

Abstract
Human cytomegalovirus (HCMV) is the leading viral cause of birth defects and life-threatening lung-associated diseases in premature infants and immunocompromised children. Although the fetal lung is a major target organ of the virus, HCMV lung pathogenesis has remained unexplored, possibly as a result of extreme host range restriction. To overcome this hurdle, we generated a SCID-hu lung mouse model that closely recapitulates the discrete stages of human lung development in utero. Human fetal lung tissue was implanted into severe combined immunodeficient (CB17-scid) mice and inoculated by direct injection with the VR1814 clinical isolate of HCMV. Virus replication in the fetal lung was assessed by the quantification of infectious virus titers and HCMV genome copies and the detection of HCMV proteins by immunohistochemistry and Western blotting. We show that HCMV efficiently replicated in the lung implants during a 2-week period, forming large viral lesions. The virus productively infected alveolar epithelial and mesenchymal cells, imitating congenital infection of the fetal lung. HCMV replication triggered apoptosis near and within the viral lesions and impaired the production of surfactant proteins in the alveolar epithelium. Our findings highlight that congenital and neonatal HCMV infection can adversely impact lung development, leading to pneumonia and acute lung injury. We have successfully developed a small-animal model that closely recapitulates fetal and neonatal lung development and provides a valuable, biologically relevant tool for an understanding of the lung pathogenesis of HCMV as well as other human respiratory viruses. Additionally, this model would greatly facilitate the development and testing of new antiviral therapies for HCMV along with select human pulmonary pathogens.
INTRODUCTION
Human cytomegalovirus (HCMV) infection of the fetus and premature infants is a major global health problem, and in the United States, HCMV is the leading viral cause of birth defects, affecting about 1% of all live births (16). In congenital infection, HCMV spreads extremely efficiently into fetal organs following fetal viremia. Although pneumonitis is rarely the sole manifestation of congenital HCMV disease in the neonate (62), histological examinations of HCMV-infected fetuses identified the lung as a major target organ, with a large number of HCMV-infected cells (8, 25). The main burden of lung-associated HCMV diseases occurs during postnatal infection in premature infants (3, 22, 34, 65) and in immunocompromised children (predominantly after organ or bone marrow transplantation) (27, 30, 67). Some authors reported high rates (up to 37%) of postnatal cytomegalovirus transmission from seropositive breastfeeding mothers to premature infants (31, 41, 69). A significant proportion of infected infants develop pneumonitis along with hepatosplenomegaly, lymphadenopathy, enteritis, and aseptic meningitis (12, 36). Although breast milk containing HCMV is not considered dangerous to healthy full-term infants, the risk of development of early and symptomatic postnatal HCMV infection is proportional to the degree of the infant's immaturity (69). Premature infants who experience postnatal HCMV infection often develop acute respiratory distress syndrome (ARDS) and bronchopulmonary dysplasia (41, 56). Despite the morbidity and mortality associated with fetal and neonatal HCMV infection, little is known about the pathogenic mechanisms that trigger HCMV-associated lung diseases.
HCMV has a very diverse tropism in its host, infecting most cells types, including epithelial cells, endothelial cells, fibroblasts, smooth muscle cells, and various hematopoietic cells (reviewed in reference 58). During congenital infection, the fetal lungs are particularly targeted by HCMV (8, 25), and reductions in the levels of secretion of pulmonary surfactant, a complex substance that decreases surface tension, thus preventing alveolar collapse, into the amniotic fluid suggest that the virus negatively affects fetal lung maturation (50).
The lung is among the last fetal organs to mature, both structurally and functionally. Based on anatomical and histological characteristics, fetal lung development can be divided into five distinct stages: embryonic (3 to 7 weeks), pseudoglandular (7 to 16 weeks), canalicular (16 to 24 weeks), saccular (24 to 36 weeks), and alveolar (36 weeks through infancy) (52). The major airways, the bronchial tree, and a portion of the respiratory parenchyma are formed during the pseudoglandular stage. Type I and type II lung epithelial cells begin to differentiate in the canalicular stage and then continue to develop and mature during the saccular stage, becoming the most abundant epithelial cells in the lung. True alveoli first appear before birth at the alveolar stage, and their formation continues during infancy. The alveolar surface of the lung is formed by two types of epithelial cells, the type I pneumocyte and the type II pneumocyte. In the mature lung, most of the alveolar surface is covered by type I cells. These flattened, highly differentiated cells serve as a thin barrier between blood vessel endothelial cells and inhaled air. Type II cells serve as progenitor cells for type I pneumocytes, facilitating the clearance and repair of any injured area of the lung. The main function of type II cells is the secretion of lung surfactant (1), a lipoprotein complex containing glycerophospholipids, cholesterol, surfactant protein A (SP-A), SP-B, SP-C, and SP-D. Lung surfactant forms a thin layer at the interface of liquid and inhaled air that reduces surface tension along the alveolar epithelium. Premature infants are usually born during the saccular stage of lung development, when surfactant production by type II pneumocytes and the differentiation of type I pneumocytes are not sufficient for adequate gas exchange. The immature preterm lung is especially vulnerable to injury, and survival at birth depends on adequate lung development in utero (11).
HCMV is a uniquely human pathogen, and there is an urgent need for the development of novel models of human fetal organs that accurately reproduce the hallmarks of congenital infection and could facilitate the study of CMV pathogenesis and the evaluation of antiviral drugs. To date, a limited number of animal models have been developed for the long-term study of HCMV pathogenesis. These systems include CB17-scid mice for the study of HCMV replication in human fetal thymus and liver tissues coimplanted under the kidney capsule (43), retinal tissue implanted in the anterior chamber of the mouse eye (7, 33), and human foreskin fibroblasts seeded onto a biodegradable gelatin matrix and implanted subcutaneously (9). The nonobese diabetic (NOD)-scid IL2Rγ−/− humanized mouse model was developed for the study of HCMV latency and reactivation in human CD34+ hematopoietic cells (61) and for the identification of viral genes involved in viral replication and dissemination (66). In addition, CBA/lac mice have been used to establish a model of human fetal lung development (15), and fetal lungs implanted subcutaneously or under the kidney capsule of NCr-nu mice differentiated extensively, recapitulating human lung development in utero (49).
Here we developed a SCID-hu mouse lung model to study the effects of HCMV infection on the developing fetal lung. We found that HCMV efficiently replicated in the lung implants, forming large viral lesions within 7 days. We provide clear evidence that HCMV productively infects alveolar epithelial and mesenchymal cells, imitating congenital infection of the fetal lung. Furthermore, virus replication triggered apoptosis in epithelial and mesenchymal cells within the viral lesion and impaired the secretion of essential surfactant proteins by alveolar epithelial cells. During congenital HCMV infections, these processes could result in fetal lung immaturity and neonatal respiratory diseases, including acute lung injury (ALI) and ARDS. Currently developed humanized mouse models for the long-term replication of HCMV are valuable platforms for drug development, but none of them utilize key target organs of congenital and neonatal HCMV infection, as we report here. Because human lung maturation and differentiation in the SCID-hu mouse are considerably similar to normal human intrauterine lung development (49), this small-animal model allows the study of the congenital and neonatal lung pathogenesis of HCMV as well as human respiratory viruses.
MATERIALS AND METHODS
Ethics statement.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (43a). The protocol was approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco (approval number AN081969-03A). All surgery was performed under ketamine-xylazine anesthesia, and all efforts were made to minimize suffering.
Virus, cells, and tissues.
VR1814, an endothelial cell-tropic clinical strain of HCMV isolated from the cervix, was adapted for growth in human umbilical vein endothelial cells (HUVEC) (28). Virus was propagated in HUVEC (Lonza), and viral stocks were prepared from supernatant virus (10). The titers of infectious virus were determined by a rapid method for the immunological detection and quantification of HCMV immediate-early (IE) proteins that has been shown to correlate with the conventional plaque assay (2, 10). Neonatal human dermal fibroblasts (NHDF-Neo) (Lonza) were used as the indicator monolayer for the assay, and virus titers are expressed as infectious units (IU) (10). Primary human pulmonary alveolar epithelial cells (HPAEpiC) were purchased from ScienCell Research Laboratories (Carlsbad, CA). Lung tissues (18 to 24 gestational weeks [g.w.]) obtained from human fetuses after surgical termination of pregnancy were acquired from Advanced Biosciences Resources (Alameda, CA), with informed consent obtained according to local, state, and federal regulations.
SCID-hu lung mice.
For the in vivo transplantation of fetal lung tissue, 6- to 8-week-old male CB17-scid mice (C.B-Igh-1b/IcrTac-Prkdcscid; Taconic) were used. Lung tissue was carefully separated from major blood vessels and airways, dissected into 2-mm2 pieces, and implanted under the kidney capsular membrane (40, 64). Mice were maintained under specific-pathogen-free conditions for various times from 1 to 14 days, at which time the kidney containing the lung implant was collected. For the drug treatment experiments, mice received 100 mg/kg of body weight/day of ganciclovir (Cytovene) intraperitoneally once daily or various doses (3 to 300 mg/kg/day) of valganciclovir (Valcyte) orally twice daily beginning 1 day before inoculation until implant collection.
Virus inoculation and titration.
Four weeks (cohorts B and D), 6 weeks (cohort C), 10 weeks (cohort E), and 21 weeks (cohort F) after lung implantation, mice were anesthetized, and implants were surgically exposed and directly inoculated with 5.1 to 6.1 log10 IU. Cohort A (55 weeks after implantation) included mice remaining from a different study, which was not an attempt to model lungs of older infants. At 1 day, 7 days, and 14 days after inoculation, mice were euthanized, and implants were dissected, flash-frozen, and stored at −80°C for virus titration or were fixed for immunohistochemistry. For virus titration, frozen implants were placed into 0.5 ml of Dulbecco's modified Eagle's medium (DMEM) containing 1% fetal bovine serum (FBS) (HyClone) and homogenized by sonication. Viral titers were determined for each implant as described above and are expressed as IU. The limit of detection was 1.0 log10 IU/implant.
Quantitative PCR detection of HCMV genomic DNA.
DNA was extracted from 100 μl of tissue homogenate by using a DNeasy blood and tissue kit from Qiagen according to the manufacturer's instructions. The quality of the purified DNA was controlled by the amplification of human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by conventional PCR (54). The quantification of the HCMV genome was accomplished by using 50 ng of DNA for the amplification of the HCMV major immediate-early (MIE) gene (21) with the StepOnePlus real-time PCR system (Applied Biosystems) using Ex Taq master mix (TaKaRa Bio).
Immunoblotting.
Proteins were extracted from lung implants or cells (HPAEpiC and NHDF) with cell extraction buffer (Invitrogen) containing 1 mM phenylmethylsulfonyl fluoride (PMSF) and a protease inhibitor cocktail (Sigma). Protein samples were loaded onto 12% Tris-glycine gels (50 μg for implants and 10 μg for cells), underwent SDS-PAGE under reducing conditions, and were then electroblotted onto polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories). After blocking for 1 h with 5% nonfat dry milk in Tris-buffered saline (TBS) containing 0.1% Tween 20, the membranes were incubated with primary antibodies from Abcam specific for human SP-A (rabbit polyclonal), cytokeratin 19 (CK19) (rabbit monoclonal), human cytomegalovirus glycoprotein B (gB) (2F12, mouse monoclonal), and GAPDH (mouse monoclonal) at 4°C overnight. Secondary antibodies labeled with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG were obtained from Sigma, and goat anti-rabbit IgG was obtained from Jackson ImmunoResearch. Signals were detected by using ECL Western blotting detection reagents and Hyperfilm obtained from Amersham.
Immunohistochemistry and apoptotic cell labeling.
Lung implants were fixed in 3.7% formaldehyde and infiltrated with 5 to 15% sucrose, followed by embedding in an optimal-cutting-temperature compound and freezing in liquid nitrogen (23). For double immunolabeling, cells were simultaneously incubated with primary antibodies from various species and with secondary antibodies labeled with fluorescein isothiocyanate (FITC) or tetramethyl rhodamine isothiocyanate (TRITC), obtained from Jackson ImmunoResearch. Primary antibodies included mouse monoclonal antibodies specific for the HCMV IE (Millipore) and gB (Abcam) proteins, rabbit monoclonal antibodies to human CK7 and CK19 (Abcam), and rabbit polyclonal antibodies specific for human SP-A, SP-B, and SP-C (Millipore) and Fas (Sigma-Aldrich). Nuclei were counterstained with diamidino-2-phenylindole (DAPI). The specificity of each immunohistochemical reaction was verified by using nonimmune rabbit or mouse IgG (Vector Laboratories) as the primary antibody. Apoptotic cells were identified by the terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) method, using an in situ cell death detection kit that labels DNA strand breaks with fluorescein (Roche Diagnostics). Formalin-fixed tissue sections were deparaffinized in xylene, rehydrated in graded alcohols, and stained with hematoxylin and eosin (H&E). Tissue sections were analyzed with a Leica DM6000 B microscope, and images were acquired by using a QImaging Retiga EXi Fast camera. Additional detailed tissue analysis was performed by using a Leica SP5 laser confocal microscope.
Data analysis.
To quantify the fluorescence intensity of TUNEL signals from apoptotic cells and the immunolabeling of cells expressing SP-A or SP-C, all experimental and image acquisition parameters were kept constant throughout each experiment. The fluorescence intensity was analyzed as the mean gray value per pixel in the background-subtracted images by using ImageJ software. The mean gray value of the TUNEL signal was determined for 6 to 17 images of randomly selected representative areas of viral lesions in 4 infected implants and 5 mock-infected implants obtained from two different lung samples (cohorts A and B). For SP-A immunolabeling, we morphologically identified 6 to 14 representative alveolar sacs in each image and quantified the intensity of the fluorescence in 5 to 8 images from 2 infected and 5 mock-infected implants (cohorts A and B). For SP-C immunolabeling, we identified 4 to 14 representative areas of type II pneumocyte clusters in each image and quantified the intensity of the fluorescence in 5 to 7 images from 2 infected and 5 mock-infected implants (cohorts A and B). Statistical analysis was performed by using the nonparametric Mann-Whitney U test and Spearman's rank correlation coefficient.
RESULTS
Human fetal lung tissue implanted into CB17-scid mice extensively differentiates.
Since the lung is a major site of HCMV infection in fetuses and premature infants (8, 25), we developed a SCID-hu lung mouse model for the long-term study of congenital HCMV infection. To ensure the successful engraftment of the human tissue in the mice, we first carried out a thorough immunohistochemical analysis of human fetal lung tissue (18 to 24 g.w.) implanted under the kidney capsule of CB17-scid mice. Within a period of 5 weeks in vivo, the implants recapitulated human lung development up to the late saccular stage (24 to 36 weeks) (Fig. 1A and B). Notably, premature infants are usually born during the saccular stage of lung development. At this stage, the fetal lung implants displayed well-developed bronchioles and alveolar saccules divided into subsaccules or primitive alveoli (Fig. 1A and B). The size of the interstitial tissue was reduced compared to that of lung tissue before engraftment and contained numerous capillaries that provide the blood supply for the growth and development of the implant. Based on morphological criteria and immunostaining for cytokeratin 19, a marker of epithelial cells, most of the alveolar surface was covered by at least two types of epithelial cells: type II pneumocytes, with a cuboidal shape, and flattened, presumably pre-type I pneumocytes (Fig. 1C and D). Because the major function of alveolar cells is the secretion of lung surfactant, we further immunostained tissue sections for the surfactant proteins SP-A, SP-B, SP-C, and SP-D. Strong immunoreactivity to SP-A (Fig. 1E) and SP-C (Fig. 1F) was observed for type II pneumocytes. Flattened, presumably pre-type I pneumocytes were negative for SP-C, but their apical surfaces stained positive for SP-A. We speculate that these pneumocytes were at intermediate stages of differentiation into putative SP-negative type I cells. Moderate immunoreactivity to SP-B and SP-D (not shown) was observed for type II pneumocytes. Staining was performed by using tissue sections from 10 implants obtained from 3 different fetal lungs. These results indicated that human fetal lung tissue implanted under the kidney capsule of CB17-scid mice rapidly grows and differentiates, expressing the characteristic components of pulmonary surfactant proteins. Moreover, the in vivo maturation of fetal lung xenografts from the canalicular to the saccular stage ideally positions this system for the study of HCMV pathogenesis in congenital and neonatal infection.
Fig 1.
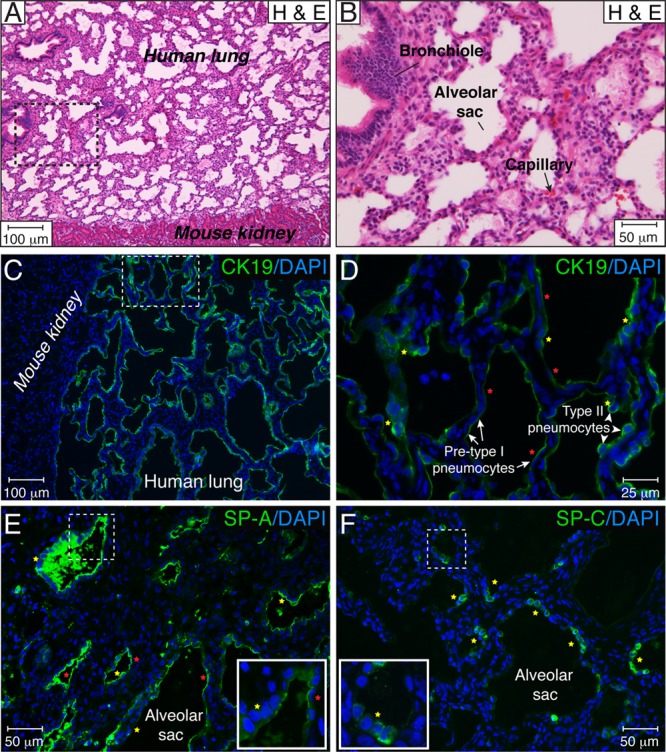
Extensive differentiation of human fetal lung tissue implanted under the kidney capsule of CB17-scid mice. (A and B) Fetal lung tissue implanted under the mouse kidney capsule developed over a period of 5 weeks. Histological tissue sections were stained with hematoxylin and eosin (H & E). The area outlined by the dotted lines in panel A is magnified in panel B. (C and D) The alveolar surface of the lung is formed by at least two types of epithelial cells, type II pneumocytes, with a cuboidal shape (yellow stars), and flattened, presumably pre-type I pneumocytes (red stars). Alveolar pneumocytes were visualized by CK19 immunostaining, and nuclei were counterstained with DAPI. The area outlined by the dotted lines in panel C is magnified in panel D. (E and F) During lung implant development, type II pneumocytes (yellow stars) robustly produced SP-A (E) and SP-C (F). Flattened, presumably pre-type I pneumocytes (red stars) were negative for SP-C (F), but their apical surface stained positive for SP-A (E), suggesting a premature phenotype of the type I cells. The dotted square shows the area of the inset. These results are representative of those observed for 10 implants each from three different lungs developed over 4 to 6 weeks.
HCMV replicates in fetal lung tissue implanted into CB17-scid mice.
Next, we investigated HCMV replication in fetal lung implants. First, we compared the titers of infectious progeny during a 2-week period after virus inoculation (Fig. 2A). As expected for the eclipse phase of infection, there was no infectious virus detected 1 day after inoculation. The virus titer then increased by 4.2 and 4.5 log10 IU per implant at day 7 and day 14, respectively (P = 0.03). In contrast, the number of HCMV DNA genomes did not change as dramatically (Fig. 2B). At day 1, the levels of HCMV genomes were 0.9 log10 IU lower than the 8.8 log10 copies inoculated and then increased by 1 log10 IU by day 14, and mock-infected implants had no detectable HCMV infection (Fig. 2A and B). Importantly, virus titers at days 7 and 14 were positively correlated with the number of HCMV DNA copies per implant (Spearman's rho = 0.87; P = 0.0008) (Fig. 2C), and the kinetics of virus replication were similar in two different mouse cohorts (Fig. 2A and D). The magnitude of virus replication was influenced by the amount of HCMV inoculated (Fig. 2E), and the mean virus yield (3.9 log10 IU per implant) exceeded the low-inoculum dose (2.8 log10 IU per implant) at 14 days (Fig. 2F). To ensure that the infectious titers detected at 14 days represented de novo HCMV replication, and not simply the retention of the virus inoculum, we treated mice intraperitoneally with ganciclovir (Fig. 2G) and orally with valganciclovir (Fig. 2H). Compared to untreated mice, ganciclovir caused a 2.3 log10 decrease in the viral titer, and 2 out of 7 mice had no detectable infectious virus. We also observed dose-dependent reductions in viral titers when mice were treated with valganciclovir. It should be noted that HCMV was isolated from all 7 mice in both cohorts in the absence of ganciclovir or valganciclovir treatment. Altogether, these data indicate that HCMV efficiently replicated in the fetal lung implants of untreated mice over 14 days, producing infectious viral progeny.
Fig 2.

HCMV efficiently replicates in human fetal lung tissue implanted into CB17-scid mice. (A) Lung implants (cohort A; 24 mice) were inoculated with 6.1 log10 IU of VR1814 per implant (arrow). Mice were euthanized for tissue collection, and virus titers (IU/implant) were determined at days 1, 7, and 14 after inoculation. The HCMV titer increased significantly from day 1 to days 7 and 14 (P < 0.05 by Mann-Whitney U test). (B) HCMV DNA in a lung implant from cohort A was quantified by real-time PCR. The HCMV genome number in the virus inoculum was 8.8 log10 copies per implant. The numbers of HCMV DNA copies/implant at 14 days were significantly higher than those at day 1 (P < 0.05 by Mann-Whitney U test). (C) The relationship between infectious units and HCMV DNA in implants from cohort A was significant by the Spearman rank correlation coefficient (r2 = 0.87; P = 0.0008). (D) Lung implants (cohort B; 15 mice) were inoculated with 5.7 log10 IU of VR1814 per implant (arrow), and virus titers were determined at days 1, 7, and 14 after inoculation. (E) Lung implants (cohort E; 20 mice) were inoculated with 2.8 and 5.8 log10 IU of VR1814 per implant, and virus titers were determined 14 days after inoculation. The HCMV titer was significantly higher with higher virus inocula (P < 0.05 by Mann-Whitney U test). (F) Kinetics of HCMV replication in lung implants (cohort E; 20 mice) inoculated with 2.8 log10 IU (dotted lines) and 5.8 log10 IU (solid lines). (G) Lung implants (cohort C; 14 mice) were inoculated with 5.4 log10 IU of VR1814 per implant (arrow), and mice were treated intraperitoneally once daily with ganciclovir beginning the day before HCMV inoculation. Compared to untreated mice, ganciclovir treatment significantly decreased lung virus titers (P < 0.001 by Mann-Whitney U test). (H) Lung implants (cohort D; 40 mice) were inoculated with 5.8 log10 IU of VR1814 per implant (arrow), and mice were treated orally twice daily with valganciclovir beginning the day before inoculation. Compared to untreated mice, valganciclovir treatment significantly decreased the titer of virus at doses of 100 and 300 mg/kg/day (P < 0.05 [by Mann-Whitney U test]). The bar represents the median value. The dotted line is the limit of detection for HCMV IU (1 log10 IU/implant) (A and D to H) or HCMV DNA (1 log10 HCMV copies/implant) (B).
We further assessed HCMV replication in fetal lung xenografts by the cellular localization of viral proteins by immunohistochemistry and fluorescence microscopy (Fig. 3). Since the fluorescence intensity of HCMV IE protein immunostaining under low-power magnification (×50) was superior to the staining for the late HCMV glycoprotein gB, we used IE immunostaining to topographically demonstrate the branches of virus spread (Fig. 3A and B). As indicated by IE protein expression, the virus formed a large viral lesion (1.1 mm in diameter) 7 days after inoculation with elongated (0.5- to 0.7-mm) distal branches of the lesion, which suggested rapid virus spread.
Fig 3.
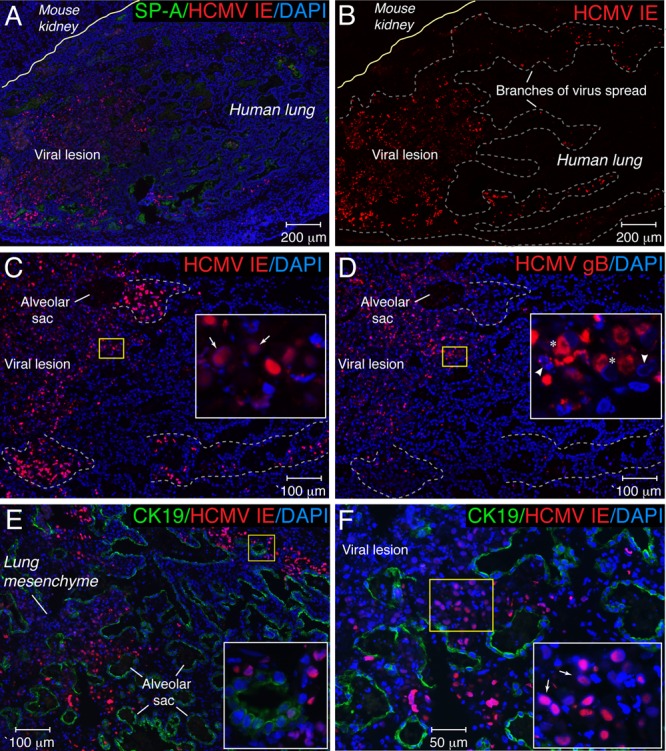
HCMV proteins were expressed in distinct patterns in lung implants 7 days after virus inoculation. Histological sections of lung implants were immunostained for HCMV IE and SP-A (A), HCMV IE (B and C), HCMV gB (D), and HCMV IE and the epithelial cell marker CK19 (E and F). Nuclei were counterstained with DAPI. Panel B represents the red channel of the RGB (red, green, blue) image shown in panel A. In panels A and B, the yellow line separates human lung tissue from mouse kidney. In panel B, the dotted gray line outlines the viral lesion and distal branches of virus spread. In panels C and D, immunohistochemical analysis of HCMV IE and gB expressions in serial tissue sections from HCMV-infected implants is shown. The first section was stained for HCMV IE (C), and the next consecutive section (10 μm deeper) was stained for HCMV gB (D). An alveolar sac located in each consecutive implant section permits comparisons of the locations of IE- and gB-expressing cells. Branches of virus spread represented by IE+ gB− cells are outlined by the light-gray dotted lines. In panels C to F, the areas outlined by the yellow boxes are magnified in the insets. In panels C and F, the arrow shows intranuclear inclusion bodies that contained IE protein. In panel D, the asterisk shows the accumulation of gB in the cytoplasm of infected cells, the right arrowhead shows chromatin condensation, and the left arrowhead shows nucleus fragmentation.
To determine whether virus truly spread in the implants, we performed an immunohistochemical analysis on serial tissue sections from HCMV-infected implants. The first section was stained for HCMV IE to identify infected cells (Fig. 3C), and the next consecutive section (10 μm deeper) was stained for HCMV gB to identify cells in the late stage of the infection (Fig. 3D, asterisks). Under higher-power magnification (×100), the same alveolar sac was located on each consecutive implant section, and images were obtained for a comparison of the locations of IE- and gB-expressing cells. Infected IE-positive (IE+) and gB+ cells had similar locations in the center of the viral lesion (Fig. 3C and D, insets). In contrast, distally infected IE+ gB-negative (gB−) cells at the periphery of the viral lesion (Fig. 3C and D, outlined by dotted lines) suggested viral spread. It is notable that the color of the merged nuclei (red for IE and blue for DAPI) of IE+ gB+ cells (Fig. 3C, inset) was more purple than that of IE+ gB− cells, indicating that infected cells with large intranuclear inclusion bodies (Fig. 3C, arrows) in the center of the viral lesion had weaker IE expression. Conversely, the accumulation of gB in the cytoplasm of these IE+ gB+ cells (Fig. 3D, asterisks) was particularly striking. HCMV produced cytopathic changes in the lung mesenchyme and alveolar saccules that were outlined by cytokeratin 19 (Fig. 3E and F). Distinguishing intranuclear inclusion bodies (Fig. 3F, arrows) that contained IE protein were detected frequently in the lung mesenchyme.
Taken together, these results indicate that HCMV actively replicates in the SCID-hu lung, producing spreading infectious viral progeny.
HCMV infection of SCID-hu fetal lung triggered epithelial and mesenchymal apoptosis.
In the center of the viral lesion, nuclei counterstained with DAPI showed intense fluorescence morphologically corresponding to chromatin condensation and nucleus fragmentation—key morphological features of apoptosis (Fig. 3D, inset, arrowheads). This observation prompted us to examine apoptosis in HCMV-infected lung implants by the TUNEL method. Seven days after inoculation, a strong TUNEL reaction was detected in the viral lesions (Fig. 4A). The vast majority of TUNEL-positive epithelial and mesenchymal cells were localized to the area of the most tissue damage (Fig. 4B, merged aqua color). In this area, we identified infected epithelial cells simultaneously expressing HCMV IE and showing TUNEL reactivity in the nucleus (Fig. 4B, inset). On the other hand, many IE-positive cells at the periphery of viral lesions were TUNEL negative but surrounded by uninfected apoptotic cells (Fig. 4C). The uninfected bystander cells often revealed an induced expression of Fas, an important mediator of apoptotic cell death (Fig. 4D). Essentially no apoptotic cells were found in mock-infected implants (data not shown) or in implants from ganciclovir-treated mice (Fig. 4E), suggesting that the inhibition of viral replication precludes HCMV-induced apoptosis. Next, we assessed the level of apoptosis by the quantification of the TUNEL labeling intensity. The TUNEL labeling intensity in the area of HCMV lesions (mean gray value, 9.7) was approximately 11 times higher than that in mock-infected implants (mean gray value, 0.9) (cohort A) (Fig. 4F). Although moderate HCMV infection was accompanied by a lower level of apoptosis, the TUNEL labeling intensity was also elevated compared to that of mock-infected implants (P < 0.05) (cohort B) (Fig. 4F). These results indicate that HCMV infection induces apoptosis in epithelial and mesenchymal cells in the SCID-hu fetal lung.
Fig 4.

HCMV infection triggered apoptosis in epithelial and mesenchymal cells in human fetal lung tissue. Apoptotic cells were labeled with FITC by a TUNEL assay (A to C and E) or identified by laser confocal microscopy and immunostaining for Fas (D). HCMV-infected cells were identified by the expression of HCMV IE. Nuclei were counterstained with DAPI. In panel A, the dotted gray line separates human lung tissue from mouse kidney. The gray boxes outline areas 1 and 2 that are magnified in panels B and C, respectively. The yellow squares in panels B and C show the areas of the insets. Insets represent the merged and single images of triple labeling. The arrowheads show TUNEL-positive uninfected bystander cells in panel C and Fas-positive uninfected bystander cells in panel D. In panel E, lung implants (cohort F; 8 mice) were inoculated with 5.8 log10 IU of VR1814 per implant, and mice were treated intraperitoneally once daily with ganciclovir (50 mg/kg) beginning the day before HCMV inoculation. Apoptotic cells were assessed by a TUNEL assay 14 days after virus inoculation. Data for cohort A (A to D and F), cohort B (F), and cohort F (E) are shown. In panel F, the fluorescence intensity of TUNEL labeling is expressed as an image mean gray value. For cohort A, 10 images of viral lesions from one infected implant and 10 images from three mock-infected implants were quantified. For cohort B, 17 images from three infected implants and 6 images from two mock-infected implants were quantified. Error bars represent standard errors of the means. As indicated by the fluorescence intensity of TUNEL labeling, the number of apoptotic cells in the area of the viral lesion was significantly higher than in mock-infected implants (P < 0.0001 [cohort A]; P < 0.05 [cohort B] [by Mann-Whitney U test]). These results are representative of those observed for 4 infected implants and 5 mock-infected implants obtained from two different lungs (cohorts A and B).
HCMV impaired secretion of surfactant proteins by alveolar epithelial cells.
Since primary HCMV infection was reported previously to be associated with a decreased level of secretion of fetal pulmonary surfactant into amniotic fluid (50), we investigated whether HCMV infection of the fetal lung affects the expression of surfactant proteins. First, we examined which types of cells in the SCID-hu lung implant are the targets of HCMV infection. Tissue sections from 9 implants obtained from 3 different fetal lungs were coimmunostained for HCMV IE and CK19, a marker of epithelial cells (Fig. 5). We identified two types of cells infected by HCMV: CK19+ alveolar epithelial cells (pre-type I and type II pneumocytes) (Fig. 5A) and CK19-negative mesenchymal cells (Fig. 5B). Desquamated infected epithelial cells originating from pneumocytes were frequently found floating in the alveolar space (Fig. 5A, insets). Next, we investigated whether HCMV infection disturbs surfactant protein production by the alveolar epithelium. Primary cultures of HPAEpiC comprised of alveolar type I and type II cells were maintained for 5 days in specialized alveolar epithelial cell medium obtained from ScienCell, according to the manufacturer's instructions. The cells were then infected with VR1814 at a multiplicity of infection (MOI) of 1. NHDF were used as a control and were inoculated at an MOI of 0.1. Four days after inoculation, all cells were fixed for fluorescence immunohistochemistry or lysed for immunoblot analysis. We found that HPAEpiC were highly susceptible to HCMV infection, expressing IE proteins (Fig. 6A to C) as well as late gB (Fig. 6D). As anticipated, HPAEpiC but not NHDF expressed high levels of the epithelial cell marker CK19 (Fig. 6A and D). Although confocal microscopy revealed very low levels of SP-A and SP-B expression in some uninfected HPAEpiC, the proteins were not detectable by Western blotting (Fig. 6B to D). These results verified that HPAEpiC differentiate in vitro to become type I alveolar epithelial cells (24), which do not proliferate in culture and do not express surfactant proteins. This lack of surfactant protein expression resulting from HPAEpiC differentiation in vitro makes this system unusable for studies of the impact of HCMV infection on lung surfactant production. To do so, we next evaluated the impact of HCMV infection on SP-A production in fetal lung implants. Productive virus replication in the implants was confirmed by the detection of HCMV gB in tissue extracts by Western blotting (Fig. 6E). Infected implants, as well as mock-infected implants and preimplantation fetal lung tissue, expressed CK19 at various levels depending on the quantity of alveolar epithelial cells present. Monomeric SP-A (28 to 36 kDa) and nonreducible SP-A dimers (64 kDa) were detected in the implants but not in the 20-g.w. fetal lung before implantation. In contrast, medium-molecular-mass SP-A oligomers (>100 kDa) were abundant both in the fetal lung before implantation and in the implants. Analysis of tissue extracts from several implants showed that SP-A expression was highly variable and often not detectable in both mock- and HCMV-infected implants. The SP-A expression level was highest in the infected and mock-infected implants shown in Fig. 6E. The caveat of this approach is that the total level of SP-A detected by Western blotting represents the molecular description of the tissue overall: a mixed population of both infected and uninfected type II pneumocytes producing SP-A. To overcome this difficulty, we applied a semiquantitative analysis of immunofluorescence by the intensity of SP-A immunostaining, which allows the examination of specific cells of interest. The colocalization of HCMV IE and SP-A revealed virtually no SP-A production in the center of viral lesions (Fig. 7A), and the lumens of alveolar saccules were packed with detached alveolar epithelial cells that had no detectable SP-A (Fig. 7A, inset, yellow star). In contrast, the periphery of viral lesions was surrounded by saccules, with the alveolar epithelium actively secreting SP-A (Fig. 7A, white stars). Detailed colocalization analysis by confocal microscopy demonstrated that alveolar pneumocytes often lost SP-A expression as a result of direct HCMV replication or indirectly through the influence of infected neighboring mesenchymal cells (Fig. 7B). Lower SP-A expression levels were frequently detected in alveolar pneumocytes adjacent to the infected IE-positive mesenchymal cells (Fig. 7B, inset). To compare the levels of SP-A production by alveolar pneumocytes in infected and mock-infected implants, we quantified the intensity of the fluorescence of SP-A expression in the alveolar sac. We found that near the viral lesion, the level of SP-A production in the alveolar epithelium decreased by more than half (P < 0.0001) (Fig. 7E), and similarly, HCMV also affected the production of SP-B (Fig. 7C). Most dramatically, however, HCMV affected the production of SP-C (Fig. 7D), the surfactant protein expressed exclusively by type II pneumocytes (Fig. 1F). We noted occasional HCMV infection of this type of cell at the periphery of viral lesions (Fig. 7D, inset, white star), but the majority of type II pneumocytes in the infected area of the lung showed little, if any, SP-C production (Fig. 7D, yellow stars). Our observations suggest that HCMV can directly infect type II pneumocytes as well as indirectly affect their function, possibly through the infection of bystander mesenchymal cells. For the quantitative evaluation of SP-C production, type II pneumocytes were morphologically identified, as shown in Fig. 1D. The fluorescence intensities of the SP-C immunolabeling of representative clusters of these cells in infected and mock-infected implants were quantified. We found that the pneumocyte SP-C expression level in HCMV-infected SCID-hu lung tissue was 2.6 times lower than that in mock-infected controls (P < 0.0001) (Fig. 6F). Taken together, these results showed that HCMV infection of SCID-hu fetal lung tissue significantly impaired the secretion of surfactant proteins by alveolar epithelial cells and suggest that this may play a role in fetal lung immaturity.
Fig 5.

HCMV targets alveolar epithelial and mesenchymal cells in human fetal lung tissue. Histological sections of lung implants 7 days after virus inoculation were coimmunostained for HCMV IE and CK19. Nuclei were counterstained with DAPI. Alveolar epithelial (A) and mesenchymal (B) cells were found to be infected by HCMV. These results are representative of those observed for 9 infected implants obtained from three different lungs (cohorts A, B, and D).
Fig 6.
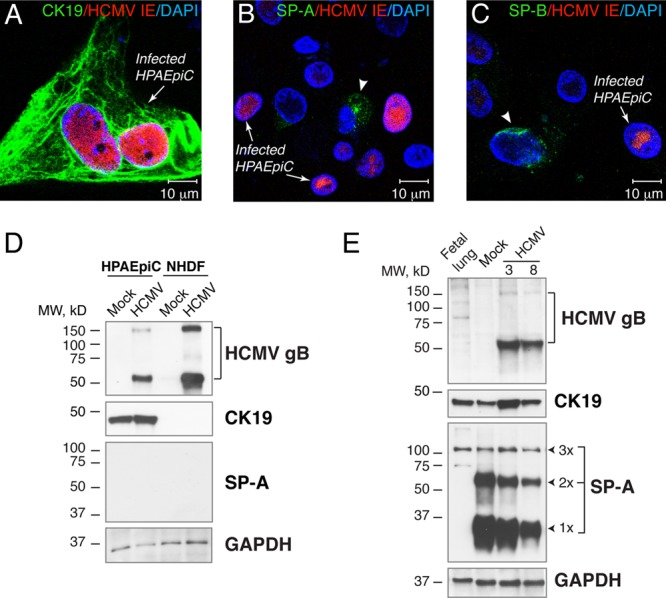
HCMV productively infects primary human pulmonary alveolar epithelial cells (HPAEpiC) and lung implants. (A to C) HPAEpiC were maintained in vitro for 5 days, inoculated with VR1814 at an MOI of 1, and fixed and costained for HCMV IE and CK19 (A), SP-A (B), and SP-B (C) 4 days after inoculation. The arrows show infected cells, and the arrowheads show cells expressing SP-A (B) or SP-B (C). Nuclei were counterstained with DAPI. (D and E) Western blot analysis of HCMV gB, CK19, and SP-A in HPAEpiC inoculated with VR1814 at an MOI of 1 and NHDF inoculated at an MOI of 0.1 (D) or lung implants (cohort F) inoculated with HCMV at 5.8 log10 IU/implant (E). Total proteins from 10 μg for cells and 50 μg for implants or preimplantation lungs (20 weeks) were subjected to 12% SDS-PAGE under reducing conditions and subsequently electroblotted. The expression level of GAPDH was used as a loading control. The results are representative of those observed for 16 infected and mock-infected implants obtained from two different lungs (cohorts A and F) (E). 1×, SP-A monomer (28 to 36 kDa); 2×, SP-A dimer (64 kDa), 3×, SP-A oligomer (>100 kDa); fetal lung, preimplantation lung (20 g.w.); 3 and 8, mouse 3 and mouse 8.
Fig 7.
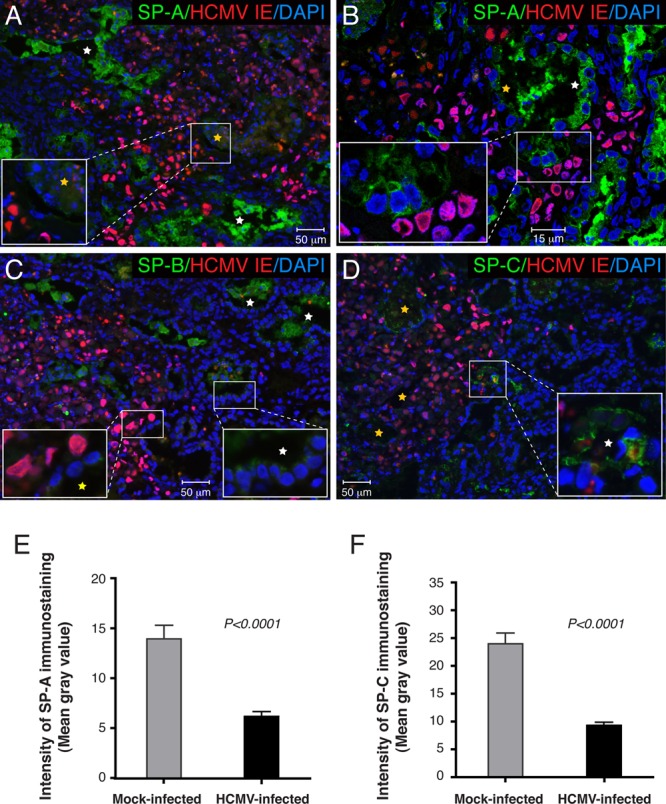
HCMV infection impaired surfactant protein expression in alveolar epithelial cells. (A to D) Histological sections of lung implants (cohort B) 7 days after virus inoculation were coimmunostained for HCMV IE and SP-A (A and B), SP-B (C), and SP-C (D). Nuclei were counterstained with DAPI. (A) Alveolar epithelial cells, identified by CK19 staining of the adjacent sections, had less SP-A secretion in the center of the viral lesion (inset, yellow star) but not in the periphery (white star). (B) Analysis of an alveolar duct at the edge of the viral lesion by laser confocal microscopy revealed diminished SP-A expression levels in pneumocytes proximal (yellow star) but not distal (white star) to the lesion. (C) SP-B was produced by the lung implant at a lower level than SP-A (inset, white star), and its levels further decreased in infected pneumocytes (inset, yellow star). (D) Type II pneumocytes expressing SP-C were not clearly evident in infected lung implants. Rare SP-C-positive type II pneumocytes (white star) were detected at the edge of the viral lesion but not in the center (yellow stars). (E and F) Representative data for SP-A (E) and SP-C (F) immunolabeling quantifications. Fourteen areas of the viral lesion from 1 implant and 14 areas from 3 mock-infected implants were quantified. Error bars represent standard errors of the means. As indicated by the fluorescence intensity, the expression levels of SP-A and SP-C were significantly reduced in HCMV-infected lung implants (P < 0.0001 by Mann-Whitney U test). These results are representative of those observed for 2 infected implants and 5 mock-infected implants obtained from two different lungs (cohorts A and B).
DISCUSSION
Here we report a new SCID-hu lung mouse model for the long-term study of congenital HCMV infection. There are several aspects of this system that make it unique. First, unlike all currently used humanized models for HCMV, this model utilizes the human fetal lung, one of the major targets of HCMV in fetuses and premature infants (8, 25). Second, fetal lung development in the SCID-hu mouse closely resembles human lung development in utero (49). Third, we show here that this system is highly permissive for HCMV replication, enabling the study of virus pathogenesis and antiviral treatment in both epithelial and mesenchymal lung cells in vivo. HCMV indeed replicated in the implants by the following criteria: (i) virus titers at days 7 and 14 positively correlated with HCMV DNA copies per implant (Spearman's rho = 0.87; P = 0.0008) (Fig. 2C); (ii) virus yield at day 14 exceeded the amount of virus inoculated when a low inoculum (2.8 log10 IU) was used (Fig. 2F); (iii) an inhibitor of HCMV DNA synthesis, ganciclovir, significantly reduced viral titers; and (iv) HCMV gB expression was detected by both immunofluorescence microscopy (Fig. 3D) and Western blotting (Fig. 6E).
Given the inherent difficulties in studying virus pathogenesis in human lung, the sole study of HCMV infection in alveolar epithelial cells in vitro was reported more than 3 decades ago (42). Much of our knowledge about the special role of the lung in CMV pathogenesis thus comes from studies of murine CMV (MCMV) infection, where the lung was identified as a major site of MCMV latency and reactivation (4). In a mouse model of interstitial pneumonia, CD8+ T cells were found to control MCMV replication during acute infection (63), and furthermore, antiviral memory-effector T cells persisting in the lung prevented reactivation from viral latency (51, 63). Although the mouse model of interstitial pneumonia is of tremendous value for studies of MCMV pathogenesis in the lung, the pathogenic mechanisms and development of HCMV pneumonia during congenital infection and perinatal infection in premature infants and immunocompromised older children have not been well studied. To address this, we have successfully developed a SCID-hu lung model that opens a new avenue for investigations of HCMV pathogenesis in the human lung and for the design of effective and safe therapies to prevent and treat HCMV disease.
Our study showed that HCMV efficiently replicates in the SCID-hu lung, forming distinct large viral lesions at 1 week of inoculation (Fig. 3A and B). Although viral titers in lung implants (Fig. 2A and D) were similar to those reported previously for Thy/Liv implants (43), the viral spread in the lung implants was particularly striking. Given the 3- to 4-day HCMV replication cycle, the area of the viral lesion not only was large 7 days after inoculation but also included elongated plaque branches in the periphery, suggesting rapid virus spread (Fig. 3A and B). The virus spread in those branches was verified by the prominent expression of HCMV IE but not late gB (Fig. 3C and D, outlined by dotted lines). In our experiments, HCMV replication was inhibited by treatment of the mice with both ganciclovir and valganciclovir. It is important to note that ganciclovir inhibits HCMV DNA synthesis and suppresses virus replication but does not eliminate the virus (18, 20, 72). We were intrigued by our similar observation that ganciclovir and valganciclovir treatment of SCID-hu mice significantly reduced but did not eliminate infectious virus in the lung implants (Fig. 2G and H). It appears that this model system closely imitates natural HCMV infection, and our finding that HCMV productively infected lung epithelial and mesenchymal cells in the SCID-hu lung is notable. The staining pattern that we observed in the lung implants, including intranuclear inclusion bodies (Fig. 3C and F, arrows) and desquamated alveolar epithelial cells (Fig. 5A), has remarkable similarities to data from previous reports on the distribution of HCMV-infected cells in the human lung (25, 59). The population of HCMV-infected cells in the lung during natural infection includes alveolar epithelial, mesenchymal, and endothelial cells. Interestingly, alveolar epithelial cells were reported to be the main target for HCMV infection in the lung. These cells seemed to be late-stage infected cells that had detached from the alveolar basement membrane and were found floating in the alveolar lumen. Our results are consistent with those previously reported observations (Fig. 5A, insets).
Our finding that HCMV infection triggered apoptosis in epithelial and mesenchymal cells within the viral lesions was especially surprising. HCMV is a slowly replicating virus that persistently infects humans. To keep infected cells alive and metabolically active, the virus has evolved unique strategies to escape cell death by blocking apoptosis (26, 55, 60, 73). Instead, our observations were entirely the opposite. Initially, we morphologically revealed DAPI-labeled nucleus fragmentation in the viral lesions, suggesting apoptosis (Fig. 3D, left arrowhead), and then visualized apoptotic cells by TUNEL labeling and quantified their numbers (Fig. 4). Essentially no apoptotic cells were detected in mock-infected lung implants or when virus replication in the implants was inhibited by ganciclovir; however, HCMV inoculation resulted in viral lesions that were seemingly underlined by apoptotic cells.
Lung epithelial cell apoptosis has been extensively studied in the context of acute lung injury (reviewed in reference 5). ALI is a syndrome characterized by acute inflammation and lung tissue injury resulting in increased vascular permeability and an influx of inflammatory cells. Clinically severe ALI is manifested as ARDS, and ARDS caused by HCMV in immunocompetent infants was described recently (14), even though pneumonitis is a common clinical manifestation of HCMV infection in premature infants (3, 22, 34, 65) and immunocompromised children (27, 30). HCMV was also detected by histopathology in 30% of cases of critically ill patients with ARDS (46). Thus, it would be interesting to consider that a respiratory syndrome associated with HCMV infection of the fetal lung could be accompanied by lung epithelial apoptosis. This notion is supported by several previously reports: (i) HCMV infection of neural precursor cells prevented their differentiation and induced apoptosis (44); (ii) in a rat kidney model of chronic allograft rejection, CMV infection induced tubular apoptosis through the tumor necrosis factor alpha (TNF-α)–TNF-R1 pathway (35); and (iii) extensive analysis by in situ TUNEL assays of 70 transbronchial biopsy samples from 25 pulmonary allograft recipients showed an increased density of apoptotic cells in HCMV pneumonitis that correlated with acute allograft rejection (32). Altogether, those previously reported observations suggest that HCMV infection in the lung may trigger apoptosis, resulting in epithelial cell damage, increased epithelial cell permeability, the production of inflammatory cytokines, and neutrophil recruitment.
The mechanism responsible for epithelial apoptosis in ALI is not completely understood, although roles for Fas/Fas ligand (FasL) have been proposed. Soluble FasL shed into the alveolar fluid of patients with ARDS induced the Fas-dependent apoptosis of epithelial cells (38). Others have reported that HCMV-infected human retinal pigment epithelial cells secreted a large amount of FasL, which in turn induced the apoptosis of bystander cells (13). HCMV IE2 induction of FasL-mediated apoptosis in the human retina was suggested to be a potential mechanism for the pathogenesis of HCMV retinitis. In accordance with that report, we detected TUNEL labeling in both infected and bystander cells (Fig. 4A to C). Furthermore, we found that Fas was induced in uninfected cells surrounding the large viral lesion (Fig. 4D). We speculate that infected epithelial and/or mesenchymal cells secrete HCMV-induced factors that trigger apoptosis in bystander cells, and we anticipate that future studies will lead us to the molecular mechanism of this HCMV-induced lung pathology.
It is equally interesting to consider a role of surfactant proteins in the modulation of lung apoptosis. SP-A is an inhibitor of apoptosis in fibroblasts and epithelial cells (68, 71). Patients with early ARDS have low concentrations of SP-A and SP-B in bronchoalveolar lavage fluid (29). On the other hand, apoptosis in the lung epithelium is a sign of ARDS (5, 39). Importantly, decreased fetal pulmonary surfactant secretion into amniotic fluid was detected in congenital HCMV infections (50). In agreement with that observation, our results indicate that HCMV infection of alveolar epithelial cells impairs the production of SP-A, SP-B, and SP-C. One consequence of the decrease in the amount of surfactant proteins during HCMV infection could be the induction of apoptosis in the lung. It seems likely that those most vulnerable to HCMV infection would be premature infants with low levels of production of antiapoptotic surfactant proteins. Indeed, premature infants with postnatally acquired HCMV infection frequently develop pneumonitis (22, 45, 65), and the risk of developing early and symptomatic infection is directly proportional to the degree of the infant's maturity (69).
Another consequence of decreased levels of surfactant proteins due to HCMV infection could be fetal lung immaturity. Reductions of fetal lung maturity indices, including the lecithin-to-sphingomyelin (L/S) ratio, the presence of phosphatidylglycerol (PG), and lamellar body (LB) counts, were observed for amniotic fluid from pregnant women with primary HCMV infection (50). The authors of that study proposed that the retardation of fetal lung maturity could be a result of the direct HCMV infection of pneumocytes secreting surfactant proteins. Here, using the SCID-hu mouse lung model, we report that HCMV infects alveolar pneumocytes and mesenchymal cells (Fig. 5). We also report for the first time that infection of these cells is associated with significant reductions in the levels of production of SP-A, SP-B, and SP-C (Fig. 7). These surfactant proteins are lung-specific gene products (70), and the main producers of surfactant proteins in the lung are alveolar type II pneumocytes (1). These cells have a number of essential functions, including the secretion of lung surfactant that facilitates the reduction of surface tension at the alveolar air-liquid interface. Type II pneumocytes also regulate the functions and activities of innate immune cells participating in the lung immune host defense (reviewed in reference 48). Furthermore, these cells serve as progenitor cells for type I pneumocytes and replenish the alveolar epithelium after lung injury (17, 47). Importantly, alveolar type II pneumocytes isolated from human lung tissue and cultured in vitro lose their characteristic phenotype and differentiate into type I cells within a 10-day period (24). For this reason, our finding that primary HPAEpiC lacked SP-A and SP-B expression during 9 days of culture was not surprising. These results clearly demonstrated that the SCID-hu lung model presents a novel and unique system for the investigation of the impact of HCMV infection on the production of surfactant proteins by type II pneumocytes. Analysis of the total level of SP-A by Western blotting (Fig. 6E) demonstrated that fetal lung tissue implanted under the mouse kidney capsule differentiated extensively. The SP-A monomer (28 to 36 kDa) is expressed at very low or undetectable levels in 16- 23-g.w. lungs (37), and accordingly, we were able to detect only the medium-molecular-mass SP-A oligomer in 20-g.w. fetal lungs before implantation. In utero, an age-dependent increase in the number of type II pneumocytes producing surfactant proteins occurs between 22 and 29 g.w. (6), and SP-A levels show a marked increase with advancing gestational age (53). Our data reflect the observations that the differentiation of fetal lung tissue in SCID mice during a period of 23 weeks results in the abundant production of SP-A monomers and dimers, in addition to the >100-kDa oligomer observed in preimplantation lungs.
Although immunoblot analysis of the total level of SP-A allowed the evaluation of fetal lung differentiation in vivo, this approach produced a molecular description of the lung tissue overall and not the specific area of virus replication. As we have demonstrated, levels of SP-A, -B, and -C were diminished in the area of the large lesion and were sometimes enhanced in the surrounding area (Fig. 7A to D). As a result, the decrease of total SP-A levels in infected implants was not consistent. Therefore, semiquantitative analysis of immunofluorescence by quantifying the intensity of immunostaining allowed us to examine the specific cells of interest and demonstrated that HCMV infection impaired the secretion of surfactant proteins by alveolar epithelial cells. It is intriguing that type II pneumocytes maintain their specific alveolar type II phenotype and plasticity to respond to lung injury through interactions with bystander mesenchymal cells (19, 57). Because we found that HCMV infects alveolar pneumocytes as well as mesenchymal cells, we speculate that the virus may impair surfactant protein secretion directly by the infection of alveolar pneumocytes or indirectly through the infection of mesenchymal cells. We anticipate that future studies will better define the precise molecular mechanisms for these effects.
In summary, we have developed a new in vivo model for the long-term study of human congenital CMV infection in SCID-hu mice. This model utilizes a major natural HCMV reservoir, the fetal lung, thus enabling the study of virus pathogenesis during congenital and neonatal infection, and it represents a valuable, clinically relevant tool for the development of new antiviral therapies.
ACKNOWLEDGMENTS
We thank Mary E. Moreno, Sofiya Galkina, Jose Rivera, Ukina Sanford, and Olga Kosikova for expert technical assistance with the mice and Mike McCune and Anna Bakardjiev for their constructive comments on the manuscript.
This work was supported in part by the AIDS Research Institute at UCSF and the Harvey V. Berneking Living Trust. We have no conflict of interest.
Footnotes
Published ahead of print 12 September 2012
REFERENCES
- 1. Andreeva AV, Kutuzov MA, Voyno-Yasenetskaya TA. 2007. Regulation of surfactant secretion in alveolar type II cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 293:L259–L271 doi:10.1152/ajplung.00112.2007 [DOI] [PubMed] [Google Scholar]
- 2. Andreoni M, Faircloth M, Vugler L, Britt WJ. 1989. A rapid microneutralization assay for the measurement of neutralizing antibody reactive with human cytomegalovirus. J. Virol. Methods 23:157–167 [DOI] [PubMed] [Google Scholar]
- 3. Ballard RA, Drew WL, Hufnagle KG, Riedel PA. 1979. Acquired cytomegalovirus infection in preterm infants. Am. J. Dis. Child. 133:482–485 [DOI] [PubMed] [Google Scholar]
- 4. Balthesen M, Messerle M, Reddehase MJ. 1993. Lungs are a major organ site of cytomegalovirus latency and recurrence. J. Virol. 67:5360–5366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bardales RH, Xie SS, Schaefer RF, Hsu SM. 1996. Apoptosis is a major pathway responsible for the resolution of type II pneumocytes in acute lung injury. Am. J. Pathol. 149:845–852 [PMC free article] [PubMed] [Google Scholar]
- 6. Betz P, et al. 1992. Determination of fetal age by immunohistochemical estimation of surfactant-producing alveolar type II cells. Forensic Sci. Int. 53:193–202 [DOI] [PubMed] [Google Scholar]
- 7. Bidanset DJ, Rybak RJ, Hartline CB, Kern ER. 2004. Efficacy of ganciclovir and cidofovir against human cytomegalovirus replication in SCID mice implanted with human retinal tissue. Antiviral Res. 63:61–64 [DOI] [PubMed] [Google Scholar]
- 8. Bissinger AL, Sinzger C, Kaiserling E, Jahn G. 2002. Human cytomegalovirus as a direct pathogen: correlation of multiorgan involvement and cell distribution with clinical and pathological findings in a case of congenital inclusion disease. J. Med. Virol. 67:200–206 [DOI] [PubMed] [Google Scholar]
- 9. Bravo FJ, Cardin RD, Bernstein DI. 2007. A model of human cytomegalovirus infection in severe combined immunodeficient mice. Antiviral Res. 76:104–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Britt WJ. 2010. Human cytomegalovirus: propagation, quantification, and storage. Curr. Protoc. Microbiol. 14:14E.13 doi:10.1002/9780471729259.mc14e03s18 [DOI] [PubMed] [Google Scholar]
- 11. Chakraborty M, McGreal EP, Kotecha S. 2010. Acute lung injury in preterm newborn infants: mechanisms and management. Paediatr. Respir. Rev. 11:162–170 [DOI] [PubMed] [Google Scholar]
- 12. Cheong JL, Cowan FM, Modi N. 2004. Gastrointestinal manifestations of postnatal cytomegalovirus infection in infants admitted to a neonatal intensive care unit over a five year period. Arch. Dis. Child. Fetal Neonatal Ed. 89:F367–F369 doi:10.1136/adc.2003.032821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chiou SH, et al. 2001. Up-regulation of Fas ligand expression by human cytomegalovirus immediate-early gene product 2: a novel mechanism in cytomegalovirus-induced apoptosis in human retina. J. Immunol. 167:4098–4103 [DOI] [PubMed] [Google Scholar]
- 14. Choi J, et al. 2008. Acute respiratory distress syndrome by cytomegalovirus infection in an immunocompetent infant. Pediatr. Pulmonol. 43:824–827 [DOI] [PubMed] [Google Scholar]
- 15. Cobb LM. 1975. Growth and development of human fetal trachea and lung in immune-deprived mice. Thorax 30:357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Colugnati FA, Staras SA, Dollard SC, Cannon MJ. 2007. Incidence of cytomegalovirus infection among the general population and pregnant women in the United States. BMC Infect. Dis. 7:71 doi:10.1186/1471-2334-7-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crosby LM, et al. 2011. Balance of life and death in alveolar epithelial type II cells: proliferation, apoptosis, and the effects of cyclic stretch on wound healing. Am. J. Physiol. Lung Cell. Mol. Physiol. 301:L536–L546 doi:10.1152/ajplung.00371.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crumpacker CS. 1996. Ganciclovir. N. Engl. J. Med. 335:721–729 [DOI] [PubMed] [Google Scholar]
- 19. Deimling J, et al. 2007. Mesenchymal maintenance of distal epithelial cell phenotype during late fetal lung development. Am. J. Physiol. Lung Cell. Mol. Physiol. 292:L725–L741 doi:10.1152/ajplung.00221.2006 [DOI] [PubMed] [Google Scholar]
- 20. Demmler GJ. 2003. Congenital cytomegalovirus infection treatment. Pediatr. Infect. Dis. J. 22:1005–1006 [DOI] [PubMed] [Google Scholar]
- 21. Demmler GJ, Buffone GJ, Schimbor CM, May RA. 1988. Detection of cytomegalovirus in urine from newborns by using polymerase chain reaction DNA amplification. J. Infect. Dis. 158:1177–1184 [DOI] [PubMed] [Google Scholar]
- 22. Fischer C, et al. 2010. Severe postnatally acquired cytomegalovirus infection presenting with colitis, pneumonitis and sepsis-like syndrome in an extremely low birthweight infant. Neonatology 97:339–345 [DOI] [PubMed] [Google Scholar]
- 23. Fisher S, Genbacev O, Maidji E, Pereira L. 2000. Human cytomegalovirus infection of placental cytotrophoblasts in vitro and in utero: implications for transmission and pathogenesis. J. Virol. 74:6808–6820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fuchs S, et al. 2003. Differentiation of human alveolar epithelial cells in primary culture: morphological characterization and synthesis of caveolin-1 and surfactant protein-C. Cell Tissue Res. 311:31–45 [DOI] [PubMed] [Google Scholar]
- 25. Gabrielli L, et al. 2009. Histological findings in foetuses congenitally infected by cytomegalovirus. J. Clin. Virol. 46(Suppl 4):S16–S21 doi:10.1016/j.jcv.2009.09.026 [DOI] [PubMed] [Google Scholar]
- 26. Goldmacher VS, et al. 1999. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. U. S. A. 96:12536–12541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Graham SM. 2003. Impact of HIV on childhood respiratory illness: differences between developing and developed countries. Pediatr. Pulmonol. 36:462–468 [DOI] [PubMed] [Google Scholar]
- 28. Grazia Revello M, et al. 2001. In vitro selection of human cytomegalovirus variants unable to transfer virus and virus products from infected cells to polymorphonuclear leukocytes and to grow in endothelial cells. J. Gen. Virol. 82:1429–1438 [DOI] [PubMed] [Google Scholar]
- 29. Greene KE, et al. 1999. Serial changes in surfactant-associated proteins in lung and serum before and after onset of ARDS. Am. J. Respir. Crit. Care Med. 160:1843–1850 [DOI] [PubMed] [Google Scholar]
- 30. Haastrup E, Muller K, Baekgaard H, Heilmann C. 2005. Cytomegalovirus infection after allogeneic stem cell transplant in children. Pediatr. Transplant. 9:734–740 [DOI] [PubMed] [Google Scholar]
- 31. Hamprecht K, et al. 2001. Epidemiology of transmission of cytomegalovirus from mother to preterm infant by breastfeeding. Lancet 357:513–518 [DOI] [PubMed] [Google Scholar]
- 32. Hansen PR, Holm AM, Svendsen UG, Olsen PS, Andersen CB. 1999. Apoptosis in acute pulmonary allograft rejection and cytomegalovirus infection. APMIS 107:529–533 [DOI] [PubMed] [Google Scholar]
- 33. Kern ER, Rybak RJ, Hartline CB, Bidanset DJ. 2001. Predictive efficacy of SCID-hu mouse models for treatment of human cytomegalovirus infections. Antivir. Chem. Chemother. 12(Suppl 1):149–156 [PubMed] [Google Scholar]
- 34. Koklu E, Karadag A, Tunc T, Altun D, Sarici SU. 2009. Congenital cytomegalovirus infection associated with severe lung involvement in a preterm neonate: a causal relationship? Eur. J. Pediatr. 168:1409–1412 [DOI] [PubMed] [Google Scholar]
- 35. Krogerus L, Soots A, Loginov R, Bruggeman C, Lautenschlager I. 2008. CMV increases tubular apoptosis through the TNF-alpha-TNF-R1 pathway in a rat model of chronic renal allograft rejection. Transpl. Immunol. 18:232–236 [DOI] [PubMed] [Google Scholar]
- 36. Kumar ML, et al. 1984. Congenital and postnatally acquired cytomegalovirus infections: long-term follow-up. J. Pediatr. 104:674–679 [DOI] [PubMed] [Google Scholar]
- 37. Liley HG, et al. 1987. Surfactant protein of molecular weight 28,000-36,000 in cultured human fetal lung: cellular localization and effect of dexamethasone. Mol. Endocrinol. 1:205–215 [DOI] [PubMed] [Google Scholar]
- 38. Matute-Bello G, et al. 1999. Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J. Immunol. 163:2217–2225 [PubMed] [Google Scholar]
- 39. Matute-Bello G, Martin TR. 2003. Science review: apoptosis in acute lung injury. Crit. Care 7:355–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McCune JM, et al. 1988. The SCID-hu mouse: murine model for the analysis of human hematolymphoid differentiation and function. Science 241:1632–1639 [DOI] [PubMed] [Google Scholar]
- 41. Meier J, et al. 2005. Human cytomegalovirus reactivation during lactation and mother-to-child transmission in preterm infants. J. Clin. Microbiol. 43:1318–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Michelson-Fiske S, Arnoult J, Febvre H. 1975. Cytomegalovirus infection of human lung epithelial cells in vitro. Intervirology 5:354–363 [DOI] [PubMed] [Google Scholar]
- 43. Mocarski ES, Bonyhadi M, Salimi S, McCune JM, Kaneshima H. 1993. Human cytomegalovirus in a SCID-hu mouse: thymic epithelial cells are prominent targets of viral replication. Proc. Natl. Acad. Sci. U. S. A. 90:104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43a. Odeberg J, et al. 2006. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J. Virol. 80:8929–8939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Odeberg J, et al. 2006. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J. Virol. 80:8929–8939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Okulu E, et al. 2012. Severe postnatal cytomegalovirus infection with multisystem involvement in an extremely low birth weight infant. J. Perinatol. 32:72–74 [DOI] [PubMed] [Google Scholar]
- 46. Osawa R, Singh N. 2009. Cytomegalovirus infection in critically ill patients: a systematic review. Crit. Care 13:R68 doi:10.1186/cc7875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Otto WR. 2002. Lung epithelial stem cells. J. Pathol. 197:527–535 [DOI] [PubMed] [Google Scholar]
- 48. Pastva AM, Wright JR, Williams KL. 2007. Immunomodulatory roles of surfactant proteins A and D: implications in lung disease. Proc. Am. Thorac. Soc. 4:252–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pavlovic J, et al. 2008. Differentiation of xenografted human fetal lung parenchyma. Early Hum. Dev. 84:181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Piazze J, et al. 1999. The effect of primary cytomegalovirus infection on fetal lung maturity indices. Early Hum. Dev. 54:137–144 [DOI] [PubMed] [Google Scholar]
- 51. Podlech J, Holtappels R, Pahl-Seibert MF, Steffens HP, Reddehase MJ. 2000. Murine model of interstitial cytomegalovirus pneumonia in syngeneic bone marrow transplantation: persistence of protective pulmonary CD8-T-cell infiltrates after clearance of acute infection. J. Virol. 74:7496–7507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Potter EL, Loosli CG. 1951. Prenatal development of the human lung. AMA Am. J. Dis. Child. 82:226–228 [PubMed] [Google Scholar]
- 53. Pryhuber GS, Hull WM, Fink I, McMahan MJ, Whitsett JA. 1991. Ontogeny of surfactant proteins A and B in human amniotic fluid as indices of fetal lung maturity. Pediatr. Res. 30:597–605 [DOI] [PubMed] [Google Scholar]
- 54. Radonic A, et al. 2004. Guideline to reference gene selection for quantitative real-time PCR. Biochem. Biophys. Res. Commun. 313:856–862 [DOI] [PubMed] [Google Scholar]
- 55. Reboredo M, Greaves RF, Hahn G. 2004. Human cytomegalovirus proteins encoded by UL37 exon 1 protect infected fibroblasts against virus-induced apoptosis and are required for efficient virus replication. J. Gen. Virol. 85:3555–3567 [DOI] [PubMed] [Google Scholar]
- 56. Sawyer MH, Edwards DK, Spector SA. 1987. Cytomegalovirus infection and bronchopulmonary dysplasia in premature infants. Am. J. Dis. Child. 141:303–305 [DOI] [PubMed] [Google Scholar]
- 57. Shannon JM, Hyatt BA. 2004. Epithelial-mesenchymal interactions in the developing lung. Annu. Rev. Physiol. 66:625–645 [DOI] [PubMed] [Google Scholar]
- 58. Sinzger C, Digel M, Jahn G. 2008. Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 325:63–83 [DOI] [PubMed] [Google Scholar]
- 59. Sinzger C, et al. 1995. Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 76(Pt 4):741–750 [DOI] [PubMed] [Google Scholar]
- 60. Skaletskaya A, et al. 2001. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. U. S. A. 98:7829–7834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Smith MS, et al. 2010. Granulocyte-colony stimulating factor reactivates human cytomegalovirus in a latently infected humanized mouse model. Cell Host Microbe 8:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stagno S. 1995. Cytomegalovirus, p 312–353 In Remington JS, Klein JO. (ed), Infectious diseases of the fetus and newborn infant, 4th ed W. B. Saunders Co., Philadelphia, PA [Google Scholar]
- 63. Steffens HP, Kurz S, Holtappels R, Reddehase MJ. 1998. Preemptive CD8 T-cell immunotherapy of acute cytomegalovirus infection prevents lethal disease, limits the burden of latent viral genomes, and reduces the risk of virus recurrence. J. Virol. 72:1797–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stoddart CA, et al. 2001. Impaired replication of protease inhibitor-resistant HIV-1 in human thymus. Nat. Med. 7:712–718 [DOI] [PubMed] [Google Scholar]
- 65. Takahashi R, et al. 2007. Severe postnatal cytomegalovirus infection in a very premature infant. Neonatology 92:236–239 [DOI] [PubMed] [Google Scholar]
- 66. Umashankar M, et al. 2011. A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog. 7:e1002444 doi:10.1371/journal.ppat.1002444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. van Burik JA, Lawatsch EJ, DeFor TE, Weisdorf DJ. 2001. Cytomegalovirus enteritis among hematopoietic stem cell transplant recipients. Biol. Blood Marrow Transplant. 7:674–679 [DOI] [PubMed] [Google Scholar]
- 68. Vazquez de Lara L, et al. 2000. Surfactant components modulate fibroblast apoptosis and type I collagen and collagenase-1 expression. Am. J. Physiol. Lung Cell. Mol. Physiol. 279:L950–L957 [DOI] [PubMed] [Google Scholar]
- 69. Vochem M, Hamprecht K, Jahn G, Speer CP. 1998. Transmission of cytomegalovirus to preterm infants through breast milk. Pediatr. Infect. Dis. J. 17:53–58 [DOI] [PubMed] [Google Scholar]
- 70. Weaver TE, Whitsett JA. 1991. Function and regulation of expression of pulmonary surfactant-associated proteins. Biochem. J. 273(Pt 2):249–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. White MK, Baireddy V, Strayer DS. 2001. Natural protection from apoptosis by surfactant protein A in type II pneumocytes. Exp. Cell Res. 263:183–192 [DOI] [PubMed] [Google Scholar]
- 72. Whitley RJ, et al. 1997. Ganciclovir treatment of symptomatic congenital cytomegalovirus infection: results of a phase II study. National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. J. Infect. Dis. 175:1080–1086 [DOI] [PubMed] [Google Scholar]
- 73. Zhu H, Shen Y, Shenk T. 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 69:7960–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]