

Abstract
Nonstructural protein 5A (NS5A) of hepatitis C virus (HCV) is an indispensable component of the HCV replication and assembly machineries. Although its precise mechanism of action is not yet clear, current evidence indicates that its structure and function are regulated by the cellular peptidylprolyl isomerase cyclophilin A (CyPA). CyPA binds to proline residues in the C-terminal half of NS5A, in a distributed fashion, and modulates the structure of the disordered domains II and III. Cyclophilin inhibitors (CPIs), including cyclosporine (CsA) and its nonimmunosuppressive derivatives, inhibit HCV infection of diverse genotypes, both in vitro and in vivo. Here we report a mechanism by which CPIs inhibit HCV infection and demonstrate that CPIs can suppress HCV assembly in addition to their well-documented inhibitory effect on RNA replication. Although the interaction between NS5A and other viral proteins is not affected by CPIs, RNA binding by NS5A in cell culture-based HCV (HCVcc)-infected cells is significantly inhibited by CPI treatment, and sensitivity of RNA binding is correlated with previously characterized CyPA dependence or CsA sensitivity of HCV mutants. Furthermore, the difference in CyPA dependence between a subgenomic and a full-length replicon of JFH-1 was due, at least in part, to an additional role that CyPA plays in HCV assembly, a conclusion that is supported by experiments with the clinical CPI alisporivir. The host-directed nature and the ability to interfere with more than one step in the HCV life cycle may result in a higher genetic barrier to resistance for this class of HCV inhibitors.
INTRODUCTION
Hepatitis C virus (HCV) is a positive-strand RNA virus encoding a polyprotein that is cleaved into 10 proteins, including three structural proteins (core, E1, and E2), a putative ion channel (p7), and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (42). Propelled by the development of both subgenomic replicons (8, 31) and a cell culture-based infection system (HCVcc) (9, 29, 49, 54), research has garnered a wealth of information on the role of each of these proteins in the life cycle of the virus. The structural proteins and p7 are needed for virion assembly but not RNA replication, whereas the nonstructural proteins are probably involved in both replication and infectious virus production. In addition to the requisite protease and polymerase activities, HCV also encodes proteins that perform specialized functions, such as deforming the membranes (to generate a microenvironment for RNA replication) or scaffolding (to bring the appropriate proteins together for virion assembly) (14, 24, 37, 41).
HCV infects ∼3% of the world's population and is a major risk factor for liver cirrhosis and hepatocellular carcinoma (5, 47). Current treatment comprises pegylated interferon (IFN), the nucleoside analog ribavirin, and recently approved direct-acting antivirals (DAAs) that inhibit the HCV NS3 protease. Despite the success of this triple therapy, drug resistance against the DAAs develops quickly (38), presumably because of both the error-prone nature of the viral polymerase (NS5B) and the high replication rate in vivo. Host-targeting agents (HTAs) represent an alternative to DAAs, with an expected high genetic barrier to resistance because of the uncoupling of the selection pressure (on the virus) and the direct drug target (from the host). The leading HTAs that have progressed into clinical trials for HCV treatment are nonimmunosuppressive derivatives of cyclosporine (CsA), such as alisporivir (ALV; also known as DEB-025) (17), SCY-635 (23), and NIM-811 (27). These compounds inhibit the host proteins cyclophilins, primarily cyclophilin A (CyPA) (52), to block HCV replication and are collectively called cyclophilin inhibitors (CPIs). Several viral proteins (NS2, NS5A, and NS5B) have been implicated in the action of CyPA as a cellular cofactor for HCV (12, 16, 50), but NS5A is likely the direct binding partner of CyPA (15, 21, 22). NS5A is an essential factor in the HCV life cycle which participates in both viral replication and assembly (3, 4, 45). It interacts with several viral proteins involved in both stages of the HCV life cycle, as well as with myriad host factors (1, 7, 11, 13, 53). The interaction between NS5A and CyPA depends on both the proline residues in NS5A domain II (21, 22) and the peptidylprolyl isomerase (PPIase) active site of CyPA (51). Accordingly, mutations of amino acid residues in the active site of CyPA abolish its ability to facilitate HCV replication (10, 26, 30).
The structural and functional consequences of CyPA-NS5A binding are not yet clear. CyPA binding to, and presumably isomerization of, NS5A domain II has been proposed to change the local conformation of the CyPA-binding site (51), which in turn can “activate” NS5A to maintain the integrity of the viral replication complex (30). In addition, Anderson et al. (2) reported a distinct change of localization and trafficking of lipid molecules upon CPI treatment. Because NS5A localizes to lipid droplets (LDs), possibly to influence viral assembly, CPIs might affect NS5A trafficking and HCV assembly as well (12, 26). The effect on NS5A subcellular localization may not be specific to CPIs, however, as another NS5A inhibitor, BMS-790052, whose target is a different domain of NS5A (domain I), also induces major changes in NS5A localization in treated cells (28, 43). NS5A is an RNA-binding protein (23a, 23b) and the crystal structures of domain I hint at an RNA-binding property for NS5A at this domain (32, 46), whereas studies using recombinant proteins indicate that all three domains of NS5A contribute to RNA binding (18). These results, along with a recent report that recombinant CyPA can influence RNA binding by NS5A domain II (19), have been obtained outside infected cells by use of a reconstituted system. The significance of RNA binding by NS5A remains unknown, as does its potential regulation by CyPA in infected cells. In the study reported here, we demonstrated that RNA binding by NS5A in HCVcc-infected cells is sensitive to CPI treatment and that sensitivity of RNA binding is correlated with previously characterized CyPA dependence or CsA sensitivity of HCV mutants. We also revealed that part of the difference in CPI sensitivity between a subgenomic and a full-length replicon of strain JFH-1 resulted from an additional role of CyPA in HCV assembly, which was further validated with ALV treatment.
MATERIALS AND METHODS
Compounds, cell lines, and antibodies.
CsA was purchased from Alexis Corporation (San Diego, CA), and ALV was provided by the Novartis Institutes for Biomedical Research (Cambridge, MA). Huh-7.5 cells were provided by Charles Rice (Rockefeller University) and Apath LLC (St. Louis, MO). The CyPA-KD cell line (sh-A161) has been described previously (52). Anti-FLAG (rabbit and mouse) antibodies (Sigma) were used for Western blotting and immunofluorescence assay of FLAG-tagged NS5A. Commercial antibodies included anti-NS3 and anti-NS5A (BioFront Technologies), rabbit anti-green fluorescent protein (anti-GFP) (Invitrogen), anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) (Santa Cruz Biotechnology), rabbit anti-CyPA (Enzo Life Sciences), and mouse anti-J2 (English and Scientific Consulting Bt., Hungary).
Plasmids and cloning.
JFH-FLAG was generated by insertion of a FLAG tag, which replaced the amino acid sequence QPPPQGGGVAPG in domain III by introduction of NdeI and AvrII sites at the ends of the sequence. Replicon constructs pSGR2a and pFGR2a were provided by Takaji Wakita and have been described previously (25).
Cell lysate preparation.
Huh-7.5 cells infected with HCVcc at 90 to 95% infection were washed and collected. Cell pellets were resuspended in the lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1 mM dithiothreitol [DTT], 1 mM phenylmethylsulfonyl fluoride [PMSF], 1× protease inhibitor cocktail), to a concentration of 107 cells/ml. After incubation on ice for 15 min, cells were passed eight times through a 22G needle attached to a 1-ml syringe. Cell lysates were subjected to centrifugation at 8,000 × g for 2 min, and supernatants were collected for immunoprecipitation.
RNA and protein immunoprecipitation.
FLAG immunoprecipitation (FLAG-IP) was conducted with a 20-μl suspension of anti-FLAG M2 beads (Sigma). Anti-NS5A and anti-GFP pulldown assays were performed with protein G-plus-A agarose (Calbiochem/Fisher) beads that were incubated with 5 μg of the respective antibody. Beads were washed and mixed with 150 μl of the lysate and 100 μl of NET-2 buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.05% NP-40) in the presence of 5 μg bovine serum albumin (BSA), single-stranded DNA (ssDNA), and 80 U RNasin (Promega) for 3 h at 4°C. Afterward, beads were washed seven times with 500 μl of NET-2 buffer and divided into two sets for RNA and protein extractions. Protein samples were treated with SDS sample loading buffer at 95°C before being loaded for Western blotting. RNA samples were treated with DNase I, and then RNA was extracted with TRIzol (Invitrogen) according to the manufacturer's protocol. RNA pellets were resuspended in 20 μl of water and used for quantitative reverse transcription-PCR (qRT-PCR) analysis.
Strand-specific RT-PCR.
Total RNA was subjected to strand-specific cDNA synthesis with the following HCV-specific primers: 5′-GGGTCCAGGCTGAAGTCGAC-3′ (recognizing the positive strand) and 5′-GCTGTGCCCCAGACCTATCAG-3′ (recognizing the negative strand). The resulting cDNAs were then amplified with the following PCR primers directed at the NS3 region: 5′-CTACCTCCATTCTCGGCATCGG-3′ (forward) and 5′-CGGGATGGGGGGTTGTCACTG-3′ (reverse).
Immunostaining.
Cells were plated on slides and treated with compounds before being fixed with 4% paraformaldehyde. Anti-mouse–fluorescein isothiocyanate (FITC) (1:500), anti-rabbit–tetramethyl rhodamine isocyanate (TRITC) (1:200), anti-rabbit–FITC (1:200), anti-mouse–Cy3b (1:200), and anti-mouse–TRITC (1:40) were purchased from Sigma. Boron-dipyrromethene (BODIPY [493/503]) was purchased from Invitrogen and was used according to the manufacturer's protocol. Colocalizations were analyzed from confocal images taken with a Leica TCS SP2 AOBS microscope. Images were processed with LCS AF Lite software.
Colocalization coefficient.
The colocalization coefficient was analyzed with the JACop plug-in in the Image J program, using Costes's randomization. Pearson's (r) and Mander's (M1 and M2) coefficients were calculated from two separate fields, each containing multiple cells.
Colony formation assays.
In vitro transcription and colony formation assays for both subgenomic and full-length replicons in CyPA-KD cells were performed as described previously (52). To obtain colonies with viral particles produced from FGR2a cells, the supernatant collected from the FGR cells was filtered and used to infect naïve Huh-7.5 cells for 6 h, and cells were then washed and incubated in G418-containing medium for 3 weeks until the colonies were visible.
Treatment of infected cells.
Infection of Huh-7.5 cells with Gaussia luciferase (GLuc)-expressing virus was allowed to proceed until HCV NS3 antigen could be detected in >80% of cells. The cells were then treated with various concentrations of ALV for 9 h, after which the medium was removed and cells were washed with phosphate-buffered saline (PBS) three times before being placed in fresh medium. The treated cells were then allowed to “recover” for 8 h, after which virus-containing medium was collected as the “recovery 1” group. Cells were again allowed to “recover,” for an additional 8 h, and the “recovery 2” medium group was collected.
Lipid droplet purification.
Confluent T-175 flasks of JFH-FLAG-infected Huh-7.5 cells were treated with 4 μg/ml of CsA for 16 h before being harvested for purification of LDs by use of the buffers and procedures described by Sato et al. (39).
NS3 and core ELISAs.
For HCV NS3 enzyme-linked immunosorbent assay (ELISA) (BioFront Technologies), cell lysates of infected or replicon cells were prepared according to the manufacturer's instructions. Briefly, ∼1 × 106 cells were resuspended in 0.5 ml of lysis buffer and mixed by rotation for 30 min at 4°C. The samples were then centrifuged at 18,000 × g for 5 min, and 200 μl of the clarified lysate was used for ELISA. Analysis of core levels in cell culture supernatant was performed with an HCV antigen ELISA kit (Ortho-Clinical Diagnostics, Japan) according to the manufacturer's instructions.
RESULTS
Detection of NS5A-RNA interaction in HCVcc-infected cells.
One of the proposed functions of NS5A is RNA binding during either replication, virion encapsulation, or both. To study the potential effect of CPIs on the RNA-binding properties of NS5A in a cell culture system, we engineered a FLAG-tagged HCVcc and developed a coupled IP and RT-PCR method to detect and quantify RNA binding by NS5A in HCVcc-infected cells. A FLAG epitope tag was inserted into a region at the C terminus of NS5A (Fig. 1A, top panel) that has been shown to tolerate insertions without affecting HCVcc replication or infectivity (6). The FLAG-tagged virus (JFH-FLAG) was fully infectious, and immunostaining with an anti-FLAG antibody clearly identified HCVcc-infected cells (Fig. 1A, bottom panel). Furthermore, immunoprecipitation with anti-FLAG antibody-conjugated beads but not control IgG beads efficiently pulled down NS5A. CyPA was detected in the FLAG-IP complex, but only when the cells were not treated with CsA (Fig. 1B). These results suggest that the FLAG-IP procedure was able to purify NS5A complexes from infected cells that were relevant for CyPA's action. We then determined if HCV RNA could be detected specifically in the FLAG-IP complex. As shown in Fig. 1C, A strong HCV RNA signal was detected in the FLAG-IP complex only for cells infected with JFH-FLAG virus, not for cells infected with untagged JFH-1 virus, despite similar levels of HCV RNA and protein in the input samples. Quantitative RT-PCR analysis confirmed that less than 0.02% of the input HCV RNA could be detected in the FLAG-IP complex from the JFH-1-infected cell lysate, whereas 2.7% of the total HCV RNA was detected in the FLAG-IP complex from the JFH-FLAG-infected cells. In addition, in either cell type, the percentage of a cellular mRNA (GAPDH) present in the FLAG-IP complex was very low (0.15% and 0.21%) (Fig. 1D), providing further support for the specificity of the co-IP–RT-PCR procedure.
Fig 1.
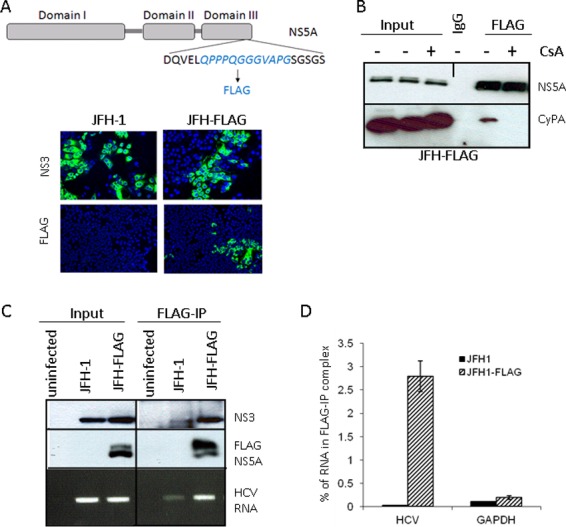
An in vivo binding assay detects NS5A-HCV RNA interaction in infected cells. (A) (Top) Schematic representation of JFH-FLAG genome. A FLAG tag (italicized) was inserted into domain III of NS5A in the JFH-1 background. (Bottom) JFH-1- and JFH-FLAG-infected cells were immunostained with an anti-NS3 (BioFront Technologies) or rabbit anti-FLAG (Sigma) antibody. (B) CyPA coprecipitates with FLAG-NS5A from infected cell lysates in a CsA-sensitive manner. CsA treatment of JFH-FLAG-infected cells was done for 18 h at a concentration of 1 μg/ml. We also included 0.1% formaldehyde in the IP mixture to stabilize the CyPA-NS5A interaction. (C) Specificity of HCV RNA pulldown from infected cell lysate by use of anti-FLAG beads. HCV RNA was detected by RT-PCR analysis of RNA immunoprecipitated from cells infected by JFH-1 or JFH-FLAG. A background band of lesser intensity that migrated slightly faster than the NS3 band was also detected in the JFH-1 sample after FLAG-IP; the identity and cause of this band are unknown. (D) Quantitative RT-PCR analysis of HCV RNA and a cellular mRNA (GAPDH) present in the FLAG-IP complex. Percentages of precipitated RNA versus input RNA were plotted.
CPIs interfere with RNA binding by NS5A.
We then measured the effect of CsA treatment on NS5A's RNA-binding activity by using the co-IP–RT-PCR method. Dosage (1 μg/ml or 0.83 μM) and treatment times (8 h) were carefully optimized so that CsA would not significantly reduce the viral RNA level at the time of cell collection (Fig. 2A). A clear inhibition of RNA binding by the NS5A complex was observed in CsA-treated cells (Fig. 2B). In contrast, alpha interferon (IFN-α) or BMS-790052, a highly potent NS5A inhibitor (20), did not show the same inhibitory effect on RNA binding by NS5A (Fig. 2B). The inhibition by CsA was dosage dependent (Fig. 2C, left panel) and was not due to reduced levels of NS5A in the treated cells (Fig. 2C, right panel). These results suggest that CyPA modulates RNA binding by NS5A in HCV-infected cells. We next asked whether the CPI sensitivity of RNA binding could be modulated by the DEYN mutations that confer CPI resistance on HCVcc (51). FLAG-IP was carried out with the lysate of Huh-7.5 cells infected with a JFH-1 virus carrying both the DEYN mutations and the FLAG insertion (JFH-FLAG-DEYN), and NS5A-associated HCV RNA was quantified. In contrast to the results with wild-type JFH-FLAG, an increasing dosage of CsA did not produce any consistent inhibition of RNA binding by DEYN NS5A (Fig. 2D), even at concentrations as high as 4 μM. These results are consistent with those for CsA resistance levels in HCVcc infection (51).
Fig 2.
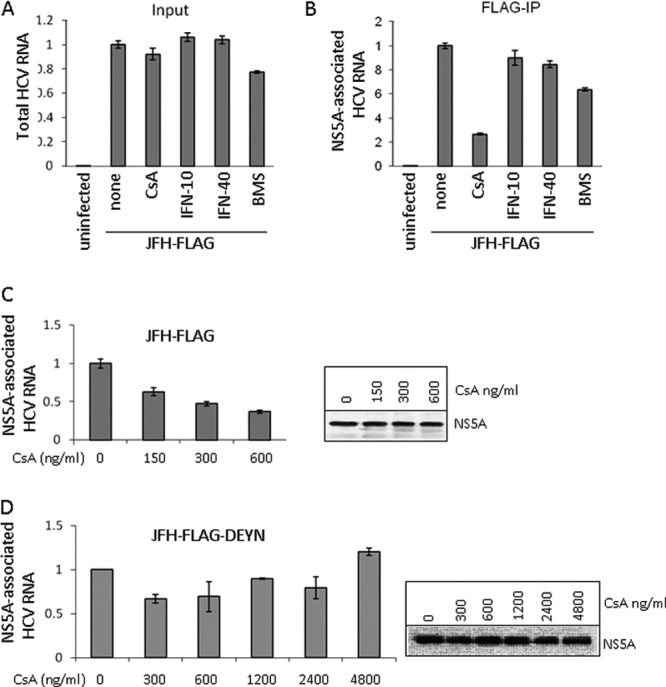
CPIs interfere with HCV RNA association with the NS5A complex. (A) Input HCV RNA levels from JFH-FLAG-infected cells exposed to various treatments for 8 h. CsA was used at 1,000 ng/ml, IFN was used at 10 U/ml and 40 U/ml, and BMS-790052 was used at 10 pM. The HCV RNA was normalized to the amount of GAPDH mRNA present in each lysate, and the untreated sample value was set to 1. (B) CsA treatment significantly reduces NS5A-associated HCV RNA. Each lysate was subjected to FLAG-IP followed by RNA recovery from IP samples and analysis by qRT-PCR. The untreated sample value was set to 1. (C) Dose-dependent inhibition of RNA binding of NS5A complex by CsA. (Left) HCV RNA detected in FLAG-IP complexes isolated from JFH-FLAG-infected cells treated with CsA at the indicated concentrations for 8 h. Percentages of precipitated RNA versus input RNA were calculated, and the untreated sample value was set to 1. (Right) Western blot analysis of NS5A levels in treated cell lysates. (D) Analysis of RNA binding by NS5A of the JFH-FLAG-DEYN virus. The RNA and NS5A protein were analyzed as described for panel C.
NS5A associates with both positive- and negative-strand RNAs in a CyPA-dependent manner.
The ability to coprecipitate HCV RNA from infected cells presented us with an opportunity to determine whether NS5A associates with positive-strand genomic RNA, the replication-intermediate negative-strand RNA, or both. We performed FLAG-IP and then used strand-specific primers in RT-PCR experiments to distinguish positive and negative strands of HCV RNA. Both positive and negative strands of RNA could readily be detected in the FLAG-IP complexes, but only when the FLAG tag was present in the virus used (Fig. 3A). Although the amount of negative-strand RNA in the infected cells was significantly lower than that of the positive-strand RNA, the association between NS5A and both strands of RNA was sensitive to CsA treatment and inhibited to similar extents (∼70%) (Fig. 3B). These results demonstrate that NS5A is associated with both positive- and negative-strand RNAs, in a CyPA-dependent manner. We also determined whether NS5A colocalized with double-stranded RNA (dsRNA), an intermediate product of RNA replication, in HCVcc-infected cells. A monoclonal antibody (J2) that specifically recognizes dsRNA in virus-infected cells (40) was used to visualize dsRNA in Huh-7.5 cells infected with JFH-FLAG. Strong, punctate staining of dsRNA could be detected in HCVcc-infected cells but not in uninfected cells. Partial overlapping between dsRNA and FLAG-NS5A was detected in untreated cells but was lost from the infected cells treated with a CPI (Fig. 3C). Colocalization coefficient analysis revealed a gradual loss of dsRNA-NS5A colocalization from 8 h to 16 h posttreatment (Fig. 3D).
Fig 3.

NS5A associates with both positive and negative strands of HCV RNA, in a CsA-dependent manner. (A) Both positive and negative strands of HCV RNA associate with the NS5A complex. Either JFH-1- or JFH-FLAG-infected cell lysate was used for FLAG-IP followed by RT-PCR using strand-specific primers for cDNA formation. (B) CsA sensitivity of NS5A binding to positive and negative strands of HCV RNA. Both the input (top) and precipitated (bottom) RNAs are shown. The untreated sample value for positive-strand RNA was set to 1. (C) Colocalization of NS5A and dsRNA is decreased by ALV treatment (300 ng/ml). (D) Colocalization coefficients of NS5A and dsRNA, with and without ALV treatment. Pearson's (r) and Mander's (M1 and M2) coefficients were calculated from two separate fields, each containing multiple cells.
Correlation between CyPA dependence of RNA binding and replication capacity.
The effect of CsA on the association between NS5A and the replication intermediates prompted us to determine whether NS5A binding in subgenomic replicons is also inhibited by CsA. Subgenomic replicons of both genotype 1b (GT1b) (SGR1b) (35) and GT2a (SGR2a) (25) were used for the study. A striking difference was observed between the CsA sensitivities of RNA binding by NS5A proteins from the two replicons. CsA effectively inhibited RNA binding in the genotype 1b system while having no effect on the 2a replicon; the BMS-790052 compound, on the other hand, did not inhibit RNA binding of either genotype (Fig. 4A). The insensitivity of NS5A RNA binding to CPI treatment in the SGR2a replicon suggested a reduced dependence of this particular replicon on CyPA for replication, which was confirmed in a colony formation assay. SGR1b was highly sensitive to CyPA knockdown and produced far fewer colonies in the knockdown cell line (sh-A161) than in the control cells (sh-Luc) (Fig. 4B, top panel), as previously reported by our group for this and an H77 (GT1a)-based subgenomic replicon (52). In contrast, the JFH-1-based SGR2a was fairly insensitive to CyPA knockdown and formed many colonies in both cell lines (Fig. 4B, bottom panel).
Fig 4.

Suppression of CyPA expression inhibits the production of infectious particles in vitro. (A) Genotypic difference in CsA sensitivity of RNA binding by NS5A. Both GT1b (SGR1b) and GT2a (SGR2a) replicon cells were treated with 1,000 ng/ml CsA for 8 h, followed by isolation of NS5A-RNA complexes. In addition, SGR1b- and JFH-1-infected cells were also treated with BMS-790052. The GT1b replicon (GS5) contains a GFP gene inserted into the NS5A region (35), and an anti-GFP antibody was used for IP. GT2a NS5A was precipitated with an anti-NS5A monoclonal antibody (4B8). (B) Genotypic difference in CyPA dependence of replication. Colony formation assays were performed using GT1 and GT2a replicons and CyPA knockdown cells. (C and D) Equivalent expression of intracellular HCV antigens in FGR2a cells with and without CyPA knockdown. (E) Reduced infectivity of the supernatant collected from FGR2a/sh-A161 cells, measured by qRT-PCR analysis of HCV RNA in the infected cells. (F) Infectivity analysis with a colony formation assay.
Suppression of CyPA expression in vitro negatively regulates the production of infectious HCV particles.
Because productive infection by particles produced from the full-length JFH-1 genome could be inhibited significantly in sh-A161 cells (51, 52), the SGR2a result shown in Fig. 4B suggested to us that CyPA may be required for an additional step in the HCV life cycle. To address this question, we used a full-length JFH-1 replicon (FGR2a) that can both be selected by antibiotics (similar to the subgenomic replicons) and produce infectious particles (similar to the wild-type full-length genome). We reasoned that if we could obtain replicon colonies by selection in sh-A161 cells, we could normalize RNA replication by expanding the selected stable cells and then measure the effect of CyPA knockdown on the assembly of infectious virions. The FGR2a RNA formed fewer colonies in the sh-A161 cells than in the control cells, as expected, due to both the presence of NS2 (12) and the additional contribution of reinfection to colony formation. After expansion, however, the expanded FGR2a/sh-A161 replicon cells expressed HCV proteins at levels similar to those in the FGR2a/sh-Luc cells (Fig. 4C), and they maintained CyPA knockdown (Fig. 4D). Sequencing results confirmed that no CsA resistance mutations had arisen in the FGR2a/sh-A161 replicon (data not shown).
We then determined if the assembly of infectious virions was affected by CyPA knockdown. We collected supernatants from equal numbers of FGR2a/sh-Luc and FGR2a/sh-A161 cells and used them to infect naïve Huh-7.5 cells. At day 12 postinfection, intracellular HCV RNA levels were measured by qRT-PCR and showed a >70% reduction in infectivity of the virus collected from the FGR2a/sh-A161 cells (Fig. 4E). Colony formation of the infected cells confirmed the difference in infectivity between viruses collected from these two cell lines (Fig. 4F). Together, these results indicate that CyPA plays a role in modulating the assembly of HCV particles produced in cell culture.
The CPI ALV inhibits production of infectious HCV particles in addition to blocking RNA replication.
We then determined if the clinical CPI ALV could inhibit HCVcc assembly. HCVcc-infected Huh-7.5 cells were treated with various amounts of ALV for 9 h and then washed thoroughly to remove the drug. Supernatants from both treated and untreated cells were collected at 8 and 16 h post-drug removal for use in the test of infectivity (Fig. 5A). Because the HCVcc used in this experiment was tagged with GLuc, whose expression and secretion have been demonstrated to reflect the intracellular RNA replication level (36), we were able to measure the effect of ALV on RNA replication with the same supernatants as those used for infection. ALV treatment did not significantly affect the GLuc readings in the supernatant collected after the shorter recovery period. The higher doses (400 and 1,200 ng/ml) of ALV, however, reduced GLuc readings after the longer recovery period (Fig. 5B), manifesting the well-documented inhibitory effect of CPIs on RNA replication. We used the supernatants with comparable GLuc readings for further analysis of core secretion and virion infectivity. CPI treatments that did not inhibit RNA replication still interfered with core secretion from these cells (Fig. 5C), suggesting a suppression of viral assembly. Furthermore, when these supernatants were used to infect naïve Huh-7.5 cells, a correlation between samples with reduced core secretion and those with reduced infection efficiency was observed (Fig. 5D and E). Finally, quantification of both extracellular and intracellular levels of infectivity for the same cells revealed similar reductions of viral titers in both compartments (Fig. 5F), ruling out an inhibitory effect on virion release. To exclude the possibility that the infectivity inhibition observed was due to the carryover of residual ALV in the supernatants, we applied the same supernatants to a Con1-based replicon (GS5) that is highly sensitive to CsA (35). Incubating the GS5 cells with the treated supernatants did not inhibit expression of GFP (not shown) or NS3 (Fig. 5G) in these cells at any of the concentrations used originally. These results confirm that interfering with CyPA function through chemical means can specifically suppress the assembly of infectious particles of HCVcc in vitro.
Fig 5.

The CPI ALV inhibits the production of infectious particles in addition to blocking RNA replication. (A) Outline of treatment and recovery conditions for measuring the effect of ALV on HCV assembly. The treatment time was carefully optimized to minimize inhibition of RNA replication. (B) Secreted luciferase activities (GLuc) in the supernatants of treated cells. Samples 1 through 6 showed no reduction with ALV treatment and were used in further analyses of infectivity. (C) Extracellular core levels in the supernatants of treated cells. The untreated sample levels were set to 100%. (D) HCV infectivity in the supernatants of treated cells. The infections were allowed to proceed for 48, 72, or 120 h before infected cell lysates were analyzed for luciferase activity. (E) Intracellular NS3 levels further support the reduction of infectivity in supernatants of treated cells. (F) Similar reductions of HCVcc infectivity intracellularly and in the culture medium upon CPI treatment. Both cell lysate and culture medium of the ALV-treated cells (D and E) were used to infect naïve Huh-7.5 cells, and GLuc activity of the infected cells was determined 5 days later. (G) Lack of carryover ALV in the supernatant. GS5 cells were treated with the same supernatants as in panel E for 120 h, and intracellular NS3 expression is shown.
CPIs perturb the intracellular localization of NS5A and core proteins.
We next determined if CPIs such as ALV and CsA disrupt the subcellular localization of HCV proteins, especially the direct binding partner of CyPA, the NS5A protein, and the main viral assembly protein, HCV core. We biochemically isolated LD fractions and observed an inhibitory effect of CsA on the association between NS5A and LDs. The association between core and LDs was not suppressed (Fig. 6A). Immunofluorescence staining with a polyclonal anti-FLAG antibody of Huh-7.5 cells infected with JFH-FLAG virus showed the expected punctate staining of NS5A. Both NS5A and core proteins colocalized with staining by BODIPY (Fig. 6B and C, panels a to c), a lipid dye that specifically stains LDs, consistent with the expected roles of these proteins in HCV virion assembly (4, 44), which occurs at or around these organelles (34). Treatment with CsA specifically removed NS5A from LDs without affecting the colocalization of core and LDs (Fig. 6B and C, panels d to f). In addition, the colocalization between core and NS5A around the LDs in the infected cells was also reduced by CsA treatment (Fig. 6D). In contrast, treating the cells with the same concentration of CsA did not affect the colocalization of NS5A and NS3 (Fig. 6E). The colocalization between NS5A and LDs in cells infected with the DEYN virus was resistant to CsA treatment (Fig. 6F). Disruption of NS5A's colocalization with core and LDs in the infected cells could contribute to the reduced infectivity of HCVcc particles produced from CPI-treated cells. Consistent with this, NS5A localization in relation to the LDs in a subgenomic replicon where no structural proteins or viral particles were present was distinct. Instead of fully surrounding the LDs, the NS5A protein localized to discrete spots on the outer edge of the LDs (Fig. 6F).
Fig 6.

CsA perturbs intracellular localization of NS5A around LDs. (A) Inhibition of NS5A's association with LDs upon CsA treatment. The effect of CsA (4 μg/ml for 16 h) on colocalization in JFH-FLAG-infected cells is shown for NS5A with LDs (B), core with LDs (C), NS5A with core (D), and NS5A with NS3 (E). (F) DEYN mutations restore the LD association of NS5A in the presence of CsA treatment. (G) Distinct localization of NS5A in a subgenomic replicon (GS5) that is not affected by CsA treatment. Confocal images were taken on a Leica TCS SP2 AOBS microscope and were processed using LCS AF Lite software (Leica).
DISCUSSION
The ability of NS5A to bind viral RNA is suggested by structural information and supported by in vitro binding experiments with synthetic RNAs. Herein we show that the NS5A complex binds to HCV RNA in HCVcc-infected cells and that this binding is sensitive to CPIs. Treatment with CsA or ALV significantly reduces the association between HCV RNA and NS5A, in a dosage-dependent manner. Although our assay cannot differentiate direct from indirect binding due to the need to analyze authentic, replicating HCV RNA in the infected cells, the correlation between the CPI sensitivity of RNA binding and HCV infection provides the first line of functional evidence that RNA binding by NS5A is important for the HCV infection cycle.
The relative contributions of the three domains of NS5A toward RNA replication and virion assembly are distinct. While domains I and II are likely to be required for replication, domain III is essential for efficient production of infectious particles but dispensable for genome replication (4, 44). These observations suggest that domains I and II are sufficient to mediate RNA binding without domain III's contributions, at least for the RNA-binding function that is involved in replication. How domains I and II coordinate with each other to achieve RNA binding is not clear, but because the major CyPA-binding sites lie outside domain I (21), our current study suggests that perturbation of domain II's structure can significantly affect the overall RNA binding by the full-length NS5A protein in infected cells. On the other hand, the highly potent HCV inhibitor BMS-790052, which most likely targets domain I of NS5A, had no significant effect on RNA binding. The lack of effect on RNA binding of BMS-790052 treatment could be due to the relative insensitivity of JFH-1 to this compound, although the RNA binding of a more sensitive (∼10 times more sensitive) 1b replicon was not inhibited by BMS-790052 either. Thus, it is possible that domain II itself comprises the major RNA-binding activity in infected cells and that CPIs directly abolish RNA binding by domain II. Perhaps the intrinsically disordered domain II can somehow adopt a conformation capable of binding nucleic acids upon interaction with CyPA. Alternatively, CyPA binding to domain II can result in conformational changes in domain I that are necessary for the latter to physically interact with HCV RNA, presumably via the RNA-binding groove revealed by crystal structures. We attempted to address whether domain I or II individually could bind HCV RNA in cells by use of a vaccinia virus-expressing system where the HCV RNA and proteins are expressed in the absence of RNA replication. We did not detect any specific NS5A-RNA interaction with the replication-deficient constructs expressed by vaccinia virus, using the same FLAG-IP methodology as that used here (data not shown). Potential technical issues (e.g., interference from vaccinia virus infection) notwithstanding, these results are consistent with the hypothesis that RNA binding by NS5A is dependent upon active RNA replication. Note that we found that both the positive and negative strands of HCV RNA bound to the NS5A complex and were equally affected by CPI treatment.
The CPI sensitivity of RNA binding by NS5A correlated well with that of viral replication. For example, JFH-1-based SGR2a cells were largely insensitive to CyPA knockdown (Fig. 4B) and only modestly sensitive to CsA treatment (12). In contrast, the GT1b SGR was highly sensitive to CsA treatment or CyPA knockdown. These replication phenotypes were directly correlated with the CsA sensitivity of RNA binding by the NS5A complex in each system. Consistent with a previous report (12), we observed that higher concentrations of CsA (e.g., 4 μM) and longer treatment durations (e.g., 40 h) could inhibit SGR2a replication. Nevertheless, the treatment used in this study (0.83 μM for 8 h) (Fig. 4A) clearly differentiates SGR1b from SGR2a, as well as SGR2a from full-length JFH-1 virus. Furthermore, a CsA-resistant JFH-1 mutant containing the DEYN mutations in domain II of NS5A (51) showed no sensitivity to CsA for RNA binding, even at a concentration of 4 μM and in the full-length background (Fig. 2). These results support the notion that disruption of NS5A's RNA-binding activity is an integral part of a CPI's mechanism of action.
In addition to being part of the replication complex and associating with other members of the replication complex, including NS5B, NS3, and HCV RNA, NS5A also interacts with the core protein and assists in viral assembly (33). The drastic difference in CyPA dependence between the JFH-1 SGR and the full-length virus raised the possibility that CyPA plays a role in NS5A's function in viral assembly. Experiments with RNA interference and CPI treatment both confirmed this prediction and pointed to a role of CyPA in the production of infectious virions. In the full-length FGR system, where the replication levels were normalized, a reduction of infectious virus secreted into the medium was detected in the CyPA knockdown cells, and treating preinfected Huh-7.5 cells with ALV led to suppression of core secretion and infectivity under conditions that did not inhibit RNA replication. Although the perturbation of RNA binding is also compatible with assembly inhibition (e.g., CPIs may interfere with the transition of RNA from replication to packaging, a process proposed to be assisted by NS5A), a more direct mechanism may be at play. Both NS5A and core were extensively colocalized with LDs in HCVcc-infected cells. Upon CPI treatment, core remained associated with LDs, while the majority of NS5A no longer colocalized with core protein or the LDs. The disruption of NS5A and core colocalization and the previously reported perturbation of LDs (2) by CPI treatment can all contribute to an inhibition of assembly efficiency. Given the role of CyPA in RNA replication, a more detailed dissection of its function in HCV assembly can potentially benefit from an in vitro system where replication and assembly are uncoupled, such as the one recently described by Triyatni et al. (48).
Overall, our results suggest that CyPA modulates several aspects of NS5A function, including RNA binding and LD association. CPIs interfere with these essential functions of NS5A to suppress both replication and assembly of HCV. Because the CPIs directly target the host cell as opposed to the virus, and because they can inhibit more than one step of the viral life cycle, these compounds may present a higher genetic barrier to the development of drug resistance.
ACKNOWLEDGMENTS
We thank Takaji Wakita, Kai Lin, Brett Lindenbach, and Charles M. Rice for reagents; Vaithilingaraja Arumugaswami for helpful discussions regarding the design of the JFH-FLAG construct; Brian Washburn, Kristina Poduch, Henry Grise, and Ruth Didier for technical assistance; and Anne B. Thistle for proofreading the manuscript.
A.N. was supported in part by a postdoctoral fellowship from the Leukemia and Lymphoma Society. This research was supported by NIH grant R01 AI079150 (H.T.) and also in part by a grant from the Novartis Institutes of Biomedical Research (J.M.R. and H.T.).
Footnotes
Published ahead of print 12 September 2012
REFERENCES
- 1. Ahn J, et al. 2004. Systematic identification of hepatocellular proteins interacting with NS5A of the hepatitis C virus. J. Biochem. Mol. Biol. 37:741–748 [DOI] [PubMed] [Google Scholar]
- 2. Anderson LJ, Lin K, Compton T, Wiedmann B. 2011. Inhibition of cyclophilins alters lipid trafficking and blocks hepatitis C virus secretion. Virol. J. 8:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Appel N, Pietschmann T, Bartenschlager R. 2005. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J. Virol. 79:3187–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Appel N, et al. 2008. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 4:e1000035 doi:10.1371/journal.ppat.1000035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Armstrong GL, et al. 2006. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann. Intern. Med. 144:705–714 [DOI] [PubMed] [Google Scholar]
- 6. Arumugaswami V, et al. 2008. High-resolution functional profiling of hepatitis C virus genome. PLoS Pathog. 4:e1000182 doi:10.1371/journal.ppat.1000182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benga WJ, et al. 2010. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 51:43–53 [DOI] [PubMed] [Google Scholar]
- 8. Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974 [DOI] [PubMed] [Google Scholar]
- 9. Cai Z, et al. 2005. Robust production of infectious hepatitis C virus (HCV) from stably HCV cDNA-transfected human hepatoma cells. J. Virol. 79:13963–13973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chatterji U, et al. 2009. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J. Biol. Chem. 284:16998–17005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen YJ, et al. 2010. Heat shock protein 72 is associated with the hepatitis C virus replicase complex and enhances viral RNA replication. J. Biol. Chem. 285:28183–28190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ciesek S, et al. 2009. Cyclosporine A inhibits hepatitis C virus nonstructural protein 2 through cyclophilin A. Hepatology 50:1638–1645 [DOI] [PubMed] [Google Scholar]
- 13. de Chassey B, et al. 2008. Hepatitis C virus infection protein network. Mol. Syst. Biol. 4:230 doi:10.1038/msb.2008.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Egger D, et al. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76:5974–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fernandes F, Ansari IU, Striker R. 2010. Cyclosporine inhibits a direct interaction between cyclophilins and hepatitis C NS5A. PLoS One 5:e9815 doi:10.1371/journal.pone.0009815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fernandes F, et al. 2007. Sensitivity of hepatitis C virus to cyclosporine A depends on nonstructural proteins NS5A and NS5B. Hepatology 46:1026–1033 [DOI] [PubMed] [Google Scholar]
- 17. Flisiak R, Jaroszewicz J, Flisiak I, Lapinski T. 2012. Update on alisporivir in treatment of viral hepatitis C. Expert Opin. Invest. Drugs. 21:375–382 [DOI] [PubMed] [Google Scholar]
- 18. Foster TL, Belyaeva T, Stonehouse NJ, Pearson AR, Harris M. 2010. All three domains of the hepatitis C virus nonstructural NS5A protein contribute to RNA binding. J. Virol. 84:9267–9277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Foster TL, Gallay P, Stonehouse NJ, Harris M. 2011. Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J. Virol. 85:7460–7464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gao M, et al. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grise H, Frausto S, Logan T, Tang H. 2012. A conserved tandem cyclophilin-binding site in hepatitis C virus nonstructural protein 5A regulates alisporivir susceptibility. J. Virol. 86:4811–4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hanoulle X, et al. 2009. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J. Biol. Chem. 284:13589–13601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hopkins S, et al. 2012. The cyclophilin inhibitor SCY-635 suppresses viral replication and induces endogenous interferons in patients with chronic HCV genotype 1 infection. J. Hepatol. 57:47–54 [DOI] [PubMed] [Google Scholar]
- 23a. Huang L, et al. 2012. Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J. Biol. Chem. 280:36417–36428 [DOI] [PubMed] [Google Scholar]
- 23b. Hwang J, et al. 2012. Hepatitis C virus nonstructural protein 5A: biochemical characterization of a novel structural class of RNA-binding proteins. J. Virol. 84:12480–12491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jirasko V, et al. 2010. Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLoS Pathog. 6:e1001233 doi:10.1371/journal.ppat.1001233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kato T, et al. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817 [DOI] [PubMed] [Google Scholar]
- 26. Kaul A, et al. 2009. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 5:e1000546 doi:10.1371/journal.ppat.1000546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lawitz E, et al. 2011. Safety, pharmacokinetics, and antiviral activity of the cyclophilin inhibitor NIM811 alone or in combination with pegylated interferon in HCV-infected patients receiving 14 days of therapy. Antiviral Res. 89:238–245 [DOI] [PubMed] [Google Scholar]
- 28. Lee C, et al. 2011. The hepatitis C virus NS5A inhibitor (BMS-790052) alters the subcellular localization of the NS5A non-structural viral protein. Virology 414:10–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lindenbach BD, et al. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626 [DOI] [PubMed] [Google Scholar]
- 30. Liu Z, Yang F, Robotham JM, Tang H. 2009. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J. Virol. 83:6554–6565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lohmann V, et al. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113 [DOI] [PubMed] [Google Scholar]
- 32. Love RA, Brodsky O, Hickey MJ, Wells PA, Cronin CN. 2009. Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J. Virol. 83:4395–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McLauchlan J. 2009. Hepatitis C virus: viral proteins on the move. Biochem. Soc. Trans. 37:986–990 [DOI] [PubMed] [Google Scholar]
- 34. Miyanari Y, et al. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089–1097 [DOI] [PubMed] [Google Scholar]
- 35. Nelson HB, Tang H. 2006. Effect of cell growth on hepatitis C virus (HCV) replication and a mechanism of cell confluence-based inhibition of HCV RNA and protein expression. J. Virol. 80:1181–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Phan T, Beran RK, Peters C, Lorenz IC, Lindenbach BD. 2009. Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 83:8379–8395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Popescu CI, et al. 2011. NS2 protein of hepatitis C virus interacts with structural and non-structural proteins towards virus assembly. PLoS Pathog. 7:e1001278 doi:10.1371/journal.ppat.1001278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sarrazin C, Hezode C, Zeuzem S, Pawlotsky JM. 2012. Antiviral strategies in hepatitis C virus infection. J. Hepatol. 56(Suppl 1):S88–S100 [DOI] [PubMed] [Google Scholar]
- 39. Sato S, et al. 2006. Proteomic profiling of lipid droplet proteins in hepatoma cell lines expressing hepatitis C virus core protein. J. Biochem. 139:921–930 [DOI] [PubMed] [Google Scholar]
- 40. Schonborn J, et al. 1991. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res. 19:2993–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stapleford KA, Lindenbach BD. 2011. Hepatitis C virus NS2 coordinates virus particle assembly through physical interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 85:1706–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tang H, Grisé H. 2009. Cellular and molecular biology of HCV infection and hepatitis. Clin. Sci. 117:49–65 [DOI] [PubMed] [Google Scholar]
- 43. Targett-Adams P, et al. 2011. Small molecules targeting hepatitis C virus-encoded NS5A cause subcellular redistribution of their target: insights into compound modes of action. J. Virol. 85:6353–6368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tellinghuisen TL, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 4:e1000032 doi:10.1371/journal.ppat.1000032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tellinghuisen TL, Foss KL, Treadaway JC, Rice CM. 2008. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J. Virol. 82:1073–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tellinghuisen TL, Marcotrigiano J, Rice CM. 2005. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435:374–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tran G. 2008. The role of hepatitis C virus in the pathogenesis of hepatocellular carcinoma. Biosci. Horiz. 1:167–175 [Google Scholar]
- 48. Triyatni M, Berger EA, Saunier B. 2011. A new model to produce infectious hepatitis C virus without the replication requirement. PLoS Pathog. 7:e1001333 doi:10.1371/journal.ppat.1001333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wakita T, et al. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Watashi K, et al. 2005. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell 19:111–122 [DOI] [PubMed] [Google Scholar]
- 51. Yang F, et al. 2010. A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS Pathog. 6:e1001118 doi:10.1371/journal.ppat.1001118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang F, et al. 2008. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J. Virol. 82:5269–5278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yi Z, et al. 2006. Subproteomic study of hepatitis C virus replicon reveals Ras-GTPase-activating protein binding protein 1 as potential HCV RC component. Biochem. Biophys. Res. Commun. 350:174–178 [DOI] [PubMed] [Google Scholar]
- 54. Zhong J, et al. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]