bushy stunt virus in vitro and in vivo. Virology 326:250–261 [DOI] [PubMed] [Google Scholar]
- 61. Rajendran KS, Nagy PD. 2006. Kinetics and functional studies on interaction between the replicase proteins of Tomato bushy stunt virus: requirement of p33:p92 interaction for replicase assembly. Virology 345:270–279 [DOI] [PubMed] [Google Scholar]
- 62. Reiss S, et al. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rieder SE, Emr SD. 2001. Isolation of subcellular fractions from the yeast Saccharomyces cerevisiae. Curr. Protoc. Cell Biol. Chapter 3:unit 3.8 [DOI] [PubMed] [Google Scholar]
- 64. Rubino L, Burgyan J, Russo M. 1995. Molecular cloning and complete nucleotide sequence of carnation Italian ringspot tombusvirus genomic and defective interfering RNAs. Arch. Virol. 140:2027–2039 [DOI] [PubMed] [Google Scholar]
- 65. Rubino L, Navarro B, Russo M. 2007. Cymbidium ringspot virus defective interfering RNA replication in yeast cells occurs on endoplasmic reticulum-derived membranes in the absence of peroxisomes. J. Gen. Virol. 88:1634–1642 [DOI] [PubMed] [Google Scholar]
- 66. Russo M, Burgyan J, Martelli GP. 1994. Molecular biology of tombusviridae. Adv. Virus Res. 44:381–428 [DOI] [PubMed] [Google Scholar]
- 67. Salonen A, Ahola T, Kaariainen L. 2005. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 285:139–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sasvari Z, Izotova L, Kinzy TG, Nagy PD. 2011. Synergistic roles of eukaryotic translation elongation factors 1Bgamma and 1A in stimulation of tombusvirus minus-strand synthesis. PLoS Pathog. 7:e1002438 doi:10.1371/journal.ppat.1002438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sasvari Z, Nagy PD. 2010. Making of viral replication organelles by remodeling interior membranes. Viruses 2:2436–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Serva S, Nagy PD. 2006. Proteomics analysis of the tombusvirus replicase: Hsp70 molecular chaperone is associated with the replicase and enhances viral RNA replication. J. Virol. 80:2162–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Serviene E, Jiang Y, Cheng CP, Baker J, Nagy PD. 2006. Screening of the yeast yTHC collection identifies essential host factors affecting tombusvirus RNA recombination. J. Virol. 80:1231–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Serviene E, et al. 2005. Genome-wide screen identifies host genes affecting viral RNA recombination. Proc. Natl. Acad. Sci. U. S. A. 102:10545–10550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shah Nawaz-Ul-Rehman M, et al. 2012. Proteome-wide overexpression of host proteins for identification of factors affecting tombusvirus RNA replication: an inhibitory role of protein kinase C. J. Virol. 86:9384–9395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sharma M, Sasvari Z, Nagy PD. 2011. Inhibition of phospholipid biosynthesis decreases the activity of the tombusvirus replicase and alters the subcellular localization of replication proteins. Virology 415:141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sharma M, Sasvari Z, Nagy PD. 2010. Inhibition of sterol biosynthesis reduces tombusvirus replication in yeast and plants. J. Virol. 84:2270–2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Stork J, Kovalev N, Sasvari Z, Nagy PD. 2011. RNA chaperone activity of the tombusviral p33 replication protein facilitates initiation of RNA synthesis by the viral RdRp in vitro. Virology 409:338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang RY, Nagy PD. 2008. Tomato bushy stunt virus co-opts the RNA-binding function of a host metabolic enzyme for viral genomic RNA synthesis. Cell Host Microbe 3:178–187 [DOI] [PubMed] [Google Scholar]
- 78. Wang RY, Stork J, Nagy PD. 2009. A key role for heat shock protein 70 in the localization and insertion of tombusvirus replication proteins to intracellular membranes. J. Virol. 83:3276–3287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang RY, Stork J, Pogany J, Nagy PD. 2009. A temperature sensitive mutant of heat shock protein 70 reveals an essential role during the early steps of tombusvirus replication. Virology 394:28–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Waters MG, Blobel G. 1986. Secretory protein translocation in a yeast cell-free system can occur posttranslationally and requires ATP hydrolysis. J. Cell Biol. 102:1543–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Weber-Lotfi F, Dietrich A, Russo M, Rubino L. 2002. Mitochondrial targeting and membrane anchoring of a viral replicase in plant and yeast cells. J. Virol. 76:10485–10496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. White KA, Morris TJ. 1994. Nonhomologous RNA recombination in tombusviruses: generation and evolution of defective interfering RNAs by stepwise deletions. J. Virol. 68:14–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. White KA, Morris TJ. 1994. Recombination between defective tombusvirus RNAs generates functional hybrid genomes. Proc. Natl. Acad. Sci. U. S. A. 91:3642–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. White KA, Nagy PD. 2004. Advances in the molecular biology of tombusviruses: gene expression, genome replication, and recombination. Prog. Nucleic Acid Res. Mol. Biol. 78:187–226 [DOI] [PubMed] [Google Scholar]