

Abstract
The E2 envelope glycoprotein of hepatitis C virus (HCV) binds to the host entry factor CD81 and is the principal target for neutralizing antibodies (NAbs). Most NAbs recognize hypervariable region 1 on E2, which undergoes frequent mutation, thereby allowing the virus to evade neutralization. Consequently, there is great interest in NAbs that target conserved epitopes. One such NAb is AP33, a mouse monoclonal antibody that recognizes a conserved, linear epitope on E2 and potently neutralizes a broad range of HCV genotypes. In this study, the X-ray structure of AP33 Fab in complex with an epitope peptide spanning residues 412 to 423 of HCV E2 was determined to 1.8 Å. In the complex, the peptide adopts a β-hairpin conformation and docks into a deep binding pocket on the antibody. The major determinants of antibody recognition are E2 residues L413, N415, G418, and W420. The structure is compared to the recently described HCV1 Fab in complex with the same epitope. Interestingly, the antigen-binding sites of HCV1 and AP33 are completely different, whereas the peptide conformation is very similar in the two structures. Mutagenesis of the peptide-binding residues on AP33 confirmed that these residues are also critical for AP33 recognition of whole E2, confirming that the peptide-bound structure truly represents AP33 interaction with the intact glycoprotein. The slightly conformation-sensitive character of the AP33-E2 interaction was explored by cross-competition analysis and alanine-scanning mutagenesis. The structural details of this neutralizing epitope provide a starting point for the design of an immunogen capable of eliciting AP33-like antibodies.
INTRODUCTION
Hepatitis C virus (HCV) infects an estimated 2 to 3% of the world population (4, 31) and is a major cause of chronic liver disease. The standard of care for chronic infection—a combination of pegylated alpha interferon and ribavirin—is effective in only 50% of patients infected with genotype 1 and is further limited by significant side effects, resistance, and high costs. This treatment has recently been updated to include two new direct-acting antivirals (DAAs), boceprevir (30) and telaprevir (36). A combination of either of these with pegylated alpha interferon and ribavirin has become the new standard therapy for patients with HCV genotype 1 infections. This approach to treatment, while improving the sustained virological response (SVR) rate compared to pegylated alpha interferon and ribavirin alone, still suffers a number of drawbacks: the regimen is restricted to patients with genotype 1 HCV infection, and there is an increased rate of adverse effects. Additionally, since the DAA treatment still requires coadministration of pegylated alpha interferon and ribavirin to reduce the risk of selecting for resistant strains (45), the problems of high cost and low tolerance associated with these drugs remain. There is therefore a pressing need to develop alternative anti-HCV therapies, particularly in the arena of preventative or therapeutic vaccines. The observation that some individuals are able to spontaneously clear HCV infection with virus-specific immune responses (37) has spurred interest in the potential of HCV vaccines, but as yet no such vaccine exists. Progress toward this goal has been hampered by a number of factors, in particular the considerable genetic diversity of HCV.
HCV, a member of the Flaviviridae family of positive-strand RNA viruses, is composed of a nucleocapsid core enveloped by a lipid bilayer in which the two surface glycoproteins, E1 and E2, are anchored. E1 and E2 exist as heterodimers and play an essential role in viral entry into target cells (11). The entry process, while not fully understood, is known to involve a number of host cell surface entry factors, including CD81, scavenger receptor class B type I (SR-BI), and the tight junction proteins Claudin 1 and Occludin (5, 13, 46, 47). E2 is a major target for neutralizing antibodies and contains hypervariable region 1 (HVR1), which is immunodominant and highly variable in sequence (22). Consequently, while antibodies to HVR1 can be neutralizing, they tend to be isolate specific and are unable to recognize E2 from other genotypes or isolates (14, 49). While more broadly neutralizing antibodies exist, the majority of these recognize conformational epitopes on E2 that are noncontiguous and therefore extremely challenging to mimic in a potential vaccine (1, 3, 18, 19).
There has therefore been a great deal of interest in neutralizing antibodies (NAbs) that are directed against conserved, linear epitopes. AP33 is a mouse monoclonal antibody (MAb) that can strongly inhibit the interaction between E2 (in various forms, including soluble E2, E1E2, and virus-like particles) and CD81 (8, 41, 42). The AP33 epitope, which spans residues 412 to 423 of HCV E2, is linear and highly conserved and encompasses a tryptophan residue that plays a critical role in CD81 recognition. Indeed, the antibody has been shown to be capable of potently neutralizing infection across all the major genotypes (20, 42). The AP33 epitope is also recognized by several other MAbs, including HCV1, 95-2, and 3/11 (6, 15).
The rational development of immunogens that might mimic such epitopes and elicit AP33-like antibodies has been stymied by the lack of detailed structural information available for the viral glycoproteins. To further understand the mechanism by which AP33 neutralizes HCV infection and to aid the development of a potential epitope vaccine, the X-ray crystal structure of the Fab portion of AP33 in complex with its epitope peptide has been determined to 1.8Å. The structure is compared to the recently described complex of a different MAb, HCV1, with a similar peptide (26). Additionally, the interaction between AP33 and E2 has been further characterized by cross-competition analyses, alanine-scanning mutagenesis of E2, and mutagenesis of AP33 at key epitope-binding residues.
MATERIALS AND METHODS
Crystallization of AP33 Fab and Fab-peptide complexes.
Purified AP33 Fab was concentrated to 4 mg ml−1 for sitting drop vapor diffusion crystal trials. Trials were carried out at 293 K using a nanodrop crystallization robot (CartesianHoneybee; Genomic Solutions). An initial hit was obtained in condition 47 of the PACT Suite crystallization screen (Qiagen). In the optimized setup, the reservoir contained 65 μl of 18% polyethylene glycol 6000 (PEG 6K), 0.1 M Tris-HCl (pH 8), and 0.2 M CaCl2, and the drop contained 0.5 μl reservoir solution and 0.5 μl Fab. Because attempts to soak the Fab crystals with peptide proved unsuccessful, cocrystallization was performed by screening around the unliganded Fab crystallization conditions. One millimolar synthesized 14-mer 8834 (IQLINTNGSWHINR) or 12-mer 8741 (GlpLINTNGSWHVN) (Enzo Life Sciences) was premixed with 4 mg ml−1 Fab in 20 mM Tris-HCl (pH 7.2). Optimized crystals for each peptide complex grew in 20% PEG 6K, 0.1 M Tris-HCl (pH 8.0), and 0.3 M CaCl2.
Data collection, structure determination, and refinement of AP33 Fab.
AP33 Fab crystals were soaked briefly in cryoprotectant containing crystallant plus 20% glycerol before flash-freezing in a nitrogen stream at 100 K. X-ray data were collected to 2.65 Å on an in-house Rigaku MicroMax-007HF rotating-anode X-ray generator and Saturn 944 charge-coupled-device (CCD) detector. Crystals belonged to space group I4122 with unit cell dimensions a = b = 90.8 Å, c = 458.7 Å, α = β = γ = 90°. Data were processed by using the software suite HKL-2000 (40), and the structure was determined by molecular replacement using the program Phaser (35). In order to sidestep the problem of an unknown elbow angle, three ensembles that encompassed the CHCL, VH, and VL regions, respectively, were used as the molecular replacement model in Phaser. These ensembles were selected on the basis of highest sequence homology to the corresponding AP33 Fab subdomain: CHCL from Fab HGR-2 F6 (PDB accession code 1DQD), VH of Fab B2B4 (3KS0), and VL from BION-1 (1EGJ). The top Phaser solution (translation function Z-score [TFZ score] = 22.9; log-likelihood gain [LLG] = 1,831) contained two copies of each ensemble that together formed two Fab molecules in the asymmetric unit. Each Fab was composed of four canonical immunoglobulin folds. The model was refined to an Rcryst of 24.7% (Rfree = 31.9%) at 2.65 Å using iterative cycles of model building in the software program Coot and refinement using the program Refmac (12, 39).
Data collection, structure determination, and refinement: AP33 Fab complex with peptide 8741.
Crystals were soaked briefly in cryoprotectant containing crystallant plus 20% glycerol before flash-freezing in a nitrogen stream. Data were collected to 1.8 Å on an in-house Rigaku MicroMax-007HF rotating-anode X-ray generator. Crystals belonged to space group C2 with the following unit cell dimensions: a = 127.6 Å, b = 56.9 Å, c = 81.8 Å, α = γ = 90°, and β = 113.9°. Data were processed using HKL-2000 (40), and the structure was determined by molecular replacement with Phaser (35), using one molecule of the unliganded AP33 Fab structure as the search model. The Phaser solution contained one AP33 Fab molecule in the asymmetric unit (TFZ score = 20.1; LLG = 732). After several cycles of refinement in Refmac (39) and model building in Coot (12), the peptide could be built unambiguously into the electron density in the antigen binding site. Rcryst and Rfree values for the refined model were 17.8% and 21.0%, respectively.
Data collection, structure determination, and refinement of AP33 Fab in complex with peptide 8834.
Crystals belonged to space group C2 with the following unit cell dimensions: a = 171.9 Å, b = 40.7 Å, c = 73.8 Å, α = γ = 90°, and β = 112.1°. Data collected in-house were processed using the software program Mosflm (33), and the structure was determined by molecular replacement with Phaser (35), using one molecule of the unliganded AP33 Fab structure as the search model. Phaser found one AP33 Fab molecule in the asymmetric unit (TFZ score = 37.2; LLG = 2021). After several cycles of refinement in Refmac (39) and model building in Coot (12), the peptide could be built unambiguously into the electron density in the antigen binding site. Further refinement and addition of waters yielded a final model with an Rcryst value of 24.4% (Rfree = 32.0%).
Statistics of the data processing and refinement of unliganded and peptide-bound AP33 Fab are detailed in Table 1.
Table 1.
X-ray data collection and refinement statistics for unliganded and peptide-bound AP33 Fab
| Statistic | Value for AP33 Faba |
||
|---|---|---|---|
| AP33 Fab | AP33 Fab complex with peptide 8834 | AP33 Fab complex with peptide 8741 | |
| Data collection | |||
| Space group | I 41 2 2 | C 2 | C 2 |
| Unit cell dimensions (Å)/(°) | a = 90.9, b = 90.9, c = 459.1 | a = 171.9, b = 40.7, c = 73.8 | a = 127.6, b = 56.9, c = 81.8 |
| α = β = γ = 90 | α = γ = 90, β = 112.1 | α = γ = 90, β = 113.9 | |
| Resolution range (Å) | 21.6–2.65 (2.8–2.65) | 20–2.5 (2.64–2.51) | 50–1.80 (1.83–1.80) |
| No. of unique observations | 27,789 | 16,455 | 49,204 |
| Completeness (%) | 97.4 (86.8) | 99.3 (96.8) | 98.4 (81.0) |
| Redundancy | 6.2 (5.8) | 3.5 (3.1) | 3.5 (2.4) |
| Rmerge (%)b | 10.0 (35.5) | 6.0 (48.0) | 3.8 (15.8) |
| 〈I/σI〉 | 12.3 (4.1) | 14 (2.3) | 45.6 (8.1) |
| Refinement | |||
| No. of Fab atoms | 6,521 | 3,283 | 3,316 |
| No. of peptide atoms | 87 | 108 | |
| Avg B factors (Å2) | |||
| Fab/peptide | 40.8e | 45.5/43.1 | 24.1/22.3 |
| Rcrystc | 24.7 | 24.4 | 17.8 |
| Rfreed | 31.9 | 32.0 | 21.1 |
| RMSD bond distance (Å) | 0.013 | 0.016 | 0.028 |
| RMSD bond angle (°) | 1.61 | 1.68 | 2.24 |
Numbers in parentheses refer to the highest-resolution shell.
Rmerge = ∑hkl ∑i | Ihkl, i − 〈Ihkl〉|/∑hkl 〈Ihkl〉, where 〈Ihkl〉 is the average of symmetry-related observations of a unique reflection.
Rcryst = (∑ | |Fo| − |Fc| |)/(∑ |Fo|).
Test set comprised 5% of reflections.
Value for Fab.
Cells.
AP33 hybridoma cells were grown in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% ultra-low-IgG fetal calf serum and gentamicin in CELLine CL 350 bioreactors (Sartorius) according to the manufacturer's instructions. Human epithelial kidney cells HEK-293T (ATCC CRL-1573), baby hamster kidney (BHK) cells, and the human hepatoma cell line Huh-7 were propagated as described previously (20, 44).
Antibodies.
The anti-E2 human monoclonal antibodies (hMAbs) CBH-4B, CBH-4D, CBH-5, CBH-7, CBH-17, HC-1, and HC-11 and an isotype-matched anti-human cytomegalovirus (HCMV) antibody, hMAb R04, have been described previously (19, 23, 25). The anti-E2 mouse MAb AP33 (41) was purified from hybridoma supernatant on a HiTrap protein G column according to the manufacturer's protocol (GE Healthcare). The Fab fragment of AP33 was made by digesting the MAb for 7 h with immobilized papain, followed by purification through a protein A column according to the manufacturer's protocol (Pierce). It was further purified for crystallization by anion exchange on a Mono Q 5/50 GL column (GE Healthcare) in 20 mM Tris (pH 8.5), using a gradient of 0 to 300 mM NaCl for elution. Biotinylation was carried out using the Immunoprobe biotinylation kit (BK101; Sigma) according to the manufacturer's instructions.
Wild-type (WT) and mutant chimeric AP33 MAbs were produced by cotransfection, using the calcium phosphate method, of 1.5 × 106 subconfluent HEK-293T cells with 8 μg each of the heavy- and light-chain expression plasmids pGID200 and pKN100. Two days after transfection, the cells were subcultured into a larger flask and allowed to grow for another 3 days, and the supernatant, which contained about 0.3 μg/ml of IgG, was harvested. The concentration of IgG was measured by enzyme-linked immunosorbent assay (ELISA).
Plasmid constructs and mutagenesis.
The variable domains of the heavy and light chains of mouse MAb AP33 were grafted onto a human IgG1 CH1+CH2+CH3 and CLκ framework in the heavy- and light-chain expression vectors pGID200 and pKN100 (a kind gift from MRC Technology, London, United Kingdom), respectively. Selected amino acids within the complementarity-determining regions (CDRs) were changed to alanine by site-directed mutagenesis, and the mutations were confirmed by nucleotide sequencing. The mutagenesis was carried out on a human-mouse chimeric MAb because this facilitated use of the mutants in other related studies.
Alanine substitution mutants of genotype 1a strain H77c E1E2 (GenBank accession no. AF009606) were constructed as previously described (25). Glycine was substituted at positions where alanine is the WT residue.
IgG capture ELISA.
Microtiter plates (Immulon II) were coated overnight with anti-human IgG (Fab-specific) antibody produced in goat (I-9010, 1:10,000; Sigma) and blocked with 4% skimmed milk– 0.05% Tween 20 in phosphate-buffered saline (PBS). Serially diluted IgG-containing medium was added, with an appropriate IgG standard alongside. The captured MAb was detected with anti-human IgG (Fc-specific) peroxidase conjugate (A-0170, 1:10,000; Sigma), followed by TMB (3,3′, 5,5′-tetramethylbenzidine, Invitrogen) substrate. Absorbance was measured at 450 nm.
Galanthus nivalis agglutinin (GNA) capture ELISA.
The assays were performed essentially as described previously (44). Soluble genotype 1a E2 was made by infecting BHK cells at 5 PFU/cell with recombinant vaccinia virus expressing amino acids (aa) 384 to 660 of genotype 1a strain H77c E2 (41). Four days after infection, the cells were harvested, washed in PBS, and resuspended in lysis buffer (40 mM Tris [pH 7.5], 1 mM EDTA, 150 mM NaCl, 1% Igepal CA-630, 20 mM iodoacetamide, and complete protease inhibitor cocktail [Roche]). Nuclei were pelleted by centrifugation at 15,000 × g, and the cytoplasmic extract containing E2660 was stored in aliquots at −20°C.
WT and mutant genotype 1a E1E2 proteins were made by transfecting HEK-293T cells, using the calcium phosphate method, with the plasmids described above. Two days after transfection, the cells were washed in PBS and the cytoplasmic extracts were prepared as described above.
Microtiter plates (Immulon II) coated with 2.5 μg/ml GNA were used to capture E2 glycoproteins from cell lysates. Serially diluted MAbs were added and incubated for 90 min. After washing, the bound antibodies were detected using an anti-species IgG-HRP, followed by TMB substrate and measurement of absorbance at 450 nm.
Competition assay.
Competition analysis was performed by modifying the GNA-E2 ELISA essentially as described previously (24). Briefly, genotype 1a strain H E1E2 glycoproteins from transiently transfected HEK-293T cell lysates were captured on Immulon II microtiter plates precoated with GNA. After overnight incubation, the plates were washed and 50 μl of competing antibody was added per well at a concentration of 40 μg/ml. After 30 min, an equal volume of biotinylated test antibody was added at four times the half-maximal effective concentration (EC50), so that the final concentration of biotinylated antibody was twice the EC50, and that of competitor was 20 μg/ml. Under these conditions, the signal obtained is closely proportional to the concentration of biotinylated antibody bound to the plate. The plates were incubated for 90 min and washed, and 100 μl of streptavidin-HRP polymer was added per well (S2438, 1:20,000; Sigma). This was followed by TMB substrate and measurement of absorbance at 450 nm.
To develop a cross-competition matrix, the mean signal obtained with each biotinylated antibody in the presence of competing antibody was expressed as a percentage of the signal obtained in the absence of competing antibody.
Protein structure accession numbers.
The coordinates and structure factors are deposited in the Protein Data Bank under accession codes 4gag, 4gaj, and 4gay.
RESULTS
Overall structure of AP33 Fab.
The Fab was numbered according to the Kabat and Wu convention (21), and the peptides according to the corresponding stretch of HCV E2 in the reference genotype 1a strain H77 polyprotein. Initially, peptide 8834, a 14-mer corresponding to residues 411 to 424 of HCV E2 (IQLINTNGSWHINR), was used as a cocrystallization partner with AP33. The structure of the complex was solved to 2.5 Å using the unliganded AP33 Fab as a molecular replacement model. However, better-quality data to 1.8 Å were obtained using the slightly shorter peptide 8741 (GlpLINTNGSWHVN), which represents residues 412 to 423 of the genotype 1a Glasgow strain (44). This peptide differs slightly from 8834 in that Q412 is a pyroglutamic acid (Glp) and I422 is a valine in 8741. Data collection and refinement statistics are detailed in Table 1. Structural comparison of the two complexes confirms that the backbones of the differing residues are similarly placed and the side chains of these residues are solvent exposed and do not make any direct interactions with AP33 Fab (data not shown). In view of this similarity and the more ordered composition of the Fab component of the higher-resolution complex, the structural analysis presented here will refer to the complex with 8741.
The Fab is composed of four canonical immunoglobulin folds (Fig. 1A), and when it is in complex with peptide 8741, clear electron density is visible for residues 1 to 214 of the light chain and residues 1 to 213 of the heavy chain, with the exception of residues 127 to 132 of the heavy chain, which are disordered. The elbow angle is 142°, which is within the range observed for Kappa Fab structures (50). In the unliganded Fab structure, the elbow angles for the two Fab molecules in the asymmetric unit are 131° and 139°, respectively; this difference likely reflects their different symmetry interactions in the crystal. The elbow angle of the Fab in complex with peptide 8834 is 138°. Previous studies have demonstrated that it is not unusual to observe variation in elbow angles for a particular Fab in different crystal forms or liganded/unliganded states (50). There are two residues, RL68 and AL51 (the subscript L or H denotes the light or heavy chain), in disallowed regions of the Ramachandran plot. The side chain of RL68 is disordered, but the main chain has clear electron density. AL51, which is clearly defined in the electron density map, is commonly observed to possess disallowed dihedral angles in Fab structures.
Fig 1.
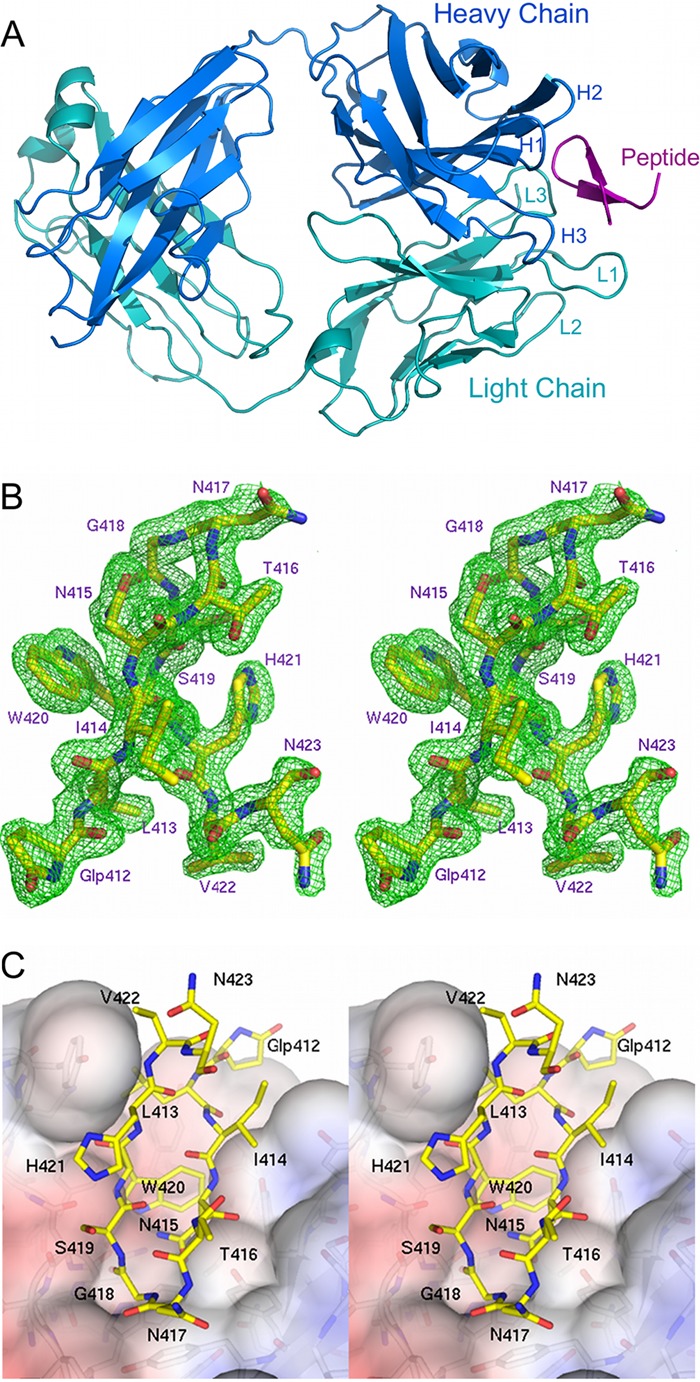
AP33 Fab in complex with its E2 epitope. (A) Overview of peptide 8741 bound in the AP33 Fab combining site. The Fab heavy and light chains are colored dark blue and cyan, respectively. The positions of the CDR loops are labeled. The peptide is colored magenta. (B) Stereoview of the Fo-Fc electron density map (green), contoured at 3σ, superimposed on the peptide. Peptide residues are numbered according to the corresponding residues in the E2 glycoprotein. (C) Stereoview of the surface of AP33 Fab (colored by electrostatic surface potential) with the peptide displayed as sticks with yellow carbon atoms. The figures were produced using the software program PyMOL (www.pymol.org).
CDR classification.
Despite their sequence variability, five of the six CDR loops of antibodies usually adopt one of a small repertoire of canonical structures (7). In AP33, the light-chain CDRs L1, L2, and L3, and the heavy-chain CDRs H1 and H2 belong to canonical classes 5,1,1,1 and 1, respectively (2, 34). The third hypervariable loop of the heavy chain of antibodies, H3, is too variable to be classified into canonical structures but does tend to adopt either a “kinked” or “extended” conformation at the base of the loop. In AP33, the heavy-chain CDR H3 exhibits the more common kinked conformation stabilized by a hydrogen bond between the nitrogen of WH103 and the carbonyl oxygen of MH100B, in agreement with its sequence-predicted structure type (29).
E2 peptide conformation.
The peptide adopts a β-hairpin structure within a pocket formed by the antibody combining site (Fig. 1A). All 12 residues of the peptide are clearly defined in the electron density (Fig. 1B) with the exception of the side chains of N417 and H421, which are partially disordered. The β-hairpin conformation of the peptide is maintained by a pair of backbone hydrogen bonds between I414 and H421 and another pair between T416 and S419 (Table 2). Additional stability of the peptide conformation is provided by a hydrogen bond formed by Oδ1 of N415 to the amide nitrogen of G418 (distance, 3.05 Å) and by a bridging water molecule positioned between the backbone oxygens of pyroglutamic acid residue 412 and V422.
Table 2.
Inter- and intramolecular hydrogen bonds formed by peptide 8741 in its complex with AP33 Faba
| Peptide atom | H-bond partner |
Distance (Å) | Comment | |
|---|---|---|---|---|
| Type | Name | |||
| Glp412 O | Water | w35 | 2.8 | Bridges to peptide (V422) |
| L413 N | Fab | WH100 OH | 2.9 | |
| L413 O | Water | w55 | 2.8 | Bridges to YH33 |
| I414 N | Peptide | H421 O | 2.8 | Peptide β-sheet interaction |
| I414 O | Peptide | H421 N | 2.9 | Peptide β-sheet interaction |
| N415 N | Fab | YH33 OH | 2.9 | |
| N415 Oδ1 | Fab | YH50 OH | 2.7 | |
| N415 O | Water | w176 | 3.0 | Bridges to SH54 via water 163 |
| T416 Oγ1 | Water | w262 | 2.9 | Bridges to CDR H2 via waters 73/57 |
| T416 N | Peptide | S419 O | 3.0 | Peptide β-sheet interaction |
| T416 O | Peptide | S419 N | 3.1 | Peptide β-sheet interaction |
| N417 O | Water | w150 | 2.8 | Bridges to DL94 via water 97 |
| G418 O | Fab | WL96 Nε1 | 2.7 | |
| W420 N | Fab | NL92 Oδ1 | 2.8 | |
| W420 Nε1 | Fab | NL91 O | 2.9 | |
| W420 O | Water | w116 | 2.8 | |
| V422 O | Water | w35 | 2.8 | Bridges to peptide (Glp412) |
Direct hydrogen bonds connecting the peptide to the Fab are highlighted in boldface.
Overview of peptide binding by AP33.
The antigen binding site is a predominantly hydrophobic cavity with aromatic residues lining the combining site. Two asparagine residues of the light-chain CDR L3 contribute some polar character and improve the charge complementarity between AP33 and the peptide (Fig. 1C). The CCP4 software program SC (32) confirms the high shape complementarity at the Fab-peptide interface, giving a shape correlation (SC) value of 0.81. Residues from CDRs L1, L3, H1, H2, and H3 directly contact the peptide. CDR L2, which generally contributes few or no interactions in other peptide-bound Fabs (53), is too distant to interact directly with the peptide.
Interactions between AP33 and the peptide.
The Fab forms six hydrogen bonds directly to the peptide (Table 2). The solvation of the peptide also appears to play a role in recognition by the antibody, being within hydrogen bonding distance of nine water molecules, of which six bridge either directly or via another water molecule to residues of the antibody CDRs (Table 2). The E2 peptide residue W420, which is 99% buried in the combining site, is involved in extensive van der Waals interactions with YH33, YH100, and FL32. IH95 and WL96 also contribute to the hydrophobic nature of the pocket. Additionally, the backbone amide and side chain Nε1 of W420 are within hydrogen-bonding distance of NL91 O and NL92 Oδ1, respectively. The side chain of the E2 residue N415 is 94% buried in the complex and forms hydrogen bonds to YH33 and YH50. Peptide residue L413 is 82% buried in the complex, while G418 is 100% buried. The main-chain amide of L413 hydrogen bonds to the side-chain hydroxyl of YH100, while the carbonyl of G418 forms a hydrogen bond with WL100 Nε1. Peptide residues Glp412, I414, T416, N417, S419, H421, V422, and N423 are solvent exposed and make no direct interactions with AP33 Fab, although the T416 side chain links to a network of waters that reaches CDR H2, and S419 is indirectly connected to DL94 via two bridging water molecules.
Comparison with MAb HCV1 bound to a similar epitope.
Recently, the crystal structure of the same region of E2 in complex with a different neutralizing antibody Fab, HCV1, was reported (26). Two crystal forms (P21 and C2) of HCV1 in complex with the epitope were described. This antibody shares little sequence homology with AP33 in the CDR regions (Fig. 2A) and consequently forms a completely different set of interactions with the epitope peptide. In the AP33 Fab structure, the peptide runs approximately parallel to the interface between the heavy- and light-chain CDRs, while in the HCV1 Fab structure the peptide lies across this interface (Fig. 2B). Interestingly, the peptide itself adopts a very similar β-hairpin conformation whether in complex with AP33 or HCV1 (Fig. 2C). The similarity is particularly apparent in the central portion of the hairpin: the backbone root mean square deviation (RMSD) for the AP33-bound and HCV1-bound peptides over residues 412 to 423 is 1.9 Å or 1.76 Å (for the P21 and C2 forms, respectively), reducing to 0.90 Å or 0.79 Å for residues 413 to 422. Consequently, the paratopes share some similarities with regard to their shape and surface charge, despite the different compositions of the antigen-binding residues. Residues that are deeply buried in AP33 Fab-peptide complex, notably L413, N415, G418, and W420, are also buried in the HCV1 Fab-peptide complex (Fig. 2D). Residues 413 to 422 bury a larger area on the surface of AP33 (559 Å2) than on the two HCV1 structures (476 Å2 and 488 Å2 for the P21 and C2 forms, respectively), suggesting a slightly more intimate association between the peptide and AP33 relative to the complex with HCV1.
Fig 2.
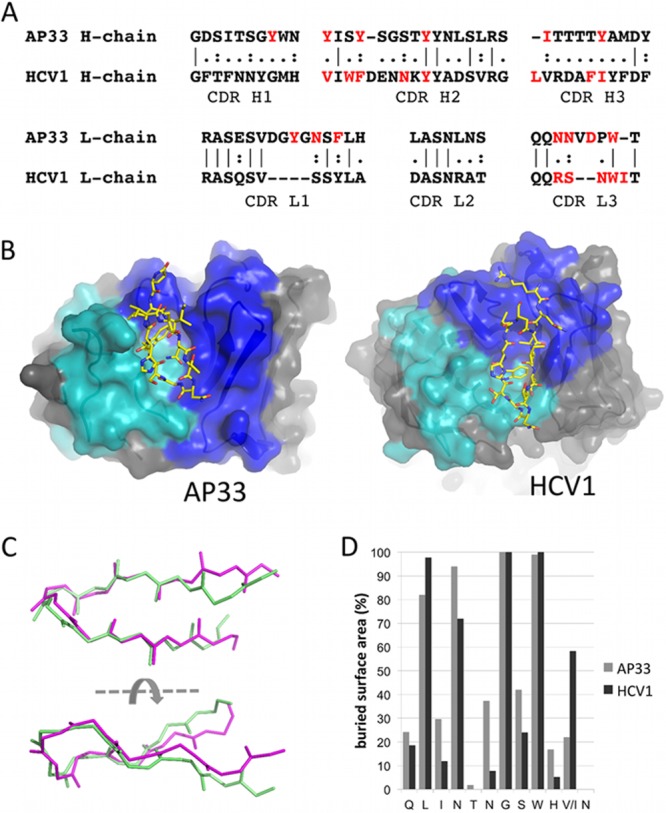
AP33 and HCV1 epitopes adopt a similar conformation. (A) Sequence alignment of AP33 and HCV1 CDRs. The residues of each Fab that interact with the epitope peptide are shown in red. (B) Orientation of the E2 peptide in AP33 (this study) and HCV1 (26) Fab combining sites. Residues of the heavy-chain CDRs are colored blue, and those of the light-chain CDRs are shown in cyan. The peptides are shown as sticks with yellow carbon atoms. (C) Superposition of E2 epitope peptides from complexes with AP33 (pale green) and HCV1 (magenta). Backbone atoms of peptide residues 412 to 422 are shown. (D) Percentage surface area of each peptide residue buried upon binding to AP33 and HCV1, calculated by the software program PISA (28).
AP33 mutagenesis.
To experimentally determine the importance of individual antibody residues for E2 binding, 15 AP33 residues in close proximity to the E2 epitope peptide (Table 3) were individually replaced with alanine, and the effect of each mutation on E2 binding was measured by ELISA. The mutants were named according to the identification and position of the amino acid residue in the wild-type (WT) sequence; for example, in YH33A, the tyrosine at position 33 in the heavy chain was changed to alanine (Table 4). Expression vectors encoding the appropriate antibody heavy- and light-chain combinations were cotransfected into HEK-293T cells. Antibody concentrations in the culture medium were measured by IgG capture ELISA, and the medium was diluted as necessary to equalize the concentrations of IgG. The WT and mutant antibodies were then tested by GNA-capture ELISA for reactivity with soluble genotype 1a E2. The binding of E2 by each mutant is shown in Table 4, expressed as a percentage of binding by WT AP33. Of the 15 mutations tested, YH33A, YH50A, YH58A, IH95A YH100A, FL32A, NL91A, and WL96A resulted in a greater than 90% reduction of E2 binding relative to that for the WT. NL92A showed a 40% reduction, while the remainder exhibited little or no reduction in E2 binding. The substitutions that reduced binding are clustered around the central part of the binding pocket (Fig. 3), whereas the substitutions that did not affect E2 binding (despite being within van der Waals range of the E2 peptide) are positioned toward the outer edges of the pocket.
Table 3.
AP33 residues in close proximity to E2 peptide 8741
| AP33 residue | No. of contacts with peptide 8741a | Buried surface area (%)b |
|---|---|---|
| H chain | ||
| YH33 | 26 | 95.2 |
| YH50 | 16 | 73.5 |
| YH53 | 3 | 25.0 |
| YH58 | 24 | 40.7 |
| IH95 | 2 | 38.0 |
| TH97 | 8 | 46.1 |
| YH100 | 31 | 49.4 |
| L chain | ||
| YL28 | 39 | 46.3 |
| NL30 | 1 | 18.3 |
| FL32 | 18 | 58.1 |
| NL91 | 13 | 40.1 |
| NL92 | 30 | 78.0 |
| VL93 | 1 | 0 |
| DL94 | 2 | 20.8 |
| WL96 | 18 | 26.0 |
Atomic intermolecular contacts between AP33 Fab residues and the E2 epitope peptide 8741 were determined using the software program CONTACT, as implemented in the CCP4 suite (54). Atoms within 4.5 Å of each other were considered to be in contact.
Percent surface area of the AP33 residue that becomes buried upon complex formation with the peptide. Buried surface areas were calculated by the software program PISA (33).
Table 4.
Reactivity of E2 with AP33 alanine substitution mutants
| AP33 mutation | Relative strength of binding to E2 (%)a |
|
|---|---|---|
| Mean | SD | |
| H chain | ||
| YH33A | 2.5 | 6.8 |
| YH50A | −1.0 | 3.9 |
| YH53A | 100.4 | 3.4 |
| YH58A | 0.7 | 2.1 |
| IH95A | 9.9 | 6.0 |
| TH97A | 100.6 | 15.5 |
| YH100A | 4.9 | 3.7 |
| L chain | ||
| YL28A | 111.9 | 16.0 |
| NL30A | 88.8 | 3.2 |
| FL32A | −3.5 | 5.0 |
| NL91A | 6.6 | 5.3 |
| NL92A | 59.8 | 4.0 |
| VL93A | 78.6 | 3.5 |
| DL94A | 74.4 | 0.2 |
| WL96A | −0.4 | 0.2 |
Binding strength of each AP33 mutant, expressed as a percentage of WT AP33 binding to soluble genotype 1a E2 in GNA ELISA. Values shown are the means and standard deviations of data from three replicate assays. Alanine substitutions that reduced E2 binding by 90% or more are displayed in boldface.
Fig 3.
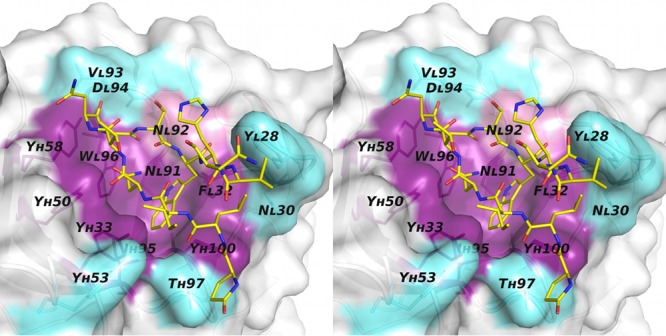
AP33 mutagenesis. Stereoview of the molecular surface of the AP33 Fab binding pocket. The positions of alanine substitutions that reduced binding by >90% are colored purple. NL92A, which reduced binding by 40%, is in pink. Positions of mutations that had little or no effect on E2 binding are colored cyan. The peptide is displayed as sticks with yellow carbon atoms.
AP33 cross-competition analysis.
The major element of the AP33 epitope clearly consists of the conserved linear sequence QLINTNGSWHIN (residues 412 to 423) within E2. However, experimental data show that AP33 binds more weakly to denatured than to native E2, indicating that optimal recognition is affected by the conformation of the glycoprotein (52). This either could be due to denaturation of the β-hairpin structure of the epitope or could indicate that the MAb has some contact residue(s) beyond the linear epitope. To address this question, a competition analysis between AP33 and a panel of well-characterized E2 human MAbs (hMAbs) was performed.
The hMAbs CBH-4B, CBH-5, CBH-7, HC-1, and HC-11 all recognize conserved, conformational epitopes on E2 and have been extensively analyzed by cross-competition analysis and by epitope mapping using a panel of E2 alanine substitution mutants (19, 23, 25). CBH-4B binds in antigenic domain A, CBH-5, HC-1, and HC-11 bind to overlapping epitopes in antigenic domain B, and CBH-7 binds in antigenic domain C. Antigenic domain B and C hMAbs inhibit E2 binding to CD81 and neutralize HCV infection, while antigenic domain A hMAbs are nonneutralizing. The binding of each biotinylated hMAb to E2 was measured in the presence of excess AP33 and vice versa: the binding of biotinylated AP33 was measured in the presence of an excess of each hMAb. The effectiveness of the assay was demonstrated by testing each biotinylated MAb in the presence of an excess of self. Surprisingly, the results showed a marked (>85%) reduction of antigenic domain B antibody binding in the presence of excess AP33 but no significant reduction of AP33 binding in the presence of an excess of any of the hMAbs (Table 5). The same one-way competition was seen with the Fab fragment of AP33 as with the whole AP33 MAb (Table 5), indicating that the AP33 epitope may overlap or be very close to antigenic domain B.
Table 5.
Competition matrix
| Competing antibody (20 μg/ml)c | Bindinga of biotinylated test antibodyb to E2 |
||||||
|---|---|---|---|---|---|---|---|
| CBH-4B | CBH-5 | HC-1 | HC-11 | CBH-7 | AP33 | AP33 Fab | |
| CBH-4B (A) | 12 | 107 | 112 | ||||
| CBH-5 (B) | 17 | 101 | 103 | ||||
| HC-1 (B) | 9 | 94 | 80 | ||||
| HC-11 (B) | 8 | 85 | 74 | ||||
| CBH-7 (C) | 4 | 109 | 112 | ||||
| AP33 | 73 | 13 | 13 | 12 | 93 | 3 | 5 |
| AP33 Fab | 95 | 13 | 13 | 13 | 81 | 5 | 8 |
| R04d | 105 | 95 | 98 | 91 | 109 | 98 | 102 |
The binding of each biotinylated antibody to E2 in the presence of competing antibody is given as a percentage of binding in the absence of competing antibody.
The concentration of each biotinylated test antibody was twice the EC50 (range, 0.05 to 1.0 μg/ml).
Letters in parentheses denote antigenic domain A, B, or C.
Isotype-matched control antibody to a cytomegalovirus protein.
E2 alanine-scanning mutagenesis.
To further investigate the possibility that there may be contact residues outside the main epitope that participate in AP33 binding, alanine substitution studies were performed in which each residue of E2 from 410 to 446, 526 to 540, 611 to 619, and 649 to 655 was individually mutated to alanine. These regions encompass residues involved in CD81 binding, recognition by broadly neutralizing antibodies, and also a region associated with an AP33-resistant virus escape mutant (10, 16). HEK-293T cells were transfected with plasmids encoding the WT and mutant E2 sequences, and the glycoproteins were recovered by cell lysis. The lysates were first normalized for E2 on the basis of reactivity with CBH-17, an antibody to an unrelated linear epitope (22), and then tested for their ability to bind to AP33 and antigenic domain A, B, and C hMAbs. As expected from the crystal structure, alanine substitution at L413, N415, G418, and W420 greatly reduced or prevented AP33 binding (Fig. 4). Greater than 40% reduction in AP33 binding to E2 with substitutions Y611A, R614A, or C652A was also observed. However, the Y611A and R614A substitutions also prevented binding by all three conformation-sensitive antibodies, and C652A reduced binding by CBH-7 and HC-11. Given that the antigenic domain A, B, and C hMAbs bind to nonoverlapping epitopes and do not share contact residues, any substitution that affects more than one hMAb is acting by disrupting E2 conformation. The observed reduction in binding of AP33 to these mutants does not therefore indicate that they are contact residues but agrees with previous data which show that AP33 binds less well to misfolded E2. Instead, alanine mutation at L654 did not alter binding to antigenic domain A and C antibodies but reduced the binding of both AP33 and the antigenic domain B hMAb HC-11 by 40%, indicating that this might be a common contact residue shared by AP33 and the antigenic domain B hMAbs.
Fig 4.

Epitope mapping of AP33 using alanine-scanning mutagenesis of E2. WT genotype 1a H77c E2 and alanine substitution mutants were expressed in HEK-293T cells. In each mutant, one amino acid residue within four selected regions of E2 (410 to 446, 526 to 540, 611 to 619, and 649 to 655) was replaced by alanine (or glycine, where alanine is the WT residue). The cell lysates were first normalized for E2 on the basis of reactivity to CBH-17, an hMAb to a linear E2 epitope. The normalized lysates were then used in GNA capture ELISA to test the binding of AP33 alongside that of the hMAbs CBH-4D, HC-11, and CBH-7, which recognize nonoverlapping conformational E2 epitopes in antigenic domains A, B, and C, respectively. Antibody binding to each mutant is expressed as a percentage of binding to WT E2. Red indicates 0 to 20%, orange 21 to 40%, yellow 41 to 60%, white 61 to 90%, and green >90% of WT binding.
DISCUSSION
There is currently great interest in neutralizing antibodies that are targeted to conserved regions on E2, such as the 412–423 epitope. This region encompasses the highly conserved E2 residue W420, which plays a critical role in CD81 recognition and serves as an important contact residue for several broadly neutralizing antibodies, including AP33 and HCV1 (reviewed by Angus and Patel and Di Lorenzo et al. [3, 10]). Antibodies to this region are relatively uncommon during natural HCV infection, indicating that it is poorly immunogenic (51).
The current study reveals the structural details of AP33 Fab in complex with its epitope peptide (Glp-LINTNGSWHVN) and allows comparison with the recently described structure of a different antibody (HCV1) in complex with a highly similar peptide (R-QLINTNGSWHIN). It is notable that the β-hairpin conformations of the peptides are very similar, despite the very different compositions of the antibody CDRs in AP33 and HCV1. This is a strong indication that the β-hairpin structure represents the conformation of this region in intact E2. Each E2 residue is buried or accessible to a similar extent whether bound to AP33 or HCV1, emphasizing the importance of residues, such as W420, which are completely buried in both complexes. It is interesting that W420 is therefore likely to be solvent exposed, despite its hydrophobic nature. However, surface-exposed tryptophans are occasionally found, particularly in proteins involved in protein-protein interactions (43, 48).
The structures of the AP33- and HCV1-bound peptide provide insights into the positioning of the epitope within the full-length glycoprotein. No high-resolution structural data currently exist for HCV E2, but Krey et al. have constructed a homology model based on related class II fusion proteins using the positions of the protein's disulfide bridges together with functional data and secondary structure predictions (27). In the homology model, the AP33/HCV1 epitope falls within the central domain (domain I), an eight-stranded β-sandwich structure with up-and-down topology that is present in all class II fusion proteins. It is interesting to note that the epitope is predicted to encompass the majority of the first two strands, B0 and C0, of domain I, in agreement with the AP33 and HCV1 Fab-peptide complexes, in which the peptide forms a β-hairpin, with G418 positioned at the turn. In the AP33 and HCV1 complexes, the side of the β-hairpin that lies opposite the antibody-binding face is glycosylated and therefore highly unlikely to be buried. If the peptide were a component of the β-sandwich, the antibody-binding face of the peptide, which is deeply buried when complexed with both AP33 and HCV1, would be inaccessible to antibody or CD81. In this situation, the AP33 and HCV1 antibodies would need to induce a conformational change in order to bind, incurring a high entropic cost. However, the fact that AP33 and HCV1 are both able to bind E2 with high affinity (26, 52; unpublished data) lends strong support to the view that the epitope exists as an exposed flap-like structure (as discussed by Kong et al. 2012 [26])rather than forming part of the β-sandwich.
In the current study, we investigated the effects of mutations within the antibody combining site. Antibody residues that were identified from the crystal structure as being in close proximity to the epitope peptide were individually replaced with alanine. AP33 mutations that had the greatest effect on E2 binding were clustered around the central portion of the binding pocket, while more distal residues, despite being within 4.0 Å of the peptide, had little or no effect on E2 binding, suggesting that their contribution to epitope recognition is minimal. It is noteworthy that the light-chain residue YL28A did not appear to affect E2 binding, even though in the complex this tyrosine appears to interact extensively with residues toward both the N and C termini of the peptide. These results confirm that the peptide structure reflects the true conformation of this region in the intact glycoprotein and suggest that the key determinants for epitope recognition are those that anchor the central and turn regions of the β-hairpin.
The structural details of the Fab-peptide complex provide further confirmation of previous data that indicated that the linear sequence QLINTNGSWHIN (E2 residues 412 to 423) comprises the major element of the AP33 epitope. This was originally established by peptide mapping (41), and the contact residues were identified by phage display and site-directed mutagenesis (52). It cannot be discounted, however, that recognition by AP33 may also involve additional E2 residues beyond those represented by the linear epitope. The observation that AP33 exhibits slightly reduced binding to denatured E1E2 suggests that there may be a modest conformational component to the interaction (42, 52).
To investigate this possibility, a cross-competition analysis with well-characterized conformation-sensitive hMAbs was performed. It was surprising to note that while AP33 competed with the antigenic domain B hMAbs CBH-5, HC-1, and HC-11, there was no reduction of AP33 binding in the presence of an excess of any of the hMAbs. There are several possible interpretations of these findings. The first is that they reflect a difference in binding strengths. An antibody with high affinity will compete effectively with a low-affinity antibody but not vice versa, with the result that nonreciprocal competition by antibodies that bind to overlapping epitopes is observed if their affinities for the antigen are markedly different. AP33 binds strongly, with an EC50 of about 0.23 nM, to genotype 1a E2 (52; unpublished data), while the apparent affinities of HC-1, HC-11, and CBH-5 are lower, with EC50 values of 1.3 nM, 2.4 nM, and 220 nM, respectively (23). Alternatively, if binding strengths are not too disparate, one-way competition can be an indication of close proximity, rather than overlap, of epitopes (24, 38). We know that the HC-11 epitope includes E2 residues 425 to 428 (25) (Fig. 4), which are directly adjacent to the epitope of AP33, and that HC-1 binding is also affected by changes in this region (25). A third possibility is that AP33, although directed primarily at the linear epitope between residues 412 and 423, has other contact residues outside this region that contribute a minor component to its epitope and are shared contact residues with antigenic domain B hMAbs. Given the conformational nature of the antigenic domain B hMAb epitopes, the availability of all discontinuous contact points may be required for binding by these antibodies, and competition for even one shared contact residue could more easily displace them. Finally, an interesting interpretation is that AP33 binding induces a conformational change in E2, as discussed above, which distorts the epitope of the antigenic domain B hMAbs sufficiently to prevent them from binding. However, this seems doubtful, since the entropic cost of such a rearrangement is likely to be prohibitively high.
To explore this further, a series of E2 alanine substitutions was tested for reactivity to AP33 and to the conformational hMAbs CBH-4D, HC-11, and CBH-7, which bind to antigenic domains A, B, and C, respectively. The results showed that E2 residues L413, N415, G418, and W420 are critical for binding, as demonstrated by a more than 73% reduction in AP33 binding of mutant compared to wild-type E2 when these residues were individually mutated to alanine. These findings correlate well with the X-ray structure, in which the corresponding four residues in the peptide are the most intimately associated with the antibody, with buried surfaces of 82%, 94%, 100%, and 99% for L413, N415, G418, and W420, respectively. Significant (>40%) reductions in binding were caused by mutations at residues 611, 614, and 652. However, these mutations also affected binding by two or three of the conformation-sensitive hMAbs, indicating that they disrupted the tertiary structure of E2. The reduction of AP33 binding by these mutations agrees with the observation that there is a conformational element to the AP33-E2 interaction. Interestingly, alanine substitution at residue L654, while not affecting antigenic domain A or domain C antibody binding, reduced binding of AP33 and HC-11 by 40%. This is the only indication of a possible contact residue shared by AP33 and the antigenic domain B hMAbs and is consistent with the observation that the adjacent residue, E655, has previously been implicated in AP33 recognition (16). We included an E655A mutant in the analysis and saw no significant reduction in AP33 binding, in agreement with findings of the previous study, in which AP33 binding was not reduced by an E655G mutation (16). In comparison to the effect of substituting the four critical residues L413, N415, G418, and W420, the reduction of AP33 binding by the L654A substitution is very modest, and therefore this is not strong evidence of a contact residue outside the linear epitope. That said, our analysis was limited to selected regions of E2, and there is still scope for a more extensive mutational analysis to definitively settle this question.
Residues N417 and N423 of E2 are glycosylated (17). The exposed nature of these two asparagine side chains in the Fab-peptide complex is consistent with the observation that glycosylation at these sites does not prevent AP33 binding (52). The structural information also sheds further light on our previous analyses of four cell culture-adaptive E2 variants that arose during extensive passaging of infected cells and exhibit enhanced in vitro replication (9). These mutations (N415D, T416A, N417S, and I422L) occur within the AP33 epitope. The study showed that in contrast to WT E2, the N415D and N417S variants were completely resistant to neutralization by AP33 and showed greatly reduced binding to AP33 by ELISA. N415 is completely buried in the structure, and it is not unexpected that mutation of this residue in a highly shape- and charge-complementary binding site would disrupt the interaction. N417, by contrast, is exposed in the complex, and its side chain makes no contact with the antibody. However, mutation of N417 to serine is likely to introduce a new potential glycosylation site at N415 (9). Since this residue plays such a critical role in recognition by AP33, glycosylation at N415 would certainly be expected to prevent AP33 binding. T416A and I422L remained highly sensitive to neutralization by AP33. These side chains make no direct contact with AP33 and would not be expected to greatly affect the interaction. A further mutation, G418D, was generated under AP33 selective pressure, and as with N415D and N417S, this mutant proved resistant to neutralization by AP33. G418, present at the turn of the β-hairpin, becomes buried upon AP33 binding, and the structure suggests that the interaction could not accommodate a larger side chain at this position, particularly as this would disrupt the β-hairpin.
The data presented here provide structural details of the AP33 HCV E2 epitope in its AP33-bound form and suggest that the key determinants for epitope recognition are those that anchor the central and turn regions of the β-hairpin. That the peptide displays a similar conformation whether bound to AP33 or HCV1 strongly suggests that this region does indeed form a β-hairpin in the intact glycoprotein, which is essential information for potential vaccine design. In terms of vaccine potential, the hairpin conformation opens up the possibility of designing a cyclized form of the peptide that may stabilize its secondary structure in solution.
Work toward the production of a peptide immunogen capable of eliciting AP33-like antibodies is ongoing.
ACKNOWLEDGMENTS
This work was supported by the Medical Research Council and the University of St Andrews, St Andrews, United Kingdom. Resources of the Scottish Structural Proteomics Facility, funded by the Scottish Funding Council, were used in the crystallization experiments.
Footnotes
Published ahead of print 19 September 2012
REFERENCES
- 1. Allander T, et al. 2000. Recombinant human monoclonal antibodies against different conformational epitopes of the E2 envelope glycoprotein of hepatitis C virus that inhibit its interaction with CD81. J. Gen. Virol. 81:2451–2459 [DOI] [PubMed] [Google Scholar]
- 2. Al-Lazikani B, Lesk AM, Chothia C. 1997. Standard conformations for the canonical structures of immunoglobulins. J. Mol. Biol. 273:927–948 [DOI] [PubMed] [Google Scholar]
- 3. Angus AG, Patel AH. 2011. Immunotherapeutic potential of neutralizing antibodies targeting conserved regions of the HCV envelope glycoprotein E2. Future Microbiol. 6:279–294 [DOI] [PubMed] [Google Scholar]
- 4. Anonymous 1999. Global surveillance and control of hepatitis C. Report of a WHO consultation organized in collaboration with the Viral Hepatitis Prevention Board, Antwerp, Belgium. J. Viral Hepat. 6:35–47 [PubMed] [Google Scholar]
- 5. Bartosch B, et al. 2003. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 278:41624–41630 [DOI] [PubMed] [Google Scholar]
- 6. Broering TJ, et al. 2009. Identification and characterization of broadly neutralizing human monoclonal antibodies directed against the E2 envelope glycoprotein of hepatitis C virus. J. Virol. 83:12473–12482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chothia C, et al. 1989. Conformations of immunoglobulin hypervariable regions. Nature 342:877–883 [DOI] [PubMed] [Google Scholar]
- 8. Clayton RF, et al. 2002. Analysis of antigenicity and topology of E2 glycoprotein present on recombinant hepatitis C virus-like particles. J. Virol. 76:7672–7682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dhillon S, et al. 2010. Mutations within a conserved region of the hepatitis C virus E2 glycoprotein that influence virus-receptor interactions and sensitivity to neutralizing antibodies. J. Virol. 84:5494–5507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Di Lorenzo C, Angus AG, Patel AH. 2011. Hepatitis C virus evasion mechanisms from neutralizing antibodies. Viruses 3:2280–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dubuisson J, et al. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 68:6147–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Evans MJ, et al. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805 [DOI] [PubMed] [Google Scholar]
- 14. Farci P, et al. 1996. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc. Natl. Acad. Sci. U. S. A. 93:15394–15399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Flint M, et al. 1999. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol. 73:6235–6244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gal-Tanamy M, et al. 2008. In vitro selection of a neutralization-resistant hepatitis C virus escape mutant. Proc. Natl. Acad. Sci. U. S. A. 105:19450–19455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goffard A, et al. 2005. Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J. Virol. 79:8400–8409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Habersetzer F, et al. 1998. Characterization of human monoclonal antibodies specific to the hepatitis C virus glycoprotein E2 with in vitro binding neutralization properties. Virology 249:32–41 [DOI] [PubMed] [Google Scholar]
- 19. Hadlock KG, et al. 2000. Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J. Virol. 74:10407–10416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iro M, et al. 2009. A reporter cell line for rapid and sensitive evaluation of hepatitis C virus infectivity and replication. Antiviral Res. 83:148–155 [DOI] [PubMed] [Google Scholar]
- 21. Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. 1991. Sequences of proteins of immunological interest, 5th ed U.S. Department of Health and Human Services/NIH, Bethesda, MD [Google Scholar]
- 22. Kato N, et al. 1993. Humoral immune response to hypervariable region 1 of the putative envelope glycoprotein (gp70) of hepatitis C virus. J. Virol. 67:3923–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keck ZY, et al. 2008. Definition of a conserved immunodominant domain on hepatitis C virus E2 glycoprotein by neutralizing human monoclonal antibodies. J. Virol. 82:6061–6066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Keck ZY, et al. 2004. Hepatitis C virus E2 has three immunogenic domains containing conformational epitopes with distinct properties and biological functions. J. Virol. 78:9224–9232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Keck ZY, et al. 2011. Mapping a region of hepatitis C virus E2 that is responsible for escape from neutralizing antibodies and a core CD81-binding region that does not tolerate neutralization escape mutations. J. Virol. 85:10451–10463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kong L, et al. 2012. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc. Natl. Acad. Sci. U. S. A. 109:9499–9504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krey T, et al. 2010. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 6:e1000762 doi:10.1371/journal.ppat.1000762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372:774–797 [DOI] [PubMed] [Google Scholar]
- 29. Kuroda D, Shirai H, Kobori M, Nakamura H. 2008. Structural classification of CDR-H3 revisited: a lesson in antibody modeling. Proteins 73:608–620 [DOI] [PubMed] [Google Scholar]
- 30. Kwo PY, et al. 2010. Efficacy of boceprevir, an NS3 protease inhibitor, in combination with peginterferon alfa-2b and ribavirin in treatment-naive patients with genotype 1 hepatitis C infection (SPRINT-1): an open-label, randomised, multicentre phase 2 trial. Lancet 376:705–716 [DOI] [PubMed] [Google Scholar]
- 31. Lavanchy D. 1999. The importance of global surveillance of influenza. Vaccine 17(Suppl. 1):S24–S25 [DOI] [PubMed] [Google Scholar]
- 32. Lawrence MC, Colman PM. 1993. Shape complementarity at protein-protein interfaces. J. Mol. Biol. 234:946–950 [DOI] [PubMed] [Google Scholar]
- 33. Leslie AGW, Powell HR. 2007. Processing diffraction data with MOSFLM. Evolving Methods Macromol. Crystallogr. 245:41–51 [Google Scholar]
- 34. Martin AC, Thornton JM. 1996. Structural families in loops of homologous proteins: automatic classification, modelling and application to antibodies. J. Mol. Biol. 263:800–815 [DOI] [PubMed] [Google Scholar]
- 35. McCoy AJ, et al. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McHutchison JG, et al. 2009. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N. Engl. J. Med. 360:1827–1838 [DOI] [PubMed] [Google Scholar]
- 37. Micallef JM, Kaldor JM, Dore GJ. 2006. Spontaneous viral clearance following acute hepatitis C infection: a systematic review of longitudinal studies. J. Viral Hepat. 13:34–41 [DOI] [PubMed] [Google Scholar]
- 38. Moore JP, Sodroski J. 1996. Antibody cross-competition analysis of the human immunodeficiency virus type 1 gp120 exterior envelope glycoprotein. J. Virol. 70:1863–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255 [DOI] [PubMed] [Google Scholar]
- 40. Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Macromol. Crystallogr. A 276:307–326 [DOI] [PubMed] [Google Scholar]
- 41. Owsianka A, Clayton RF, Loomis-Price LD, McKeating JA, Patel AH. 2001. Functional analysis of hepatitis C virus E2 glycoproteins and virus-like particles reveals structural dissimilarities between different forms of E2. J. Gen. Virol. 82:1877–1883 [DOI] [PubMed] [Google Scholar]
- 42. Owsianka A, et al. 2005. Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glycoprotein. J. Gen. Virol. 79:11095–11104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pang SS, et al. 2010. The structural basis for autonomous dimerization of the pre-T-cell antigen receptor. Nature 467:844–848 [DOI] [PubMed] [Google Scholar]
- 44. Patel AH, Wood J, Penin F, Dubuisson J, McKeating JA. 2000. Construction and characterization of chimeric hepatitis C virus E2 glycoproteins: analysis of regions critical for glycoprotein aggregation and CD81 binding. J. Gen. Virol. 81:2873–2883 [DOI] [PubMed] [Google Scholar]
- 45. Pawlotsky JM. 2011. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 53:1742–1751 [DOI] [PubMed] [Google Scholar]
- 46. Pileri P, et al. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941 [DOI] [PubMed] [Google Scholar]
- 47. Ploss A, et al. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Potter JA, Randall RE, Taylor GL. 2008. Crystal structure of human IPS-1/MAVS/VISA/Cardif caspase activation recruitment domain. BMC Struct. Biol. 8:11 doi:10.1186/1472-6807-8-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shimizu YK, et al. 1994. Neutralizing antibodies against hepatitis C virus and the emergence of neutralization escape mutant viruses. J. Virol. 68:1494–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stanfield RL, Zemla A, Wilson IA, Rupp B. 2006. Antibody elbow angles are influenced by their light chain class. J. Mol. Biol. 357:1566–1574 [DOI] [PubMed] [Google Scholar]
- 51. Tarr AW, et al. 2007. Determination of the human antibody response to the epitope defined by the hepatitis C virus-neutralizing monoclonal antibody AP33. J. Gen. Virol. 88:2991–3001 [DOI] [PubMed] [Google Scholar]
- 52. Tarr AW, et al. 2006. Characterization of the hepatitis C virus E2 epitope defined by the broadly neutralizing monoclonal antibody AP33. Hepatology 43:592–601 [DOI] [PubMed] [Google Scholar]
- 53. Wilson IA, Ghiara JB, Stanfield RL. 1994. Structure of anti-peptide antibody complexes. Res. Immunol. 145:73–78 [DOI] [PubMed] [Google Scholar]
- 54. Winn MD, et al. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67:235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]