

Abstract
Dominant-negative (DN) mutants are powerful tools for studying essential protein-protein interactions. A systematic genetic screen of the essential murine cytomegalovirus (MCMV) protein pM53 identified the accumulation of inhibitory mutations within conserved region 2 (CR2) and CR4. The strong inhibitory potential of these CR4 mutants is characterized by a particular phenotype. The DN effect of the small insertion mutations in CR2 was too weak to analyze (M. Popa, Z. Ruzsics, M. Lötzerich, L. Dölken, C. Buser, P. Walther, and U. H. Koszinowski, J. Virol. 84:9035–9046, 2010); therefore, the present study describes the construction of M53 alleles lacking CR2 (either completely or partially) and subsequent examination of the DN effect on MCMV replication upon conditional expression. Overexpression of CR2-deficient pM53 inhibited virus production by about 10,000-fold. This was due to interference with capsid export from the nucleus and viral genome cleavage/packaging. In addition, the fate of the nuclear envelopment complex in the presence of DN pM53 overexpression was analyzed. The CR2 mutants were able to bind to pM50, albeit to a lesser extent than the wild-type protein, and relocalized the wild-type nuclear envelope complex in infected cells. Unlike the CR4 DN, the CR2 DN mutants did not affect the stability of pM50.
INTRODUCTION
The production and release of infectious herpesviral particles is a multistep process that begins in the nucleus of the infected cell, where viral genomes are packaged into nucleocapsids. A recent study of pseudorabies virus (PrV) showed that the nuclear envelope breaks down to allow the capsid to exit the nucleus (23). However, many studies have shown that the widely accepted nuclear egress pathway requires capsid translocation through the nuclear membrane via primary envelopment and de-envelopment. This process is facilitated by a number of viral and host cell protein interactions. From the viral side, the two major proteins involved are the gene products of UL31 and UL34 of HSV-1 (pUL31 and pUL34, respectively) or their homologues in all other herpesviruses studied to date, which promote efficient primary capsid envelopment at the inner nuclear membrane (INM) (11, 24, 40, 50). The pUL31 homologue of murine cytomegalovirus (MCMV), pM53, is distributed throughout the nucleosol in the absence of other viral proteins. pM53 is targeted to the INM after interacting with the membrane protein pM50 (the homologue of pUL34) via a mechanism that is conserved throughout herpesvirus morphogenesis (13, 27, 31, 46, 55, 56, 62). pM50 and pM53 form the nuclear envelopment complex (NEC) and recruit other cellular and viral proteins, such as protein kinase C and pUS3 (3, 8, 22, 37, 40, 41, 47, 53), resulting in displacement of the rigid nuclear lamina and nucleocapsid budding (reviewed in references 19 and 36).
The mechanism by which the pUL31 and pUL34 homologues mediate capsid transition through the nuclear envelope is still not fully understood, but disruption of either partner usually leads to retention of viral capsids within the nucleus (5, 11, 13, 24, 31, 50, 64). Insights into this mechanism were gained from studying the PrV proteins pUL31 and pUL34, which induce vesicle formation in the nuclei of transfected cells in the absence of other viral components (20), supporting their role in the capsid envelopment process at the INM. The reciprocal binding sites, which result in NEC targeting to the INM, are well characterized in pM53 and pM50 and are conserved throughout the pUL31 family (5, 13, 14, 30, 31, 54, 56). Furthermore, recent studies of HSV-1 suggest a second essential interaction between the N-terminal domain of pUL34 and the C terminus of pUL31, which results in membrane wrapping. This interaction is distinct from that required for INM targeting (2, 48, 49).
The NEC components are crucial for viral morphogenesis in beta- and gamma-herpesviruses (5, 11, 13, 14, 24, 27, 31, 40, 50, 64). Essential genes are not easily studied using traditional approaches because mutant viruses are difficult to reconstitute. However, functional inactivation of such proteins by coexpression of dominant-negative (DN) mutants makes them amenable to comprehensive genetic analysis (18). The function of these proteins during morphogenesis can be studied using mutant viruses. In contrast to null mutants, which only reveal the dominant role of a protein, DN alleles have the potential to arrest viral pathways at different stages, thereby addressing multiple essential functions of a protein (reviewed in reference 39). Using in-depth functional knowledge and detailed experimental information regarding protein structure, such inhibitory mutants can be created by the targeted introduction of crucial, but subtle, mutations or by deleting a domain that represents an independent folding entity (18, 39). However, such data are limited for most herpesvirus proteins.
Therefore, one objective of the present study was to construct DN mutants based on protein sequence predictions. Random mutant screens of the MCMV proteins pM50 and pM53 for DN alleles revealed the accumulation of inhibitory mutations within the conserved regions (CRs) (31, 43, 51). These regions were observed by aligning the protein sequences of 36 UL31 homologues, which showed four distinct peaks with a similarity of up to 60% within the C-terminal two-thirds. Furthermore, mutants of pM53, in which the binding site for pM50 in CR1 (31) was maintained but the C-terminal regions (CR2 to CR4) were replaced with the corresponding parts from other pUL31 family members, were not able to complement wild-type (wt) pM53 function (56). Based on these data, we proposed that chimeric pM53 proteins carrying a conserved but noncomplementing domain of a virus homologue would inhibit MCMV replication.
Here, we investigate the inhibitory potential of mutants constructed by CR domain shuffling and CR deletion. The inhibitory power of the CR2 mutants was improved by these targeted mutations, thereby allowing the functional analysis of the CR2 of pM53.
MATERIALS AND METHODS
Cells and viruses.
Murine embryonic fibroblasts (MEFs) from BALB/c mice, NIH/3T3 murine fibroblasts (ATCC CRL-1658), 293 cells (ATCC CRL-1573), 293T cells (ATCC CRL-11268), and M2-10B4 bone marrow stroma cells (ATCC CRL-1972) were cultured as described previously (35, 56). U-2 OS cells (ATCC HTB-96) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 0.3% l-glutamine, and 0.05 mM nonessential amino acids (Invitrogen). All MCMV mutants were derived from the parental MCMV bacterial artificial chromosome (BAC) pSM3fr-Δ1-16-FRT (32), in which the first 16 dispensable genes at the left side are deleted and a FLP recombination target (FRT) site is inserted and which gives rise to the virus MCMV-Δ1-16-FRT. The deletion of these genes provided sufficient cloning capacity for the insertion of recombinant plasmids. This virus was extensively characterized and found to be indistinguishable from wt MCMV in tissue culture (32). MCMV BACs were reconstituted to viruses by transfecting MEFs with 1.5 μg purified BAC DNA using SuperFect Transfection Reagent (Qiagen) according to the manufacturer's instructions, and supernatants were harvested when the cells were completely lysed. Afterward, virus inocula were scaled up on M2-10B4 cells and purified as described previously (35). The infectivity of the virus stocks was quantified by a standard plaque assay on MEFs (44).
Plasmids.
For construction of M53 CR2 deletion mutants, PCRs were performed on pO6-SVT-M53 (43) using the primer pairs M53ΔCR2AB-for/M53ΔCR2A-5′rev (for primer sequences, refer to Table S1 in the supplemental material) and M53ΔCR2A-3′for/M53ΔCR2AB-rev, resulting in PCR-5′-2A and PCR-3′-2A. The amplicons were cut with AscI and PstI, respectively, and inserted into the AscI/PstI-opened pO6-SVT-M53, giving rise to pO6-SVT-M53Δ2A. Similarly, pO6-SVT-M53Δ2B was constructed using the primer pairs M53ΔCR2AB-for/M53CRΔ2B-5′rev and M53ΔCR2B-3′for/M53ΔCR2AB-rev. pO6-SVT-M53Δ2 was generated by insertion of AscI-treated PCR-5′-2A and PstI-cleaved PCR-3′-2B into AscI/PstI-opened pO6-SVT-M53. Deletion of CR3 was achieved by PCR with the primer pairs M53ΔCR2AB-for/M53ΔCR3-5′rev and M53ΔCR3-3′for/M53ΔCR2AB-rev; cleavage of amplicons with AscI and PstI, respectively; and insertion into AscI/PstI-opened pO6-SVT-M53.
To construct M53 alleles carrying domains of murine gammaherpesvirus 68 (MHV68) ORF69, CR2 of ORF69 was excised using BglII and SacII from pMA-CR2_M6MM (synthesized by GeneArt AG) and inserted into BglII/SacII-opened pL-M53 (31), giving rise to pL-M6MM. Similarly, CR3 of ORF69 was purified from pMA-CR3_ORF69 (GeneArt AG) and inserted into pL-M53 by treatment with PmlI/PstI. To generate MMM6, the N-terminal two-thirds of the M53 open reading frame (ORF) was PCR amplified from pO6-ie-M53 (31) using the primer pair CHfor/CHrev, treated with BglII/XhoI, and inserted into BglII/XhoI-opened pL-M53, giving rise to pL-MMM. M666 was transferred from pO6-ie-M666 (56) into pL-M53 by treatment with SalI and XbaI, resulting in pL-M666. The XhoI recognition site was removed by cutting with NotI and XbaI and religation subsequent to blunting with Klenow, resulting in pL-M666ΔX. From this, CR4 of ORF69 was PCR amplified using primers CR4MHVfor/LITrev, digested with PvuII/XhoI, and inserted into StuI/XhoI-opened pL-MMM, giving rise to pL-MMM6. The AscI/StuI-excised M6MM and MMM6 ORFs and the AscI/PvuII-extracted MM6M ORF were transferred into the AscI/HpaI-treated conditional expression vector pO6-SVTe (52).
All genes were transferred from the conditional pO6-SVT backbone to a constitutive expression vector by excision with AscI and StuI and insertion into the AscI/HpaI-treated pOriR6K-zeo vector (5). All M53 constructs except the s168 mutant were Flag tagged in the constitutive expression vectors, with simultaneous removal of the N-terminal epitope that is recognized by the polyclonal anti-M53 antiserum (31), by inserting the amplicon generated by PCR on pO6-SVT-Flag-ΔN-s309 (43) using the primer pair FlagΔSakM53for/FlagM53-SalIrev and treatment with KpnI and SalI prior to insertion. pO6-ie-FlagM53s168 was constructed by inserting the Acc65I/BspEI fragment of pO6-ie-M53s168 into pO6-ie-FlagM53 (both 31). To tag M53 mutants with BlaC, the small fragments of BsiWI/PshAI-cut pO6-SVT-M53Δ2, of PshAI/PstI-digested pO6-SVT-M53Δ3, and of NotI/SacII-opened pO6T-M53s309 (43) were inserted into pO6T-C-M53 (56) linearized with the appropriate enzymes.
Construction of recombinant viral BACs.
Plasmids containing the recombinant M53 alleles in the conditional-expression cassette were inserted into the parental BAC pSM3fr-Δ1-16-FRT (32) by Flp-mediated recombination, as described previously (5).
Analysis of viral growth and expression kinetics.
The growth rates of wild-type and mutant viruses were compared by infecting M2-10B4 cells with MCMV at a multiplicity of infection (MOI) of 0.1. After 30 min of incubation at 37°C, the cells were washed twice with prewarmed fully supplemented medium, and aliquots were plated in the absence and presence of 1 μg/ml doxycycline. The supernatants of the cultures were collected daily, and the viral load was quantified by plaque assay on MEFs (44).
For expression kinetics, 3.75 × 105 M2-10B4 cells were allowed to attach to cell culture plates and were infected at an MOI of 1 using centrifugal enhancement at 900 × g for 30 min. Residual virus was removed 1 h postinfection (p.i.). Cells and supernatants were harvested at 0, 1, 3, and 5 days postinfection, washed once with phosphate-buffered saline (PBS), and lysed in total lysis buffer (TLB) (62.5 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 6 M urea, 5% β-mercaptoethanol, 0.01% bromphenol blue, 0.01% phenol red). DNA was disrupted by sonication for 5 s at 30% amplitude.
Coimmunoprecipitation.
293 cells (7.5 × 105) were cotransfected using polyethyleneimine transfection reagent (PEI) (Sigma-Aldrich) with 3 to 6 μg of plasmids expressing Flag-tagged M53, M53 mutants, or M50. Cells were harvested 48 h posttransfection and washed once with PBS, and 10% of the samples were lysed in TLB to check for protein expression. DNA was disrupted by sonication for 5 s at 30% amplitude. The remaining 90% of the samples were lysed in high-salt lysis buffer (400 mM NaCl, 20 mM Tris, 1% Triton X-100) in the presence of Benzonase (Novagen) and protease inhibitor cocktail (Roche) for 90 min at 4°C.
Western blot analysis.
Cell lysates from transfected or infected cells were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and proteins were transferred onto Hybond-P membranes (GE Healthcare) in the presence of blotting buffer (25 mM Tris, 192 mM glycine, 20% methanol, pH 8.3). The membranes were blocked in Tris-buffered saline (TBS-T) (10 mM Tris-HCl, 150 mM NaCl, 0.05% Tween 20) containing 5% nonfat dry milk for 1 h at room temperature. For detection of proteins, membranes were incubated with polyclonal rat anti-M53 (31) or rabbit anti-M50 (40) antiserum in TBS-T at 4°C overnight. The immediate-early protein pp89 was detected using a monoclonal mouse antibody (Croma101; provided by S. Jonjic, University of Rijeka, Rijeka, Croatia). A monoclonal rabbit antibody (Cell Signaling) was used for detection of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and a monoclonal mouse antibody (Sigma) for detection of the Flag tag. Bound primary antibodies were reacted with horseradish peroxidase-conjugated secondary antibodies (Dianova) in TBS-T for 1 h at room temperature, and proteins were visualized using an ECL-Plus Western Blot detection kit (GE Healthcare).
Southern blot analysis.
To assess viral packaging, 1.5 × 106 M2-10B4 cells were infected at an MOI of 0.1, and cells were harvested 48 h postinfection. Total DNA was purified with the DNEasy Blood and Tissue Kit (Qiagen) according to the manufacturer's instructions using 100 μl double-distilled water (ddH2O) for elution. Using ApaLI, 6 μg DNA was digested for 16 h and separated by 0.8% agarose gel electrophoresis. The DNA was mobilized, blotted onto a Hybond-N+ membrane (GE Healthcare), and hybridized for 16 h at 60°C with the Apa2 probe, which was generated by a PCR on pSM3fr-Δ1-16-FRT using primers Apa2-for and Apa2-rev (see Table S1 in the supplemental material). The membranes were washed, and bound probe was visualized using the DIG Luminescent Detection Kit (Roche).
Confocal laser scanning microscopy.
To analyze the cellular localization of pM53 and pM50, M2-10B4 and U-2 OS cells were grown on fibronectin-coated microscopy chamber slides (ibidi). M2-10B4 cells were infected at an MOI of 0.5 in the absence and presence of doxycycline (dox), and U-2 OS cells were transfected with up to 0.5 μg of M53- and M50-expressing plasmids using FuGene HD Transfection Reagent or X-tremeGene HP DNA Transfection Reagent (both Roche) according to the manufacturer's instructions. The cells were fixed with 4% paraformaldehyde (PFA) after 36 h of incubation, permeabilized by treatment with 0.1% Triton X-100 for 15 min, blocked for 60 min with 5% donkey serum, and costained with specific polyclonal antisera against pM53 (rat) and pM50 (rabbit) or monoclonal anti-Flag M2 antibody (Sigma). These were in turn reacted with the appropriate Alexa Fluor-coupled secondary antibodies (Molecular Probes). DNA was visualized by staining with To-Pro-3 iodide (Molecular Probes). Photographs were taken on a Zeiss LSM510 with 488-, 543-, and 633-nm laser illumination and filter sets appropriate for Alexa Fluor 488 and 555 and To-Pro-3.
Transmission electron microscopy (TEM).
NIH/3T3 cells were grown on carbon-coated sapphire discs and infected at an MOI of 5 using centrifugal enhancement at 1,000 × g for 30 min. Residual virus was removed 1 h postinfection. The cells were fixed 48 h postinfection by high-pressure freezing with an HPF 01 instrument (M. Wohlwend GmbH), freeze-substituted, and plastic embedded as described previously (6). Embedded samples were sectioned and viewed on a JEOL JEM-1400 transmission electron microscope equipped with an Olympus Veleta charge-coupled device (CCD) camera at an acceleration voltage of 80 kV.
PCA.
To study protein interaction by protein complementation assay (PCA), 7.5 × 105 293T cells were cotransfected using PEI with plasmids expressing M50 and M53 versions fused to either the N-terminal or the C-terminal part of β-lactamase (BlaN and BlaC, respectively) (56). Cells were harvested 48 h posttransfection and lysed in 75 μl luciferase reporter lysis buffer (Promega) for 60 min on ice. Following lysate clearing at 21,000 × g at 4°C for 10 min, the Bla activity of 50 μl of each sample was determined on an enzyme-linked immunosorbent assay (ELISA) plate reader by nitrocefin conversion and absorption measurement at 495 nm for 30 min at 37°C.
RESULTS
Lack of the correct CR2, but not CR3, sequence results in pM53 mutants with strong inhibitory effects.
Random linker-scanning mutagenesis of pM53 identified numerous insertion mutations but few deletion mutations that were inhibitory for virus replication (43). Further analysis of the inhibitory mutations located within CR4 of pM53 showed that their overexpression resulted in a DN effect on nuclear egress and on genome cleavage/packaging. However, the inhibitory phenotype induced by individual insertion mutations in CR2 was weak. Thus, a random screen indicated a functional site in CR2 but did not produce a DN allele with sufficient inhibitory potential for phenotypic analysis.
The DN effect of a mutant protein can be improved by insertion, replacement, or deletion of functionally important amino acids or motifs. This approach is supported by previous observations for pM50 in which the deletion of a putative domain resulted in an inhibitory phenotype stronger than that observed for subtle insertion mutants (51). Furthermore, chimeric pM53 proteins comprising the pM50 binding site fused to the C termini of other pUL31 family members were not able to complement wt pM53 function (56). Based on these observations, we next constructed a set of M53 mutants in which the CRs (31) were either replaced by the respective sequences from pORF69 (the homologue in MHV68) or partly/completely deleted. Both strategies were aimed at identifying mutant proteins with strong inhibitory potential. The mutants were studied in a tet-based conditional-expression system, which allows quantitative assessment of their inhibitory potential (52). In this system, constitutive expression of the tet repressor (tetR) prevents expression of the mutant allele. This allows the reconstitution and propagation of recombinant viruses (Fig. 1A). After dox binding to the tetR, the mutant allele is expressed, and its effects can be studied in a viral context.
Fig 1.

Inhibition of MCMV replication by M53 deletion mutants. (A) Schematic representation of replication analysis. Cells were infected with MCMV mutant viruses. In the absence of doxycycline (left), the constitutively expressed tet repressor (black box) blocks transcription of the mutant gene (gray box with black line), leading to virus replication similar to that observed in the wild type. The inhibitory potential of the mutant proteins can be analyzed upon dox application (right), which allows mutant expression by tethering tetR. (B) Schematic overview of the pM53 mutants. Proteins are depicted as gray bars, and conserved regions are represented as black boxes. The white boxes labeled “69” represent sequences exchanged for the respective stretches of MHV68 ORF69. Amino acids 178 to 211 of M53 were replaced by aa 128 to 161 of ORF69 to yield M6MM, aa 213 to 294 were replaced by ORF69 aa 165 to 245 to yield MM6M, and aa 304 to 333 were replaced by aa 253 to 292 of ORF69 to yield MMM6. Deletion mutants lack aa 181 to 207 (Δ2), 181 to 197 (Δ2A), 197 to 207 (Δ2B), or 214 to 294 (Δ3). (C) M2-10B4 cells were infected with the indicated viruses at an MOI of 0.1 in the absence (black diamonds) or presence (gray squares) of 1 μg/ml doxycycline, and the viral load in the supernatant was quantified by plaque assay. The error bars represent the standard deviations of duplicate experiments using two separate clones. Dashed lines, detection limit.
In the first group of mutants, three out of the four M53 CRs were individually replaced with the corresponding sequences from ORF69 as follows: amino acids (aa) 128 to 161 of pORF69 replaced aa 178 to 211 of pM53 to yield the protein pM6MM, aa 165 to 245 of pORF69 replaced aa 213 to 294 of pM53 to yield pMM6M, and aa 253 to 292 of pORF69 replaced aa 304 to 333 of pM53 to yield pMMM6 (Fig. 1B).
To investigate the inhibitory potentials of the chimeric proteins, the constructs were inserted into MCMV BAC plasmids as additional M53 alleles (32). Recombinant viruses were reconstituted from the BACs by transfection of MEFs in the absence of dox. The viruses were named after the corresponding BAC constructs tagged with an “MCMV” prefix. To investigate the effects of the mutant alleles on virus replication, M2-10B4 cells were infected with the mutant viruses in the absence and presence of dox, and the release of infectious viruses was analyzed under multistep growth conditions (Fig. 1C, top row).
In the absence of dox, all mutant viruses produced titers comparable with those of the wt (at least 105 PFU/ml at 5 days p.i.). Neither addition of dox to wt MCMV (data not shown) nor expression of a second wt gene copy (MCMV-SVT-M53) had any effect. However, induction of the mutant alleles led to different results. Expression of pM6MM in addition to wt pM53 (MCMV-SVT-M6MM) reduced virus production by about 10,000-fold, which was a clear improvement in DN function over the 100-fold effect reported for the i207 mutant derived from a transposon insertion library (43). In addition, the presence of pMMM6 decreased virus levels by about 3 orders of magnitude (MCMV-SVT-MMM6), whereas expression of pMM6M had only a moderate effect (MCMV-SVT-MM6M). The last two findings are in line with the random library data, which show that inhibitory mutations are preferentially located within CR4 and that insertion mutations in CR3 do not inhibit wt protein function (43).
We next studied whether the strong inhibitory phenotype of the CR2 replacement mutant could also be achieved by domain deletion. For this, mutants lacking either the complete CR2 (aa 181 to 207 [Δ2]) or parts of CR2 (aa 181 to 197 [Δ2A] and aa 197 to 207 [Δ2B]) were constructed (Fig. 1B) and tested as described above. Again, virus titers in the absence of dox reached ∼105 PFU/ml (Fig. 1C, bottom row), whereas expression of pM53Δ2 severely reduced virus release by approximately 4 or 5 orders of magnitude (MCMV-SVT-Δ2). The presence of pM53Δ2A decreased virus replication to a comparable level (MCMV-SVT-Δ2A), while the inhibitory effect of pM53Δ2B was less pronounced, reducing viral titers by about 2 orders of magnitude (MCMV-SVT-Δ2B).
To test whether the inhibitory phenotype of the CR2 deletion mutants could be specifically attributed to the CR domain, an M53 deletion mutant lacking the adjoining CR3 (Δ3; aa 214 to 294) was generated. Induction of the M53Δ3 allele by dox inhibited virus replication by only about 1 order of magnitude (MCMV-SVT-Δ3), arguing against a nonspecific effect of the domain deletions (Fig. 1C). We concluded from these and previous data that deletion or alteration of CR2 and CR4, but not of CR3, results in mutant proteins with a strong DN effect on virus replication.
Analysis of the intracellular distribution and expression of the CR2 mutants of pM53.
Changes in the coding sequence may result in proteins with aberrant folding properties, which are then trapped in compartments designated for degradation. To study whether the pM53 mutants were present in the correct cellular compartment, U-2 OS cells were transiently transfected with plasmids expressing the M53 mutants and examined for protein signals 36 h posttransfection (p.t.). Confocal microscopy of the transfected cells revealed that all pM53 mutants correctly localized to the nucleus (data not shown), indicating that all mutant proteins had the potential to obstruct viral maturation in the nucleus. Thus, the functional differences between the pM53 CR2 and CR3 deletion mutants were not due to aberrant protein localization.
To interfere with viral morphogenesis, a DN protein must be present in sufficient amounts within the cell. To study whether induction of the different mutant alleles resulted in comparable levels of protein expression, M2-10B4 cells were infected at an MOI of 1 with wt MCMV, MCMV-SVT-M53, a virus carrying a second wt M53 allele, MCMV-SVT-Δ2, or MCMV-SVT-Δ3 in the absence and presence of dox. At 0, 1, 3, and 5 days p.i., cell lysates were prepared and subjected to Western blot analysis (Fig. 2). The loading control, pp89, showed comparable levels for all viruses. A faint band representing wt pM53 was observed 24 h p.i., the intensity of which increased over time (Fig. 2A). While the addition of dox had no effect on pM53 expression in cells infected with wt MCMV, pM53 expression in cells infected with MCMV-SVT-M53 was upregulated in the presence of dox (Fig. 2A). Wt pM53 was also detectable in the absence and presence of dox after infection with MCMV-SVT-Δ2 and MCMV-SVT-Δ3 (Fig. 2B, black arrowhead). Although the endogenous and mutant M53 alleles had no detection tag, the two different pM53 proteins could be distinguished due to size differences. pM53Δ2 (35 kDa) and pM53Δ3 (29 kDa) were strongly expressed upon induction by dox and could be distinguished from the 38-kDa wt pM53 (Fig. 2B, open arrowhead).
Fig 2.
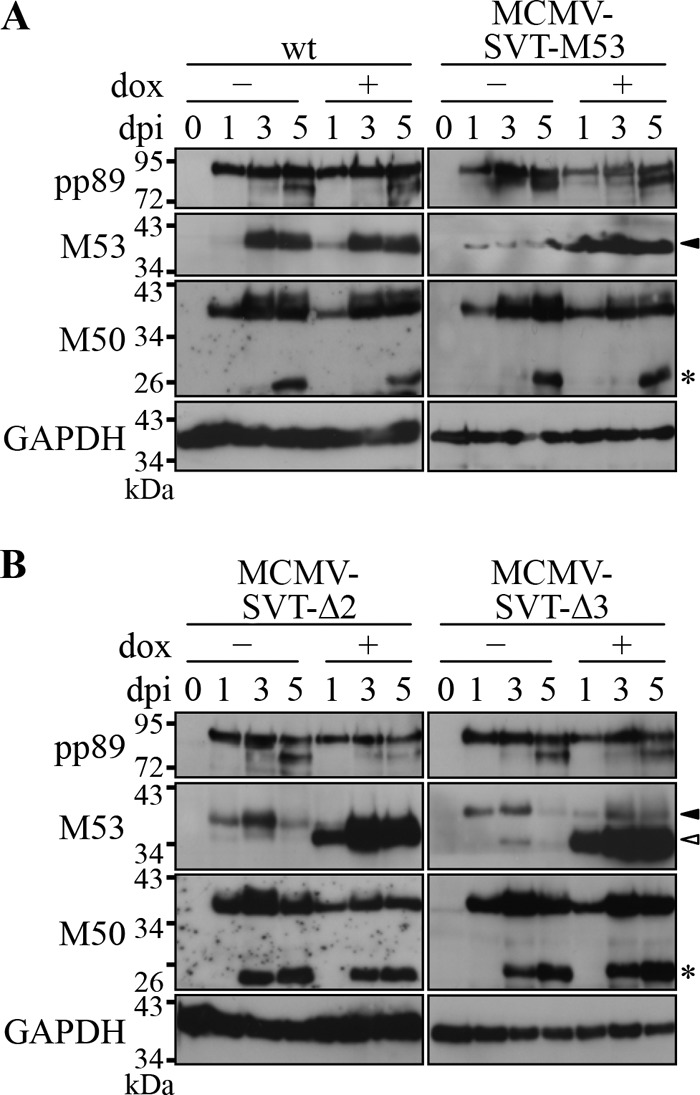
Expression of the wt MCMV NEC proteins in the presence of pM53 mutants. M2-10B4 cells were infected with the indicated viruses at an MOI of 1 in the absence (−) or presence (+) of 1 μg/ml doxycycline. Cell lysates were prepared at the indicated time points and probed for pp89, pM53, and pM50 by Western blotting with specific immune sera. GAPDH was detected using a specific monoclonal antibody. Wt pM53 is indicated by the black arrowhead and the truncated proteins pM53Δ2 and pM53Δ3 by the open arrowheads. pM50 degradation products are indicated by asterisks.
Since pM50 interacts with pM53 (31, 43), the effect of mutant pM53 overexpression on pM50 steady-state levels was also investigated. Following infection with each mutant, increasing pM50 signals were detected from 1 day p.i., accompanied by accumulating levels of pM50 degradation products. In cells infected with MCMV-SVT-Δ2 and MCMV-SVT-Δ3, the pM50 degradation products appeared earlier (Fig. 2, asterisks), but because pM53Δ3 has no inhibitory function, the early accumulation of pM50 degradation products does not explain the function of pM53Δ2.
Deletion of M53 CR2 prevents capsid egress from the nucleus.
Partial and complete deletion of M53 CR2 gave rise to DN mutants that strongly interfered with MCMV replication. The growth defects observed upon pM53Δ2 and pM53Δ2A expression were at least 100-fold stronger than the inhibitory potential of i207, the representative DN insertion mutant (harboring a mutation within M53 CR2) obtained by random linker-scanning mutagenesis (43). The strong DN effect allowed a more detailed characterization of the putative functions of the CR2.
The NEC, consisting of pM53 and pM50, is involved in MCMV genome cleavage and the nuclear egress of viral capsids (43, 56). The effect of CR2 deletions on virus morphogenesis was analyzed by TEM. NIH/3T3 cells grown on carbon-coated sapphire discs were infected at an MOI of 5 with wt MCMV, MCMV-SVT-M53, and MCMV-SVT-Δ2 in the absence and presence of dox and examined by TEM 48 h p.i. after high-pressure freezing (Fig. 3). Viral capsids were observed in the nucleus and cytoplasm of wt MCMV-infected (Fig. 3A) and MCMV-SVT-M53-infected (not shown) cells as previously described (6). In the absence of dox, the infection phenotype of MCMV-SVT-Δ2 was similar to that shown by the wt virus. However, the phenotype of MCMV-SVT-Δ2-infected, dox-treated cells differed from that of wt-infected cells. In this case, the viral capsids were trapped in the nucleus, indicating the blockade of nuclear maturation, similar to that described for the s309 mutant (43). Furthermore, the nuclear capsids in control cells were dispersed and surrounded an intranuclear structure of altered electron density reminiscent of replication compartments (25) (outlined in Fig. 3A and C). Upon dox addition, however, the majority of the nuclear capsids clustered in regularly arranged groups in the middle of these replication compartment-like structures within MCMV-SVT-Δ2-infected nuclei (outlined in Fig. 3E), possibly indicating a capsid transport defect.
Fig 3.

The M53 CR2 deletion virus shows a defect in nuclear egress. NIH/3T3 cells were infected with wt MCMV or MCMV-SVT-Δ2 at an MOI of 5 using centrifugal enhancement in the absence (A to D) and presence (E and F) of 1 μg/ml doxycycline. The cells were then fixed 48 h p.i. by high-pressure freezing, freeze-substituted, plastic embedded, and sectioned. Samples were viewed on a JEOL JEM-1400 transmission electron microscope at 80 kV. The rounded outlines indicate the approximate borders of replication compartment-like structures (A, C, and E). The boxes highlight areas of primary (A and C) and secondary (B and D) envelopment or membrane accumulation (E and F). Scale bar, 1 μm.
During nuclear morphogenesis, a complex order of events leads to mature DNA-filled capsids. Viral proteins are assembled into spherical scaffold-containing procapsids. These are believed to mature concurrently with genome packaging into C capsids, with high-density cores and a hexagonal appearance on TEM. In addition, two more products of capsid maturation can be distinguished. B capsids also contain scaffold-like procapsids, but they are icosahedral. They are thought to be the result of premature cleavage of the scaffold protein. The other form appears as A capsids, which are empty capsid shells with icosahedral symmetry. A and B capsids are believed to be nonfunctional dead-end products of capsid maturation (65), and an increase in their appearance reflects disturbance of the processes involved in capsid maturation.
To quantify the influence of pM53Δ2 expression on capsid morphogenesis, the nuclear capsids in the untreated and dox-treated wt MCMV-, MCMV-SVT-M53-, and MCMV-SVT-Δ2-infected cells seen on the TEM images were classified according to the stages described above (Fig. 4). As it is not clear whether the procapsids are preserved by the TEM preparation protocol used here and because they can be discriminated from B capsids only by their shape, we decided to classify all capsids with apparent scaffold rings into one class. Most of the capsids were classified either as procapsids/B capsids or as C capsids in wt- and MCMV-SVT-M53-infected cells in the absence and presence of dox and in cells infected with MCMV-SVT-Δ2 in the absence of dox. There were almost no A capsids. In contrast, induction of the M53Δ2 allele led to a significant increase in the number of immature A capsids, from about 1% in the absence of dox to about 7%. There was also a significant increase in the number of immature procapsids and B capsids (from about 47% to more than 80%). Accordingly, less than 10% of the nuclear capsids represented mature C capsids (Fig. 4B).
Fig 4.
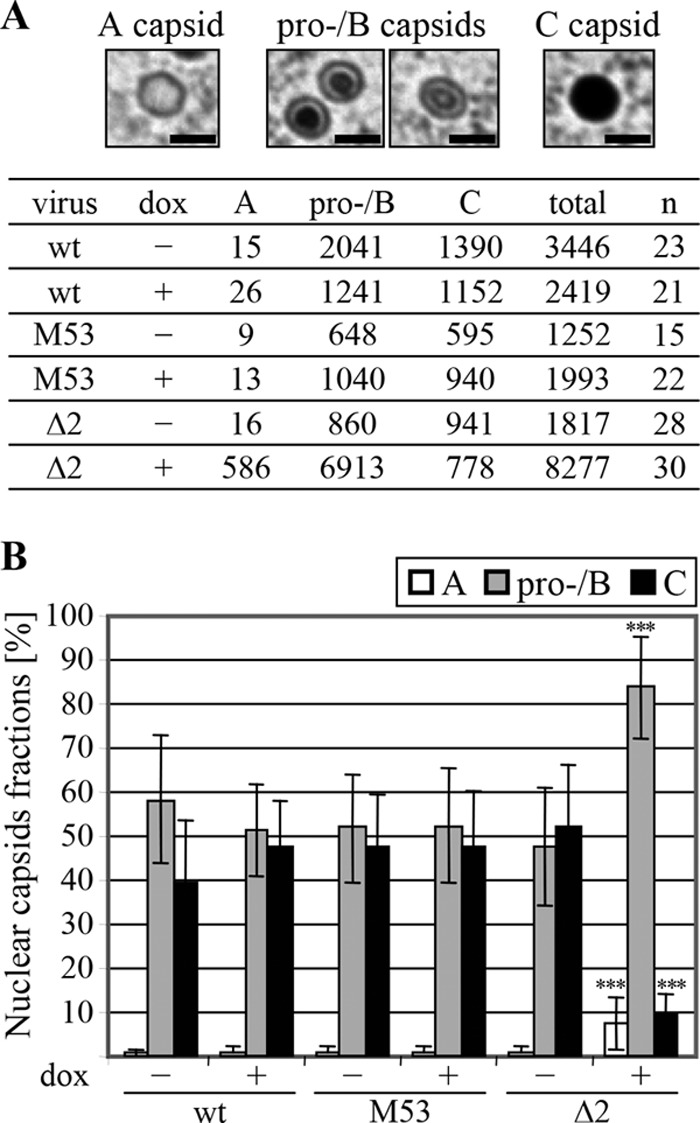
Quantification of the nuclear egress defect. NIH/3T3 cells were infected with the wt MCMV, MCMV-SVT-M53 (M53), or MCMV-SVT-Δ2 (Δ2) and treated as described in the legend to Fig. 3. (A) Nuclear capsids were categorized according to their morphogenesis stage, as shown in the images: immature, unfilled A capsids; scaffold-containing procapsids and B capsids; and mature C capsids. The capsids were counted for a given number of nuclei (n). Scale bar, 100 nm. (B) Percentage of each category of nuclear capsid. The data are expressed as the mean and standard deviation. ***, P < 0.001; unpaired t test (the two conditions for each virus were compared to each other and the wt samples; no label means no significant difference).
In addition to inhibiting capsid maturation, the DN mutant induced membrane accumulation in the nucleus, which was distinct from the membrane-surrounded vesicles observed in wt-infected cells, which are formed by invaginations of the INM during capsid egress (6). This accumulation seemed to comprise membranes folded into each other and connected by a high-staining material (compare the boxed areas in Fig. 3A and C with that in E). No capsids were found in the cytoplasm of most dox-treated MCMV-SVT-Δ2-infected cells. Furthermore, the cytoplasmic assembly and maturation site (34), where the tegument layer is added and secondary envelopment occurs, was absent in dox-treated MCMV-SVT-Δ2-infected cells (Fig. 3, right column). Instead, membrane stacks reminiscent of the Golgi apparatus were often observed, which again seemed to be connected by a high-staining material (Fig. 3F, box).
M53 CR2 deletion mutants prevent unit length genome formation.
The increased proportion of immature nuclear capsids found in dox-treated MCMV-SVT-Δ2-infected cells indicated a morphogenesis block in viral development at, or prior to, the stage of viral-DNA packaging. Therefore, we next studied genome cleavage and genome packaging. For this, M2-10B4 cells were either mock infected or infected with wt MCMV or MCMV-SVT-Δ2 in the absence and presence of dox. Total DNA was isolated 48 h p.i. and subjected to Southern blot analysis (Fig. 5). Upon replication, MCMV genomes form concatemers (34). ApaLI restriction digestion of concatemeric genomes gives rise to a 7.2-kbp control fragment and a 6.7-kbp fragment representing the uncleaved, concatemeric genome. The same fragments arise from digestion of circular BAC DNA, which mimics the concatemeric genomic form. However, restriction of unit length genomes cleaved by the viral terminase releases a 5.5-kbp fragment in addition to the 7.2-kbp control fragment (Fig. 5A).
Fig 5.
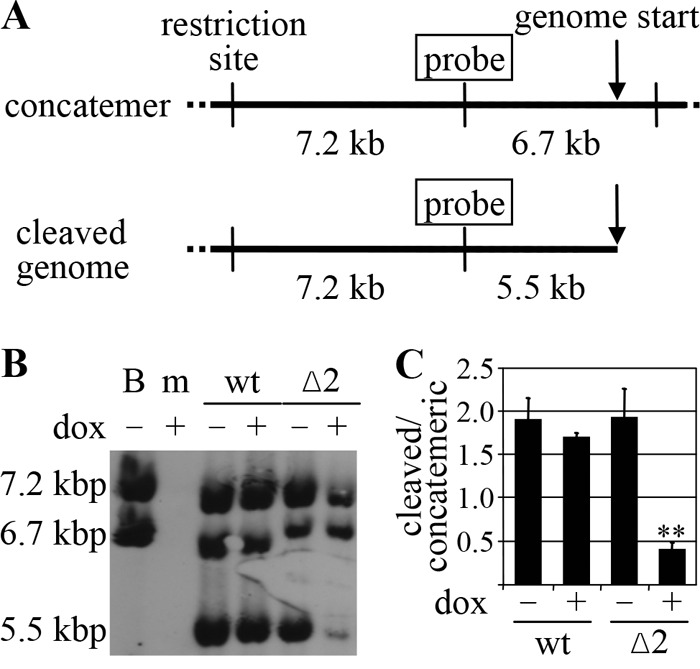
M53 CR2 deletion mutants inhibit the generation of unit length genomes. (A) Schematic representation of the Southern blot assay used to analyze MCMV genome cleavage/packaging. In their concatemeric form, MCMV genomes are connected head to tail (top, arrow). Restriction digestion releases a 7.2-kbp control fragment and a 6.7-kbp fragment, which are both recognized by the same probe. In contrast, the unit length genome (bottom) gives rise to the 7.2-kbp control fragment but also gives rise to a 5.5-kbp fragment representing genome cleavage. (B) M2-10B4 cells were mock infected (m) or infected with wt MCMV or MCMV-SVT-Δ2 (Δ2) at an MOI of 0.1 for 48 h in the absence (−) or presence (+) of 1 μg/ml doxycycline. Total DNA was purified, digested with ApaLI for 16 h, and subjected to Southern blot analysis. B, BAC DNA. (C) The chemiluminescence signal generated in the Southern blot assay was measured in a Typhoon scanner, quantified using ImageJ software (http://rsbweb.nih.gov/ij/), and then plotted as the ratio of unit length genomes to concatemeric genomes normalized to the control fragment. The error bars represent the standard deviations of four independent experiments (**, P < 0.01; paired t test).
The expected fragments of 7.2 kbp and 6.7 kbp were detected at comparable levels after ApaLI digestion of circular wt MCMV BAC DNA (Fig. 5B). The same pattern was observed for the BAC DNA of the mutant viruses (data not shown). DNA from mock-infected cells gave no signal at all, confirming the specificity of the probe. All three of the expected signals were detected after digestion of DNA from wt- and MCMV-SVT-Δ2-infected cells, although the total amount of viral DNA from dox-treated MCMV-SVT-Δ2-infected cells was less than that from nontreated MCMV-SVT-Δ2- or wt-infected cells. More importantly, expression of pM53Δ2 severely reduced the number of unit length genomes (Fig. 5B). To quantify this effect, the chemiluminescence signal was measured in a Typhoon scanner, and the ratio of cleaved to concatemeric genomes was calculated after normalization against the control fragments. In the absence of dox, MCMV-SVT-Δ2 replicated just like wt MCMV and produced unit length genomes at wt levels. In contrast, induction of pM53Δ2 reduced viral cleavage activity, resulting in a decreased number of unit length genomes and the accumulation of concatemeric DNA (Fig. 5C).
Taken together, the results obtained thus far suggest that expression of the CR2 mutants inhibits both capsid egress and genome cleavage/packaging, findings very similar to those observed for CR4 mutants (43).
The wt NEC is formed in the presence of the inhibitory pM53 mutants.
In addition to the interaction between pM53 and pM50, previous reports suggested that pM53 homologues show self-interacting homotypic activity (12, 54, 60, 61). To study the protein-protein interactions between pM53 subunits in more detail, 293 cells were cotransfected with plasmids expressing Flag-tagged wt M53, together with either wt M53 or a CR2 or CR4 deletion mutant in the presence or absence of M50. Protein complexes were then precipitated using an anti-Flag matrix and analyzed by Western blotting. In this setting, anti-M53 antiserum detected both the Flag-tagged and untagged versions of pM53. As shown in Fig. 6, all proteins of interest were expressed at comparable levels (Fig. 6A, Total). Following immunoprecipitation (IP) with the anti-Flag matrix, a signal for wt pM53 was detected, in addition to a signal for pFlagM53, whereas coprecipitation of the pM53 CR2 deletion was almost lost, giving only a minute amount of residual signal, and the CR4 truncation mutant was coprecipitated (Fig. 6A, Flag-IP, lanes 3 to 5). When pM50 was coexpressed, the same pattern was observed, i.e., the signal for wt pM53 was present, but the signals for pM53Δ2 and pM53s309 were not (Fig. 6A, Flag-IP, lanes 7 to 9). The NEC proteins, pM53 and pM50, and their homologues in other herpesviruses, interact with each other, and the complex can be precipitated via either protein partner (5, 31). In this setting, pM50 was immunoprecipitated with pFlagM53 at comparable levels in the presence of pM53, pM53Δ2, or pM53s309. This indicated that the wt NEC was formed regardless of the presence of the inhibitory deletion mutants (Fig. 6A, Flag-IP, lanes 7 to 9). Unfortunately, wt pM53 had a propensity to bind the matrix nonspecifically (Fig. 6B). Therefore, we could not clarify whether the failure to coprecipitate the deletion mutants reflected their lost capacity to interact with wt pM53 or whether the mutations simply prevented them from binding to the matrix.
Fig 6.
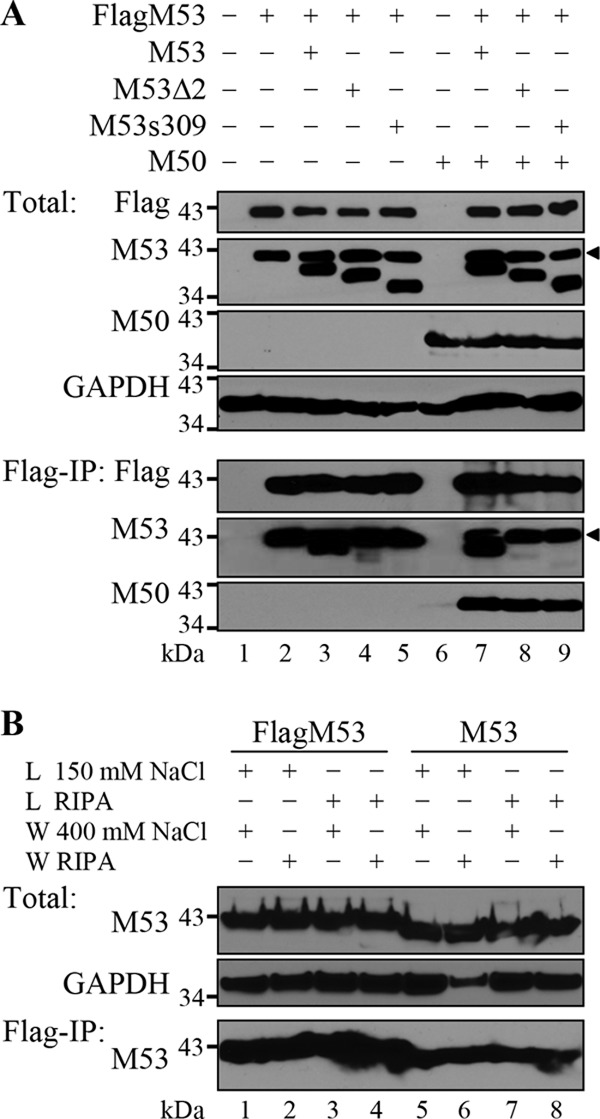
Expression of pM53 DN mutants does not influence the wt pM53-pM50 interaction. (A) 293 cells were transfected with an empty expression plasmid (lane 1) or plasmids expressing Flag-tagged M53 (lanes 2 to 5 and 7 to 9) in combination with wt M53 (lanes 3 and 7) or the mutant M53Δ2 (lanes 4 and 8) or M53s309 (lanes 5 and 9) in the absence (lanes 2 to 5) or presence (lanes 7 to 9) of M50. Cells were harvested 48 h p.t., and 10% of each sample (lysed in TLB) was used to detect total protein levels (Total). The remaining 90% was subjected to immunoprecipitation using an anti-Flag matrix (Flag-IP). pFlagM53 is indicated by a black triangle on the M53 blots. (B) 293 cells were transfected with plasmids expressing Flag-tagged (lanes 1 to 4) and untagged (lanes 5 to 8) M53. Cells were harvested 48 h p.t., and 10% of each sample was lysed in TLB and checked for M53 protein expression. GAPDH was used as the loading control (Total). The remaining 90% was subjected to immunoprecipitation with an anti-Flag matrix under different lysis (L) and washing (W) conditions. The eluates were probed for M53 (bottom; indicated by Flag-IP).
CR2 mutations affect the nuclear distribution of the NEC.
To characterize the fate of the wt NEC further, we performed immunofluorescence analysis of the intracellular localization of pM53 and pM50 in M2-10B4 cells 36 h p.i. The homologues of the HSV-1 pUL31 and pUL34 proteins form a complex at the INM, thereby affecting the curvature of the membrane and allowing capsid translocation into the cytoplasm (11, 24, 40, 50). In accordance with these reports, pM53 and pM50 colocalized at the nuclear rim to form the NEC in wt-infected cells; addition of dox did not change this localization pattern (Fig. 7Ai to Biv). A wt-like appearance was observed in the absence of dox in cells infected with MCMV-SVT-M53, a mutant expressing a second wt M53 allele (Fig. 7Ci to Civ). Overexpression of the second M53 allele increased the amount of nucleosolic pM53 protein with no apparent change in the colocalization with pM50 at the nuclear rim (Fig. 7Di to Div). In MCMV-SVT-Δ2-infected cells, pM53 and pM50 showed a pattern comparable to that observed after wt infection in the absence of dox (Fig. 7Ei to Eiv). Additionally, a few pM53 aggregates, indicative of mutant expression, were already present in the absence of dox. Upon induction of the mutant M53 allele, the normal distribution of NEC was disturbed and large aggregates containing pM53 and pM50 were formed at the nuclear periphery (Fig. 7Fi to Fiv). This granular appearance of the NEC after pM53Δ2 overexpression was similar to the accumulation of NEC in the presence of the CR4 truncation mutant s309 (43). Based on these findings, we hypothesized that the C-terminal tail of pM53 forms a functional unit and induces similar phenotypes when its overall integrity is disturbed.
Fig 7.

Cellular localization of pM50 in the presence of mutant pM53. M2-10B4 cells were infected with wt MCMV (columns A and B); MCMV-SVT-M53, a virus expressing a second wt M53 allele (columns C and D); MCMV-SVT-Δ2 (columns E and F); and MCMV-SVT-gfpSCP (columns G and H) at an MOI of 0.5 in the absence (−) or presence (+) of 1 μg/ml doxycycline and fixed 36 h p.i. The cells were costained with polyclonal antisera specific for pM53 and pM50, followed by Alexa Fluor-conjugated secondary antibodies, and analyzed under a confocal immunofluorescence microscope. DNA was visualized using To-Pro 3. Scale bar, 10 μm.
To confirm that the NEC aggregates were formed selectively after overexpression of pM53Δ2 and not as a consequence of inhibiting nuclear capsid maturation in general, M2-10B4 cells were infected with a mutant virus harboring an inducible green fluorescent protein (GFP)-coupled DN allele of the smallest capsid protein, SCP (MCMV-SVT-gfpSCP), which interferes with capsid maturation (4, 52). While the signal generated by GFP-SCP appeared as granules in the nuclear interior, pM53 colocalized with pM50 was evenly distributed at the nuclear rim, as observed for wt MCMV infection. This distribution was observed in the absence and presence of dox (Fig. 7Gi to Giv), indicating that NEC aggregation in MCMV-SVT-Δ2-infected cells was caused specifically by pM53Δ2 expression.
Many proteins use multiple protein interactions to facilitate their functions. Therefore, to examine whether NEC aggregation was the direct result of pM53Δ2 overexpression and independent of other viral proteins except pM50, U-2 OS cells were transiently transfected with plasmids expressing either M50 or M50 together with M53 or M53Δ2. The cells were then stained with polyclonal antisera against pM50 and pM53 at 36 h p.i.
When expressed individually, pM53 and pM53Δ2 were evenly distributed throughout the nucleosol (data not shown), whereas pM50 alone was observed at the nuclear membrane and in the endoplasmic reticulum (ER) (Fig. 8Ai to Aiv). Coexpression of wt pM53 and pM50 localized both proteins to the nuclear rim due to formation of the NEC (Fig. 8Bi to Biv). In the case of pM53Δ2 coexpression with pM50, a proportion of both proteins colocalized at the nuclear membrane, as did the wt proteins. However, a substantial proportion did not show the well-described nuclear-rim staining, instead forming aggregates comparable to those observed in infected cells (Fig. 8Ci to Civ). Based on these data, we concluded that overexpression of a DN CR2 mutant of pM53 affected the nuclear distribution of the wt NEC.
Fig 8.
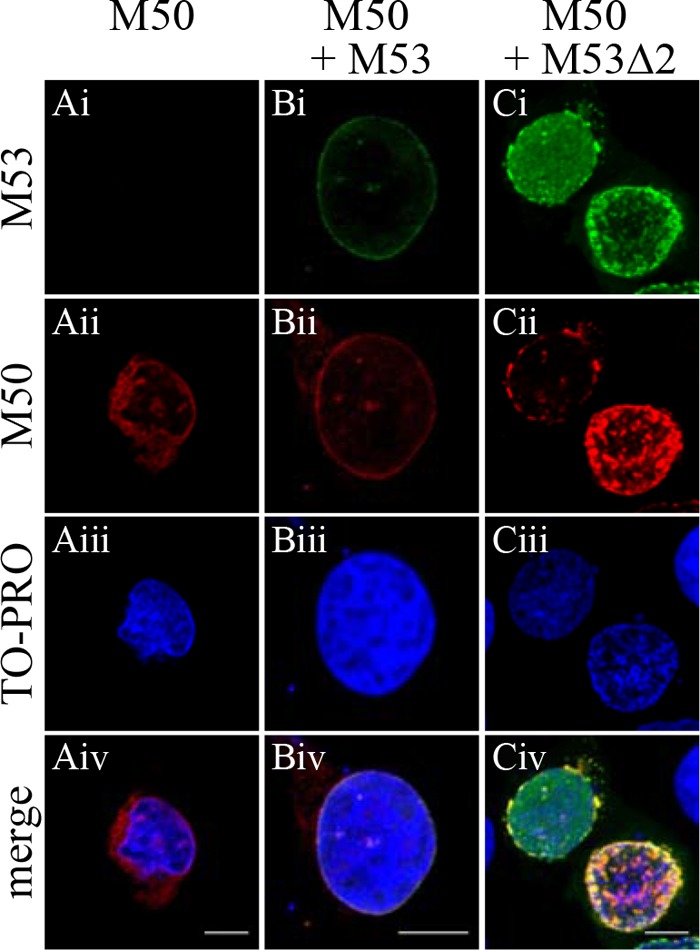
Cellular localization of pM50 in the presence of mutant pM53. U-2 OS cells were transiently transfected with plasmids expressing M50 (A), M53 and M50 (B), or M53ΔCR2 and M50 (C) and fixed 36 h p.i. The cells were costained with polyclonal antisera specific for pM53 and pM50, followed by Alexa Fluor-conjugated secondary antibodies, and analyzed under a confocal immunofluorescence microscope. DNA was stained using To-Pro 3. Scale bar, 10 μm.
pM53Δ2 binds poorly to pM50.
Since formation of NEC aggregates simply requires the presence of both pM50 and the pM53 CR2 deletion mutant, the interaction between pM53Δ2 and pM50 was analyzed in more detail. For this, 293 cells were transfected for 48 h with plasmids expressing M50, together with different Flag-tagged M53 mutants, and 90% of the cells were processed by coimmunoprecipitation with anti-Flag matrix, whereas the remaining 10% were used to check protein expression.
Western blotting (Fig. 9A) confirmed that pFlagM53 proteins and the pM50 protein were expressed at comparable levels. However, the amounts of pM50 precipitated by the pM53 proteins differed markedly. As expected, high levels of pM50 were precipitated by wt pM53 and the truncation mutant pM53s168, which consists of the variable region and the pM50 binding site in CR1. As shown previously (31), the binding-deficient mutant pM53i128 bound only background quantities of pM50. Remarkably, reduced pM50 binding was observed for the CR2 deletion mutants. The strength of pM50 binding declined from pM53Δ2B to pM53Δ2A to pM53Δ2, which precipitated almost no pM50 (Fig. 9A).
Fig 9.
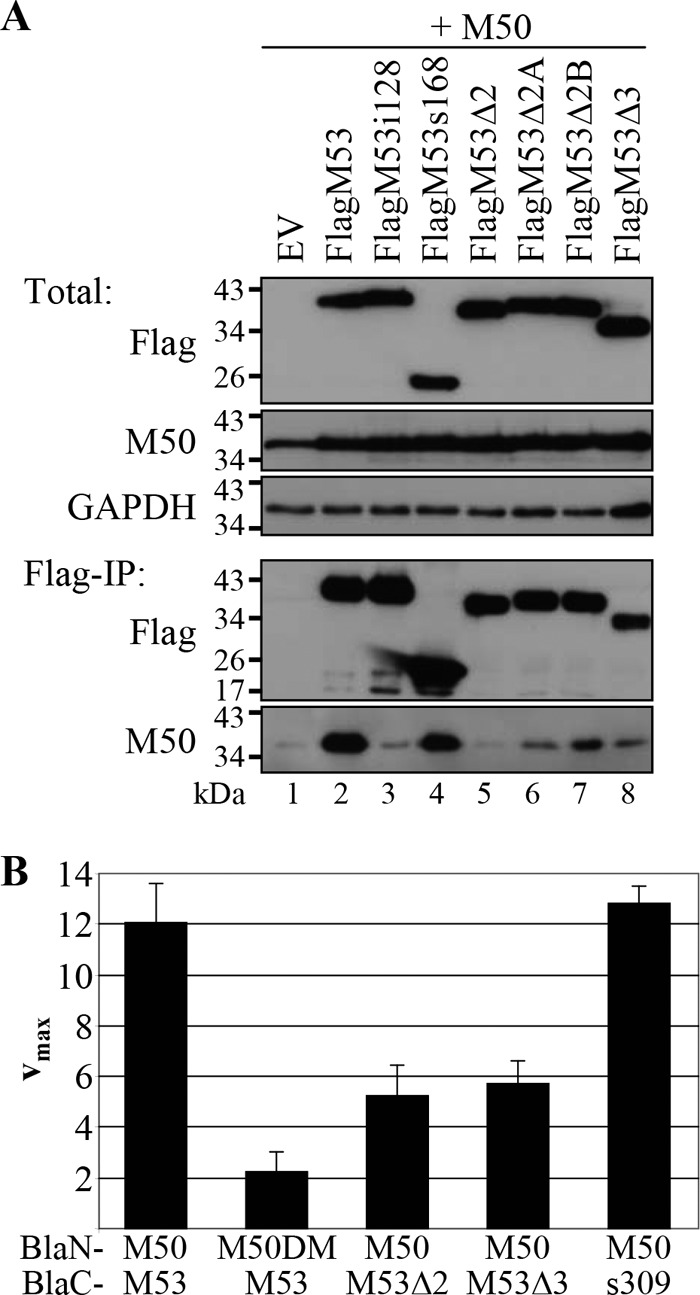
Interaction of the pM53 CR deletion mutants with pM50. (A) 293 cells were transiently transfected with plasmids expressing M50, together with different Flag-tagged M53 versions, and harvested 48 h p.t. Ten percent of each sample was lysed in TLB to detect total protein levels (Total). The remaining 90% was subjected to immunoprecipitation with an anti-Flag matrix and analyzed by Western blotting with specific immune sera (Flag-IP). The Flag signal was detected with a monoclonal anti-Flag antibody directly coupled to horseradish peroxidase. EV, empty vector. (B) 293T cells were transiently transfected with plasmids expressing BlaN-tagged M50, or M53 binding-deficient M50 (M50DM), together with different combinations of the different BlaC-tagged M53 proteins. Cell lysates were prepared 48 h p.t., and beta-lactamase activity was determined by nitrocefin conversion. Absorption was measured at 495 nm. The error bars represent the standard deviations of duplicate samples in at least three separate experiments.
To confirm that pM53Δ2 was defective in pM50 binding, the protein-protein interaction was further studied using the PCA established for the pM50-pM53 interaction (56). For this, 293T cells were transfected with plasmids expressing different combinations of M53 versions fused to the C-terminal part of the beta-lactamase (BlaC), together with different M50 versions tagged by the N-terminal Bla fragment (BlaN). When wt pM50 and pM53 interact, the two Bla fragments are in close proximity and beta-lactamase activity is restored. Cells were lysed 48 h p.t., and Bla activity was determined by nitrocefin conversion and absorbance measurement at 495 nm (Fig. 9B). The combination of pBlaC-M53 with pBlaN-M50 served as a positive control and yielded the maximum rate of hydrolysis (vmax): about 12 absorption units/min. The negative control, consisting of pBlaC-M53 and pBlaN-M50DM, a mutant unable to bind pM53 (5), generated a background signal of about 2 absorption units/min. When pBlaC-M53Δ2 was expressed together with pBlaN-M50, the nitrocefin hydrolysis rate was significantly reduced to approximately 50% of the control value, confirming the poor binding of pM53Δ2 to pM50 (as observed in the Flag-IP experiments). A similar reduction in the rate of nitrocefin conversion was observed for pBlaC-M53Δ3. In contrast, BlaC-s309, together with BlaN-M50, yielded a nitrocefin hydrolysis rate similar to the wt value, indicating an appropriate interaction between the two proteins.
In conclusion, pM53Δ2 had a reduced capacity for binding pM50. As overexpression of this DN mutant led to impaired genome cleavage/packaging and membranous intranuclear NEC accumulation, it is likely that the CR2 of M53 plays a role in coupling genome cleavage with membrane deformation. Taken together, these data show that, despite their phenotypic similarities, the pM53Δ2 and pM53Δ3 mutants are functionally different from the pM53s309 mutant.
DISCUSSION
Random screens for DN mutants are time-consuming and labor-intensive. Therefore, it was of interest to investigate whether DNs can be constructed on a predictive basis. Usually, DN mutants are designed with reference to detailed information about structure-function relationships. However, this knowledge is not yet available for most herpesvirus proteins. There is only sequence information from related viruses that might provide the basis for mutant design. Initial clues suggesting that this approach may work, however, were found in the data from a random screen of the MCMV protein pM53. Analysis of about 100 random mutants indicated the accumulation of potential DNs within two CRs of the protein, namely, CR2 and CR4, which are classified according to the degree of amino acid homology (31, 43). Thus, chimeric proteins composed of the N-terminal variable stretch harboring the nuclear localization signal (NLS) and the CR1 of pM53, which contains the pM50 binding site, still bind to pM50, even when fused to the C-terminal conserved regions of a pM53 homologue. Preliminary data show that such chimeric proteins, which harbor the conserved CR2-CR4 regions of alpha- or gammaherpesvirus proteins, bind pM50 but cannot complement the M53-null mutation. Chimeras with a shuffled CR1, however, cannot even bind pM50, suggesting that subfamily-specific features are important for full function (56).
In this study, we constructed a set of MCMV M53 mutants in which the CRs were disrupted by targeted mutagenesis. Conditional expression of these mutants in a viral context revealed their inhibitory potential. On the whole, the data observed for the designed mutants correlated with the results of the earlier random screen, indicating that an approach based on sequence comparison can be used to construct inhibitory mutants. Targeting CR2, either by domain shuffling or by deletion, gave rise to improved DN effects. These mutants blocked virus production by up to 4 orders of magnitude, an effect that was about 100-fold stronger than that observed with the insertion mutants generated in the random screen. In addition, shuffling of CR4 also produced the DN mutant expected from the data obtained from the random screen. Although mutants constructed by domain shuffling are not as inhibitory as DN mutants harboring deletions at a specific site, one benefit of this approach is that the workload is minimized and the regions of interest are identified quickly. This aspect may be of particular interest if large open reading frames need to be analyzed.
The prominent role of the pUL31 homologues is the formation of the NEC, along with pUL34 homologues, which mediate capsid budding through the nuclear envelope (3, 7, 11, 13, 14, 20, 24, 27, 28, 31, 38, 45, 46, 50, 57, 58). However, an increasing number of studies have identified additional functions for pUL31, including genome cleavage, capsid targeting to the INM, and induction of membrane curvature (43, 48, 63). Here, we constructed M53 alleles lacking CR2 to further study the putative roles of pM53 and analyzed their effects on MCMV replication using conditional expression. We observed that not only CR4-deficient (43) but also CR2-deficient pM53 significantly reduced the number of viral unit length genomes and blocked capsid export from the nucleus. Furthermore, the binding capacity of the CR2 mutant for pM50 was impaired. Nevertheless, the mutant still induced relocalization of the NEC in infected cells. This strongly indicates that mutations within this region of the protein interfere with the formation of homotypic interactions between NEC subunits, as discussed for HSV-1 (48).
Strong expression of the CR2 deletion mutant changed the distribution of the NEC and led to the formation of aggregates at the nuclear membrane. It was observed previously that pUL31 homologues are destabilized if they fail to bind to pUL34 homologues (43, 64). We proposed that the DN function of pM53 mutants was due to displacement of wt pM53 from the NEC, which renders the wt protein prone to degradation. Overexpression of pM53Δ2 did not destabilize wt pM50. This effect is quite different from the effect of the s309 CR4 mutant, which led to the degradation and loss of both wt NEC proteins (43). However, the CR2 mutant had no selective effect on wt pM53 or wt pM50 degradation. This is in line with the reduced binding capacity of pM53Δ2 for pM50. Therefore, the DN effect cannot be explained by competition between the mutant and the wt protein for incorporation into the NEC. This suggests that another, as yet unknown protein-protein interaction (mediated by pM53) is required for correct NEC formation at the nuclear rim. It might also explain why s309 has higher DN potential, because both wt pM50 and wt pM53 are degraded in cells infected with s309-expressing viruses. Studies on a charged-cluster DN mutant of HSV-1 pUL34 implicated the protein, together with CR3 of pUL31, in the induction of INM curvature (48). Notably, we did not retrieve any CR3-derived pM53 mutant with inhibitory potential from the insertion library (43) or by using the domain-shuffling approach. However, despite the lack of functional significance, we did observe a reduced binding capacity of the CR2 and CR3 mutants for pM50, which reflects the data obtained for the HSV-1 homologues. Incidentally, Roller and colleagues (48) discussed a multilevel interaction within the NEC proteins. Although the major binding motif in the MCMV NEC is clearly located in CR1, the present analysis revealed that the pM53 CR2 region also contributes to the binding of pM50.
The first UL31 deletion mutants of HSV-1 revealed a defect in viral-DNA packaging (9) that was later also confirmed for alpha-, beta-, and gammaherpesviruses (15, 21, 26, 43). In Epstein-Barr virus (EBV), deletion of the UL31 homologue, BFLF2, results in the accumulation of DNA-lacking A and B capsids, together with a defect in intranuclear migration; however, the defective particles were still exported (15). Here, we report inhibitory mutants that confirm our previous findings, i.e., that pM53 (the betaherpesvirus homologue of pUL31) is involved in the DNA cleavage/packaging process. About 91% of the nuclear capsids in MCMV-SVT-Δ2-infected cells lacked DNA, and there was a marked reduction in the number of unit length genomes. This effect alone does not explain the strong inhibitory effect but merely indicates that the NEC is involved in genome cleavage/packaging. It is possible that nuclear egress is monitored by a quality control mechanism that preferentially selects mature DNA-filled capsids for primary envelopment rather than DNA-lacking immature forms (6, 16). Recently, capsid maturation in alphaherpesviruses was shown to be linked to nuclear egress by pUL31 association with capsids. While in HSV-1 this interaction depended on pUL25 (63), a constituent of the C-capsid-specific complex (CCSC) consisting of pUL17 and pUL25, these were not required for pUL31-capsid interaction in PrV (29). Capsid-bound pUL31 was observed using a pUL34-deficient PrV, thereby stabilizing a normally transient pUL31-capsid intermediate (29). Despite its enrichment on C capsids, pUL31 was also detected on A and B capsids, although the terminase subunits, as well as the portal protein, were not required for this interaction (reference 29 and unpublished observations). In a yeast two-hybrid screen in varicella-zoster virus (VZV), pUL31 interacted with several other proteins (60). Therefore, capsid binding of pUL31 is likely mediated by at least one yet-unknown protein-protein interaction, for example, by some integral capsid protein. Although the majority of pM53 protein signal in immunofluorescence was detected at the nuclear envelope, pUL31 capsid association shows that some portion of it is present close to the replication centers, thereby supporting viral maturation. An enzymatic function for pUL31 itself has not yet been reported, indicating that the protein possibly stabilizes some other protein complex, which then efficiently executes viral-DNA cleavage and packaging. Overexpression of pM53Δ2 rapidly saturates its binding sites in pUL50, leaving a higher number of proteins free in the nucleosol to attach to capsids. There, it can interfere with DNA cleavage either by blocking terminase activity or by destabilizing a yet-uncharacterized maturation complex.
Docking of mature capsids at the INM was abrogated in an HSV-1 UL34 mutant (48), suggesting a role for pUL34 in docking selectivity. In addition, in our case, capsids docked to the INM were observed very rarely during DN mutant expression. This could be either a consequence of the impaired capsid maturation associated with reduced viral unit length genome formation or a defect associated with capsid transport to the egress sites. The first scenario would agree with previous observations showing inefficient primary envelopment of viral mutants that failed to complete the DNA-packaging process (1, 9, 10, 16, 33, 42). The second explanation is supported by the electron microscopy (EM) data from the present study; mature C capsids present in MCMV-SVT-Δ2-infected nuclei were not observed at the INM but mainly remained associated in capsid clusters formed within the nucleoplasm, arguing for a targeting deficiency caused by the CR2 mutant.
It is supposed that capsid-bound pUL31 guides mature DNA-filled capsids to the INM-bound pUL34 or pUL34-pUL31 complexes. In our case, capsid-bound pM53Δ2 bound poorly to pM50, and there was no homotypic binding to wt M53, which might account for the migration/targeting defect of the capsids. We propose that the capsid clusters observed in cells infected with the CR2 deletion mutant represent the DNA-packaging sites. These sites cannot be detected by TEM in the highly dynamic wt situation, where mature capsids are rapidly transported to the INM. Only the blockade of capsid migration by pM53Δ2 makes this process visible.
Taken together, our data both confirm and extend the current model of herpesvirus nuclear egress. During wt infection, pUL31 homologues are evenly distributed at the nuclear envelope due to pUL34 binding. However, a fraction of pUL31 proteins seems to shuttle freely within the nucleoplasm, where (in HSV-1-infected cells) it first complexes with pUL17 and pUL25. This complex then attaches to the capsid vertices (63). Although this capsid interaction has not been shown for betaherpesviruses, the involvement of pM53 in DNA cleavage/packaging points in this direction. Perhaps the structural rearrangements of the capsid, which occur subsequent to completion of the packaging process (59), are sensed by pUL31 family members. The signal could be transmitted by one of the proteins within the packaging complex or by accessory proteins involved in capsid stabilization.
Finally, membrane dynamics contribute substantially to primary envelopment of herpesviruses. When nuclear egress of the capsids is blocked, membrane stacks are formed in MCMV-SVT-Δ2-infected nuclei, as observed with other DN mutants of MCMV NEC proteins (51). These membrane aberrations are probably derived from the INM, but we did not observe vesicles similar to those monitored in the HSV-1 pUL34 CL13 mutant (49). Haugo and colleagues observed nuclear envelope blebbing, along with an electron-dense layer, upon infection with mutant UL34 HSV-1, which protruded into the cytoplasm (17). The membrane phenotypes induced by different viruses and mutants in different cell types are remarkably distinct, yet they point to the same fact, namely, that the NEC alters nuclear envelope dynamics.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sigrid Seelmeir, Simone Boos, Natalie Röder (MvP, Munich), and Eberhard Schmid (Zentrale Einrichtung Elektronenmikroskopie, Ulm) for excellent technical assistance.
This work was supported by the German Research Foundation (DFG) as part of the “Schwerpunktprogramm 1175” (SPP1175).
Footnotes
Published ahead of print 19 September 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. al-Kobaisi MF, Rixon FJ, McDougall I, Preston VG. 1991. The herpes simplex virus UL33 gene product is required for the assembly of full capsids. Virology 180:380–388 [DOI] [PubMed] [Google Scholar]
- 2. Bjerke SL, et al. 2003. Effects of charged cluster mutations on the function of herpes simplex virus type 1 UL34 protein. J. Virol. 77:7601–7610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bjerke SL, Roller RJ. 2006. Roles for herpes simplex virus type 1 UL34 and US3 proteins in disrupting the nuclear lamina during herpes simplex virus type 1 egress. Virology 347:261–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Borst E-M, Mathys S, Wagner M, Muranyi W, Messerle M. 2001. Genetic evidence of an essential role for cytomegalovirus small capsid protein in viral growth. J. Virol. 75:1450–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bubeck A, et al. 2004. Comprehensive mutational analysis of a herpesvirus gene in the viral genome context reveals a region essential for virus replication. J. Virol. 78:8026–8035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buser C, Walther P, Mertens T, Michel D. 2007. Cytomegalovirus primary envelopment occurs at large infoldings of the inner nuclear membrane. J. Virol. 81:3042–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Camozzi D, et al. 2008. Remodelling of the nuclear lamina during human cytomegalovirus infection: role of the viral proteins pUL50 and pUL53. J. Gen. Virol. 89:731–740 [DOI] [PubMed] [Google Scholar]
- 8. Cano-Monreal GL, Wylie KM, Cao F, Tavis JE, Morrison LA. 2009. Herpes simplex virus 2 UL13 protein kinase disrupts nuclear lamins. Virology 392:137–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chang YE, Van Sant C, Krug PW, Sears AE, Roizman B. 1997. The null mutant of the U(L)31 gene of herpes simplex virus 1: construction and phenotype in infected cells. J. Virol. 71:8307–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Church GA, Wilson DW. 1997. Study of herpes simplex virus maturation during a synchronous wave of assembly. J. Virol. 71:3603–3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Farina A, et al. 2005. BFRF1 of Epstein-Barr Virus is essential for efficient primary viral envelopment and egress. J. Virol. 79:3703–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fossum E, et al. 2009. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog. 5:e1000570 doi:10.1371/journal.ppat.1000570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fuchs W, Klupp BG, Granzow H, Osterrieder N, Mettenleiter TC. 2002. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 76:364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gonnella R, et al. 2005. Characterization and intracellular localization of the Epstein-Barr virus protein BFLF2: interactions with BFRF1 and with the nuclear lamina. J. Virol. 79:3713–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Granato M, et al. 2008. Deletion of Epstein-Barr virus BFLF2 leads to impaired viral DNA packaging and primary egress as well as to the production of defective viral particles. J. Virol. 82:4042–4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Granzow H, et al. 2001. Egress of alphaherpesviruses: comparative ultrastructural study. J. Virol. 75:3675–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haugo AC, Szpara ML, Parsons L, Enquist LW, Roller RJ. 2011. Herpes simplex virus 1 pUL34 plays a critical role in cell-to-cell spread of virus in addition to its role in virus replication. J. Virol. 85:7203–7215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Herskowitz I. 1987. Functional inactivation of genes by dominant negative mutations. Nature 329:219–222 [DOI] [PubMed] [Google Scholar]
- 19. Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394 [DOI] [PubMed] [Google Scholar]
- 20. Klupp BG, et al. 2007. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc. Natl. Acad. Sci. 104:7241–7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Klupp BG, Granzow H, Keil GM, Mettenleiter TC. 2006. The capsid-associated UL25 protein of the alphaherpesvirus pseudorabies virus is nonessential for cleavage and encapsidation of genomic DNA but is required for nuclear egress of capsids. J. Virol. 80:6235–6246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klupp BG, Granzow H, Mettenleiter TC. 2001. Effect of the pseudorabies virus US3 protein on nuclear membrane localization of the UL34 protein and virus egress from the nucleus. J. Gen. Virol. 82:2363–2371 [DOI] [PubMed] [Google Scholar]
- 23. Klupp BG, Granzow H, Mettenleiter TC. 2011. Nuclear envelope breakdown can substitute for primary envelopment-mediated nuclear egress of herpesviruses. J. Virol. 85:8285–8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Klupp BG, Granzow H, Mettenleiter TC. 2000. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J. Virol. 74:10063–10073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kops A, Knipe DM. 1988. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 55:857–868 [DOI] [PubMed] [Google Scholar]
- 26. Kuhn J, et al. 2008. Partial functional complementation of a pseudorabies virus UL25 deletion mutant by herpes simplex virus type 1 pUL25 indicates overlapping functions of alphaherpesvirus pUL25 proteins. J. Virol. 82:5725–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lake CM, Hutt-Fletcher LM. 2004. The Epstein-Barr virus BFRF1 and BFLF2 proteins interact and coexpression alters their cellular localization. Virology 320:99–106 [DOI] [PubMed] [Google Scholar]
- 28. Leach N, et al. 2007. Emerin is hyperphosphorylated and redistributed in herpes simplex virus type 1-infected cells in a manner dependent on both UL34 and US3. J. Virol. 81:10792–10803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leelawong M, Guo D, Smith GA. 2011. A physical link between the pseudorabies virus capsid and the nuclear egress complex. J. Virol. 85:11675–11684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liang L, Baines JD. 2005. Identification of an essential domain in the herpes simplex virus 1 UL34 protein that is necessary and sufficient to interact with UL31 protein. J. Virol. 79:3797–3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lotzerich M, Ruzsics Z, Koszinowski UH. 2006. Functional domains of murine cytomegalovirus nuclear egress protein M53/p38. J. Virol. 80:73–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maninger S, et al. 2011. M94 is essential for the secondary envelopment of murine cytomegalovirus. J. Virol. 85:9254–9267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McNab AR, et al. 1998. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J. Virol. 72:1060–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McVoy MA, Adler SP. 1994. Human cytomegalovirus DNA replicates after early circularization by concatemer formation, and inversion occurs within the concatemer. J. Virol. 68:1040–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Menard C, et al. 2003. Role of murine cytomegalovirus US22 gene family members in replication in macrophages. J. Virol. 77:5557–5570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234 [DOI] [PubMed] [Google Scholar]
- 37. Mou F, Forest T, Baines JD. 2007. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J. Virol. 81:6459–6470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mou F, Wills E, Baines JD. 2009. Phosphorylation of the UL31 protein of herpes simplex virus 1 by the US3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J. Virol. 83:5181–5191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Muhlbach H, Mohr C, Ruzsics Z, Koszinowski U. 2009. Dominant-negative proteins in herpesviruses—from assigning gene function to intracellular immunization. Viruses 1:420–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH. 2002. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297:854–857 [DOI] [PubMed] [Google Scholar]
- 41. Park R, Baines JD. 2006. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J. Virol. 80:494–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Poon AP, Roizman B. 1993. Characterization of a temperature-sensitive mutant of the UL15 open reading frame of herpes simplex virus 1. J. Virol. 67:4497–4503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Popa M, et al. 2010. Dominant negative mutants of the murine cytomegalovirus M53 gene block nuclear egress and inhibit capsid maturation. J. Virol. 84:9035–9046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reddehase MJ, et al. 1985. Interstitial murine cytomegalovirus pneumonia after irradiation: characterization of cells that limit viral replication during established infection of the lungs. J. Virol. 55:264–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reynolds AE, Liang L, Baines JD. 2004. Conformational changes in the nuclear lamina induced by herpes simplex virus type 1 require genes UL31 and UL34. J. Virol. 78:5564–5575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reynolds AE, et al. 2001. UL31 and UL34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 75:8803–8817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939–8952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Roller RJ, Bjerke SL, Haugo AC, Hanson S. 2010. Analysis of a charge cluster mutation of herpes simplex virus type 1 UL34 and its extragenic suppressor suggests a novel interaction between pUL34 and pUL31 that is necessary for membrane curvature around capsids. J. Virol. 84:3921–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roller RJ, Haugo AC, Kopping NJ. 2011. Intragenic and extragenic suppression of a mutation in herpes simplex virus 1 UL34 that affects both nuclear envelope targeting and membrane budding. J. Virol. 85:11615–11625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Roller RJ, Zhou Y, Schnetzer R, Ferguson J, DeSalvo D. 2000. Herpes simplex virus type 1 UL34 gene product is required for viral envelopment. J. Virol. 74:117–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rupp B, et al. 2007. Random screening for dominant-negative mutants of the cytomegalovirus nuclear egress protein M50. J. Virol. 81:5508–5517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rupp B, Ruzsics Z, Sacher T, Koszinowski UH. 2005. Conditional cytomegalovirus replication in vitro and in vivo. J. Virol. 79:486–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ryckman BJ, Roller RJ. 2004. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J. Virol. 78:399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sam MD, Evans BT, Coen DM, Hogle JM. 2009. Biochemical, biophysical, and mutational analyses of subunit interactions of the human cytomegalovirus nuclear egress complex. J. Virol. 83:2996–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Santarelli R, et al. 2008. Identification and characterization of the product encoded by ORF69 of Kaposi's sarcoma-associated herpesvirus. J. Virol. 82:4562–4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schnee M, Ruzsics Z, Bubeck A, Koszinowski UH. 2006. Common and specific properties of herpesvirus UL34/UL31 protein family members revealed by protein complementation assay. J. Virol. 80:11658–11666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Simpson-Holley M, Baines J, Roller R, Knipe DM. 2004. Herpes simplex virus 1 UL31 and UL34 gene products promote the late maturation of viral replication compartments to the nuclear periphery. J. Virol. 78:5591–5600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Simpson-Holley M, Colgrove RC, Nalepa G, Harper JW, Knipe DM. 2005. Identification and functional evaluation of cellular and viral factors involved in the alteration of nuclear architecture during herpes simplex virus 1 infection. J. Virol. 79:12840–12851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Trus BL, et al. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-filled HSV-1 capsids. Mol. Cell 26:479–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Uetz P, et al. 2006. Herpesviral protein networks and their interaction with the human proteome. Science 311:239–242 [DOI] [PubMed] [Google Scholar]
- 61. Vizoso Pinto MG, et al. 2011. Varicella zoster virus ORF25 gene product: an essential hub protein linking encapsidation proteins and the nuclear egress complex. J. Proteome Res. 10:5374–5382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yamauchi Y, et al. 2001. Herpes simplex virus type 2 UL34 protein requires UL31 protein for its relocation to the internal nuclear membrane in transfected cells. J. Gen. Virol. 82:1423–1428 [DOI] [PubMed] [Google Scholar]
- 63. Yang K, Baines JD. 2011. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc. Natl. Acad. Sci. U. S. A. 108:14276–14281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ye GJ, Roizman B. 2000. The essential protein encoded by the UL31 gene of herpes simplex virus 1 depends for its stability on the presence of UL34 protein. Proc. Natl. Acad. Sci. U. S. A. 97:11002–11007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yu X, et al. 2005. Dissecting human cytomegalovirus gene function and capsid maturation by ribozyme targeting and electron cryomicroscopy. Proc. Natl. Acad. Sci. U. S. A. 102:7103–7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.