

Abstract
It is widely held that any given virus uses only one type of nucleic acid for genetic information storage. However, this consensus has been challenged slightly by several recent studies showing that many RNA species are present within a range of DNA viruses that include Kaposi's sarcoma-associated herpesvirus (KSHV). RNAs extracted from purified DNA virus particles exhibit great diversity in terms of length, abundance, temporal expression, cellular localization, and coding capacity during viral infection. In addition to known RNA species, the current study showed that small regulatory RNAs were present in KSHV virions. A large number of viral and cellular microRNAs (miRNAs), as well as unusual small RNAs (usRNAs), were detected in KSHV virions by using deep sequencing. Both viral and host miRNAs detected in small RNAs extracted from KSHV virions were further shown to colocalize with KSHV virions directly by in situ hybridization (ISH)-electron microscopy (EM) (ISH-EM). Some of these miRNAs were differentially present in the host cells and KSHV virions, suggesting that they are not randomly present in KSHV virions. The virional miRNAs could be transported into host cells, and they are biologically functional during de novo viral infection. Our study revealed miRNAs and usRNAs as a novel group of components in KSHV virions.
INTRODUCTION
The human tumor virus Kaposi's sarcoma-associated herpesvirus (KSHV; also known as human herpesvirus 8 [HHV8]) is the etiological agent of KS, which is an epithelial neoplasm, as well as two other B-cell-proliferative diseases, i.e., primary effusion lymphoma (PEL) and a subset of multicentric Castleman's disease (MCD) (28). During de novo viral infection, KSHV particles enter host cells either by direct fusion between the viral envelope and the host cell plasma membrane or by internalization via macropinocytosis after binding to a cell surface receptor (reviewed in reference 8). Regardless of its mode of entry, KSHV must overcome the obstacles it encounters during transportation of virus particles from the plasma membrane to the nuclear membrane before the successful uncoating of the viral genome. The main obstacles include autophagy, apoptosis triggered by virus binding and entry, and the induction of various intrinsic, innate, and adaptive immune responses (reviewed in reference 8). The mechanisms underlying how KSHV successfully overcomes these obstacles are not fully understood at present.
Like many other herpesviruses, KSHV encodes its own viral microRNAs (miRNAs). Twelve pre-miRNAs are expressed and later are processed into mature miRNAs during viral latency (7, 14, 23, 34, 39, 44). As their cellular counterparts, these virus-encoded miRNAs have been shown to be involved in many critical processes during the viral life cycle, such as viral latency control (5, 18, 21, 22, 24, 25), viral immune evasion (21, 31), host cell cycle arrest (12), and antiapoptosis (1, 42).
It is widely considered that any given virus can be classified into a specific virus family according to its viral characteristics (http://www.ictvonline.org/virusTaxonomy.asp, http://www.web-books.com/MoBio/Free/Ch1E2.htm). Viral genome content is one of the main discriminators for virus classification (26, 38). However, this consensus has been revised somewhat, because several recent studies have shown that many RNA species, especially mRNAs, are present in the virions of some DNA viruses such as herpesviruses, including KSHV (3, 6, 9, 10, 13, 15, 36, 38, 40, 48). The RNAs extracted from these DNA virus particles exhibited great diversity in terms of length, abundance, temporal expression, intracellular localization, and coding capacity during viral infection. It is suggested that they are transported into host cells together with additional proteins and the viral genome to enhance the efficiency of viral infection. These findings illuminate virional RNAs as a novel facet of DNA virus biology. However, the exact composition of the RNAs present in DNA virus particles remains unclear. Considering the diversity of RNA species extracted from these DNA virus particles, and those DNA viruses containing RNAs all encode their own viral miRNAs (11), we hypothesized that viral miRNAs are also present in these DNA virus particles. In this study, we took KSHV as a model herpesvirus and examined the presence of KSHV miRNAs in KSHV virions.
MATERIALS AND METHODS
Cell lines.
BCBL-1, a primary effusion lymphoma cell line latently infected with KSHV, was grown in RPMI 1640 medium (HyClone) supplemented with 2 mM l-glutamine, 25 U/ml penicillin, 25 μg/ml streptomycin, and 10% fetal bovine serum. TRExBCBL-1-Rta cells (kindly provided Jae U. Jung from The Keck School of Medicine, University of Southern California) were grown in the same medium as BCBL-1 (32) with the addition of 100 μg/ml hygromycin B. Vero/rKSHV.219 (45) (kindly provided by Pinghui Feng from The Keck School of Medicine, University of Southern California), a Vero cell line that harbors recombinant KSHV genome that expresses green fluorescent protein (GFP) in viral latency and red fluorescent protein (RFP) in viral lytic replication, was maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% neonatal bovine serum, 4 μg/ml puromycin. Vero/Bac36, a Vero cell line containing bacmid, which bore the KSHV genome and expressed GFP during latency, was cultured in DMEM supplemented with 10% fetal bovine serum and 100 μg/ml hygromycin B. Human embryonic kidney 293T (HEK293T) cells were grown in DMEM supplemented with 2 mM l-glutamine, 25 U/ml penicillin, 25 μg/ml streptomycin, and 10% fetal bovine serum.
Induction of KSHV particles.
Five hundred milliliters of Vero/Bac36 or Vero/rKSHV.219 cells was induced by 25 ng/ml phorbol-12-tetradecanoate-13-acetate (TPA; Sigma-Aldrich, St. Louis, MO) and 3 mM valproic acid (VPA; Sigma-Aldrich, St. Louis, MO) for 4 to 5 days at an initial confluence of 90%. Five to 10 liters of BCBL-1 cells was induced by 25 ng/ml TPA and 3 mM VPA at a density of 0.5 × 106/ml for 6 to 7 days. Five to 10 liters of TRExBCBL-1-Rta cells was induced by 25 ng/ml TPA, 500 nm ionomycin (Sigma-Aldrich, St. Louis, MO), and 200 ng/ml doxycycline (Sigma-Aldrich, St. Louis, MO) for 6 to 7 days.
Isolation and purification of KSHV.
Culture media from induced KSHV-positive cell lines were collected and cleared by centrifugation at 4,000 × g for 30 min and then twice at 8,000 × g for 10 min to remove cells and cell debris. The supernatant was passed through 0.45-μm-pore-size filters. KSHV particles in the supernatant were then concentrated in a Beckman SW28 rotor for 2 h at 29,000 × g. The pellets were resuspended in 1× phosphate-buffered saline (PBS) overnight at 4°C. The concentrated virus particles were centrifuged twice in a Beckman SW41 rotor through 30 to 60% sucrose step gradients at 50,000 × g for 4 h. Three virus bands corresponding to A, B, and C types of KSHV particles were collected. The virus particles were then diluted with 1× PBS and further centrifuged in a Beckman SW41 rotor for 2 h at 29,000 × g. The pellets were resuspended in 1× PBS and stored at −20°C for further use.
Western blotting.
Gradient-purified viruses were first separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to a nitrocellulose membrane. KSHV ORF62, ORF45, and ORF8 were detected by immunoblot with the following antibodies: sheep polyclonal antibody to KSHV ORF62 (ab16159), mouse monoclonal antibody to KSHV ORF45 (ab36618), and rabbit polyclonal antibody to KSHV ORF8 (ab36599) (Abcam Inc., Cambridge, MA). CD63 or CD81 was detected by immunoblotting with rabbit polyclonal antibody to CD63 (b10012; Aogma Group, Shanghai, China) or mouse monoclonal antibody 5A6 to CD81 (sc-23962; Santa Cruz Biotechnology Inc., Santa Cruz, CA), respectively. Near-infrared IRDye 800-labeled secondary antibodies corresponding to primary antibodies (Li-Cor, Lincoln, NE) were used to visualize the corresponding viral antigens. Finally, membranes were scanned using the Odyssey infrared imaging system (Li-Cor, Lincoln, NE).
Negative staining.
Gradient-purified virus (3.5 μl) was transferred to a glow-discharged copper grid, and we let the virus attach to the copper for 1 min. Samples were subsequently stained for 2 min with tungstophosphoric acid after washing three times using double-distilled H2O. We dried the copper grid with filter paper and then stored the already dry copper grid in the grid box at room temperature.
ISH-EM for probing miRNA.
The basic strategy for using locked nucleic acid (LNA) probe to detect virional miRNAs by in situ hybridization-electron microscopy (ISH-EM) is that biotin-labeled LNA probe was first utilized to hybridize with corresponding miRNA. Ten-nanometer colloidal gold particle-conjugated streptavidin was then used to detect the biotin on the hybridized LNA probe. 5′Biotin-labeled LNA probes for KSHV-encoded miR-K12-4-3p, hsa-miR-21, or scramble sequence were purchased from Exiqon (Vedbaek, Denmark). Ten-nm colloidal gold particle-conjugated streptavidin was bought from Sigma-Aldrich (St. Louis, MO). Data were collected with an FEI Tecnai G2 Spirit transmission electron microscope (FEI, Netherlands) at an acceleration voltage of 100 kV.
The detailed procedure for ISH-EM probing miRNAs included the following steps. (i) The nickel grids were glow discharged at 500 V, 2 mA, for 30 s. (ii) The grids were floated with carbon film side down on 5 μl purified KSHV particles dissolved in PBS. (iii) The grids were washed with diethylpyrocarbonate (DEPC) (Invitrogen Corporation, Carlsbad, CA)-PBS three times, and then the virus particles were fixed on the nickel grids with 4% electron microscopy (EM)-grade paraformaldehyde (PFA) (Sigma-Aldrich, St. Louis, MO) and 0.05% glutaraldehyde buffered in PBS (pH 7.4) at room temperature for 15 min. (iv) The fixative was washed away with DEPC-PBS five times, and the virus particles were punctured with 0.1% Triton X-100 buffered in PBS for 10 min at room temperature. (v) The grids were rinsed with DEPC-PBS five times, and the punctured virus particles were refixed by 4% PFA for another 5 min at room temperature. (vi) The fixative was removed with rinsing in DEPC-PBS five times. The nickel grids were prehybridized in prehybridization buffer (50% formamide, 2× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 10% [wt/vol] dextran sulfate, 250 μg/ml yeast tRNA in DEPC-treated distilled H2O, pH 6.5 [prepared freshly]) at 37°C in a humidified chamber for 30 min. The solution then was incubated 10 more min in prehybridization buffer at 50°C. (vii) The biotin-labeled probe was denatured by boiling at 95°C for 5 min in prehybridization buffer with a final probe concentration of 40 nm. The diluted probe was cooled for 2 min. (viii) Fifty percent formamide in a DEPC-PBS-saturated humid chamber was added. Cutoff caps of microcentrifuge tubes were placed flat side down on a piece of parafilm overlying the filter papers. The caps were loaded with 200 μl of hybridization buffer containing the denatured probe and placed on a hot plate at 50°C, and the prehybridized grids were transferred onto the probe-containing hybridization buffer in the microcentrifuge caps. The caps were covered with 6-cm petri dishes, covered again with 9-cm petri dishes, and incubated for 2 h. (ix) The unbound probe was washed at 50°C 3 times for 5 min each with prehybridization buffer. After the second wash it was left to cool down to room temperature. (x) The probe was washed 5 times for 2 min each on drops of 0.15% glycine (wt/vol) in DEPC-PBS. (xi) The grids were incubated 4 times for 2 min each on 1% bovine serum albumin (BSA) in PBS (freshly prepared) to reduce nonspecific binding of the streptavidin during the next steps. (xii) The grid was incubated for 1 h on a 5-μl droplet of streptavidin-gold diluted (1:10 to 1:20) in 1% BSA in PBS (centrifuged at 8,000 rpm at 4°C for 2 min before use). (xiii) The grid was rinsed in 5 drops of 1% BSA in PBS five times for 1 min each and PBS drops five times for 1 min each. (xiv) The reaction was stabilized by a 5-min incubation on 4% PFA in DEPC-PBS. (xv) The grid was rinsed in 5 drops of PBS for 5 min and then washed in 10 drops of distilled H2O for 5 min. (xvi) The grids were contrasted with uranyl acetate-methylcellulose (UA-MC) (1:9), pH 4.0, for 8 min. (xvii) A wire loop was used to scoop the grid from the UA-MC, excess UA-MC was removed by dragging the loop with the grid over a filter paper, and the grid was dried in air for at least 10 min. (xviii) The grid was pinched out from the wire loop and stored on a grid box. The grid location was noted, as were the names and concentrations of labeling probes and samples. (xix) The results were visualized by using a transmission electron microscope (TEM) at an acceleration voltage of 100 kV.
Virional RNA extraction, purity, concentration, and size distribution.
Gradient-purified KSHV virions were supplemented with 20 mM Tris-HCl (pH 8.0), 5 mM NaCl, and 2.5 mM CaCl2 and then treated with micrococcal nuclease (TaKaRa Bio Inc., Shiga, Japan) and RNase A (TransGen Biotech, Beijing, China) in a final concentration of 100 U/ml at 37°C overnight. The reaction was quenched by adding 5 mM EDTA (Sigma-Aldrich, St. Louis, MO), and RNA was isolated by TRIzol LS reagent (Invitrogen, Carlsbad, CA) extraction according to the manufacturer's instructions, except that isopropyl alcohol precipitation was extended overnight. The concentration and purity of virional RNAs extracted from KSHV virions purified from different cellular backgrounds were measured by a NanoDrop 1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). RNAs were size fractionated by an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA).
RT-PCR.
To validate the quality of KSHV virional RNA, reverse transcription-PCR (RT-PCR) primers were designed for some viral transcripts previously identified in KSHV virions. Before reverse transcription, virional RNA extracted from KSHV virions was treated with RNase or left untreated. RNA extracted from BCBL-1 cells was used as a positive control. The products of RT-PCR were loaded onto a 1.5% agarose gel for electrophoresis. The specificity of primers was judged by product length and dissociation curve. The primers used in RT-PCR were the following: PAN-RT-F, 5′ACTGGGACTGCCCAGTCACC3′; PAN-RT-R, 5′GCGCCTCGAGCTACAACTGGCCTGGAGATTG3′; ORF54-RT-F, 5′AACCCCACGTGGCTCTAGCA3′; ORF54-RT-R, 5′CCCTTGAGGATGTGTCTGCG3′; ORF58-RT-F, 5′GGCAGCCAGAAAACGCCGGA3′; ORF58-RT-R, 5′TGCCAGTCACGCACTTGGCC3′; ORF59-RT-F, 5′GACAGCGTCTCGCTGACAGA3′; and ORF59-RT-R, 5′CACACGCGTGAGCTATTCGG3′.
Bulge-Loop RT-PCR and probe-specific TaqMan RT-PCR.
Two μg RNA extracted from KSHV particles was first reverse transcribed by ReverTra Ace (Toyobo, Osaka, Japan) according to manufacturer's instructions. Bulge-Loop RT-PCR was then performed according to the manufacturer's instructions (RiboBio, Guangzhou, China). In probe-specific TaqMan RT-PCR, 50 ng virional RNA was transcribed, and then RT-PCR was performed according to the manufacturer's instructions (Applied Biosystems, Foster City, CA).
Quantification of miRNA.
miRNA copy number was analyzed by probe-specific TaqMan RT-PCR using miRNA mimics (RiboBio, Guangzhou, China) as the standard. Tenfold serial dilutions of miRNA mimics (with known copy numbers of miRNA) were transcribed as the template in RT-PCR for generating standard curves. The copy numbers of each viral miRNA were calculated by reference to the standard curve.
Deep sequencing.
Small RNAs of 15 to 35 nucleotides (nt) in length from BCBL-1 cells or virional small RNAs extracted from induced supernatant of Vero/Bac36 cells, TRExBCBL-1-Rta cells, and BCBL-1 cells were used to prepare libraries by following the protocol of the Illumina library preparation procedures. The sequencing process was performed on an Illumina HiSeq 2000 platform at the Beijing Genome Institute (BGI; Shenzhen, China). Trimmed data sets were mapped against both human (UCSC Hg18; http://genome.ucsc.edu) and KSHV (accession number AF148805) genomes. Small RNA annotations were obtained from miRBase 17.0 (http://www.mirbase.org) and the UCSC Table Browser. Since the whole genome of Vero cells is not available at present, host miRNAs were mapped to the human genome without any mismatch.
Scatter plot.
To analyze the selectivity of virional miRNAs, differential expression analysis was performed between BCBL-1 virional miRNAs and miRNAs in BCBL-1 pellets by scatter plot. Deep sequencing reads for miRNAs in each sample are first normalized to obtain the expression of transcripts per million. Normalized expression equals the actual miRNA read divided by sample total clean reads times 1,000,000. Fold change was then calculated as log2(normalized read number per million in BCBL-1 virion divided by normalized read number per million in a BCBL-1 pellet). A fold change of >1 (P < 0.05) indicates data defined as upregulated. A fold change of <−1 (P < 0.05) indicates data defined as downregulated. A fold change between −1 and 1 indicates data defined as equally expressed. The P value was calculated by the following formula (2a):
where N1 represents the total reads generated from deep sequencing for sample 1, N2 represents the total reads for sample 2, x represents the read number for specific miRNA in sample 1, and y represents the read number for the same miRNA in sample 2. The next two equations allow the computation of an interval [ymin, ymax]ϵ, such as C(y ≤ ymin|x) ≤ ϵ and D(y ≥ ymax|x) ≥ ϵ. Given that an miRNA is sequenced x times in one sample, the number of occurrences (y) of this miRNA in a duplicate experiment is expected to fall within the interval [ymin, ymax]ϵ with a probability of 1 − 2ϵ.
Viral genomic DNA extraction.
Sucrose gradient-purified KSHV particles were resuspended in buffer consisting of 40 mM Tris-HCl (pH 7.4), 10 mM NaCl, 6 mM MgCl2, and 10 mM CaCl2. DNase I was added to a final concentration of 62.5 μg/ml. This suspension was incubated for 1 h at 37°C, followed by inactivation of DNase I by incubation at 65°C for 15 min. A one-fourth volume of the 5× lysis buffer (100 mM Tris-HCl [pH 7.4], 50 mM EDTA, 500 mM NaCl, 2.5% SDS) was added. Protease K was then added to a final concentration of 0.1 mg/ml. The reaction mixture was incubated at 55°C for 30 min and at 37°C overnight, followed by two phenol-chloroform extractions. Glycogen was added as a carrier, and viral DNA was precipitated with an equal volume of isopropanol, followed by an ethanol wash. The pellet was suspended in 10 mM Tris-HCl–1 mM EDTA and further treated with DNase-free RNase A (TransGen Biotech, Beijing, China).
Quantification of KSHV genome.
The quantity of KSHV genome was determined as described previously (22). Briefly, a plasmid named pGEM-T-ORFK9 containing a 184-bp fragment of KSHV ORFK9 (bp 84905 to 85088; KSHV genome accession no. AF148805) was used. Tenfold serial dilutions of pGEM-T-ORFK9 were used as the templates in real-time quantitative PCR (qPCR) for generating a standard curve. A 5-μl aliquot of the viral DNA was used for the real-time qPCR amplification of the KSHV-specific sequence within ORFK9. The copy number of viral DNA was calculated and referenced to the standard curve. The primer pair for the real-time amplification of ORFK9 was ORFK9-F (5′GTCTCTGCGCCATTCAAAAC3′) and ORFK9-R (5′CCGGACACGACAACTAAGAA3′).
Average copy number of virional miRNAs per KSHV virion.
The average copy number of virional miRNAs per KSHV virion was determined with the following formula: average copy number of virional miRNA per KSHV virion equals total miRNA copy number in KSHV virions divided by total viral genome copy number in KSHV virions.
Constructs and transfection.
Two DNA primer sequences were directly annealed together to form double-stranded DNA (dsDNA) which contains two repeated copies of sequence complementary to miR-K12-4-3p, miR-K12-5-5p, or hsa-miR-21. They were then inserted downstream of the firefly gene cassette of a reporter plasmid, pGL3 cM, which was digested with KpnI and XhoI. These three constructs were named pGL3 cM-miR-K12-4-3p-sensor, pGL3 cM-miR-K12-5-5p-sensor, and pGL3 cM-hsa-miR-21-sensor. The primer sequences were the following (miRNA sequences or sequences complementary to miRNA are underlined): miR-K12-4-3p sensor-F, 5′CTCAGCTAGGCCTCAGTATTCTAATCGTCAGCTAGGCCTCAGTATTCTAC3′; miR-K12-4-3p sensor-R, 5′TCGAGTAGAATACTGAGGCCTAGCTGACGATTAGAATACTGAGGCCTAGCTGAGGTAC3′; miR-K12-5-5p sensor-F, 5′CCTTAGGGCACCAGGGACTACCTAATCGCTTAGGGCACCAGGGACTACCTAC3′; miR-K12-5-5p sensor-R, 5′TCGAGTAGGTAGTCCCTGGTGCCCTAAGCGATTAGGTAGTCCCTGGTGCCCTAAGGGTAC3′; hsa-miR-21 sensor-F, 5′CTCAACATCAGTCTGATAAGCTAATCGTCAACATCAGTCTGATAAGCTAC3′; and hsa-miR-21 sensor-R, 5′TCGAGTAGCTTATCAGACTGATGTTGACGATTAGCTTATCAGACTGATGTTGAGGTAC3′.
HEK293 cells were transfected with 100 ng each miRNA sensor and 2 ng pRL-SV40 as a transfection efficiency control using Lipofectamine 2000 (Invitrogen, Carlsbad, CA).
UV-inactivated KSHV preparation and KSHV de novo infection.
To prepare UV-inactivated KSHV, induced supernatants filtered through 0.45-μm-pore-size membranes were inactivated in 10-cm petri dishes with UV irradiation for 2 h. Equal volumes of supernatant containing KSHV treated with UV or left untreated were incubated with HEK293T cells at 4°C for 1 h and then transferred immediately to 37°C for 10 min with brief shaking. After incubation, HEK293T cells with or without incubation at 37°C were extensively washed with 1× PBS five times, and RNA was subsequently extracted using TRIzol reagent (Invitrogen, Carlsbad, CA). For ex vivo infection, KSHV generated from induced iSLK/rKSHV.219 cells treated with or without UV was used to infect HEK293T cells pretransfected with miRNA sensor (multiplicity of infection [MOI], 5) by centrifugation at 30°C, 2,500 rpm, for 2 h. Luciferase activities were assayed using a dual-Luciferase reporter assay system (Promega, WI) 24 h after KSHV de novo infection.
RESULTS
Purification and verification of KSHV particles.
KSHV particles were first purified from the supernatant of reactivated KSHV-positive cell lines (Fig. 1A). Three milky bands were observed only in the induced supernatant after 30 to 60% sucrose gradient ultracentrifugation (Fig. 1B). According to previous studies, these three bands represent A type KSHV particles (empty virus particles, containing no scaffolding protein or viral genome in the viral capsid), B type KSHV particles (intermediate virus particles, containing scaffolding protein but no viral genome in the viral capsid), and C type KSHV particles (mature virions, containing viral genome in the viral capsid) (33). Negatively stained samples observed under an EM demonstrated that all three bands contained particles with similar size and morphology to KSHV particles (Fig. 1C). Western blotting using three antibodies specific to KSHV ORF8 (envelope glycoprotein B), ORF45 (tegument protein), or ORF62 (small capsid protein) showed that all three components of the KSHV particles could be detected in each band, suggesting that all three bands contained KSHV particles (Fig. 1D). Only viruses from the lowest band contained the KSHV genome (Fig. 1E), indicating that viruses from the lowest band shown in Fig. 1B were type C KSHV particles (mature KSHV virions) and viruses from the other two bands were B type and A type KSHV particles, respectively. Taken together, these results confirmed that the virus particles found in the three bands from the supernatant of reactivated BCBL-1 cells were A type, B type, and C type KSHV particles, respectively.
Fig 1.

Purification and verification of KSHV particles. (A) Procedures for purifying KSHV particles from induced supernatant of KSHV-positive cell lines. (B) Two liters of supernatant of uninduced BCBL-1 cells or induced BCBL-1 cells was loaded onto a 30 to 60% nonlinear sucrose gradient after condensation by ultracentrifugation. Three bands, indicated by arrows from top to the bottom, represent empty (A type), intermediate (B type), and mature KSHV particles, respectively. (C) KSHV particles negatively stained with tungstophosphoric acid were observed under an electron microscope. A scale bar is shown at the bottom or left corner of each image. (D) Immunoblot analysis with virus from the indicated bands from panel B for KSHV virion component ORF8 (glycoprotein B), ORF45 (tegument protein), and ORF62 (small capsid protein), respectively. (E) Quantification of viral genome copy using supernatant of uninduced or induced BCBL-1 cells by qPCR. Under detection indicates that the number of viral genomes is less than the detection limit of the primers, which is about 100 copies. The detailed protocol is stated in Materials and Methods. Data shown are the means ± standard deviations (SD) from three independent experiments.
RNA extracted from KSHV virions contains small RNAs.
Total RNA was then extracted from purified C type KSHV particles (termed virional RNA) after removing external nucleic acids by nuclease (Fig. 2A). The concentration and purity of virional RNA were subsequently measured by a NanoDrop 1000 spectrophotometer (NanoDrop Technologies). Typically, approximately 5 μg virional RNA can be obtained per liter of induced supernatant of BCBL-1 cells (Fig. 2B). The OD260/OD280 ratio (where OD260 is optical density at 260 nm) of virional RNA was about 2.00 (Fig. 2C), suggesting that virional RNA contained pure RNA molecules. Four viral transcripts found in KSHV virions previously (3) were used to evaluate the reliability of virional RNA quality. All four of these virional transcripts were able to be detected by RT-PCR (Fig. 2D), confirming their presence in our virional RNA sample. The detection of known viral transcripts in RNA extracted from purified KSHV virions validates the reliability of our virional RNA.
Fig 2.

KSHV virional RNA contains small RNAs. (A) Procedures for extracting RNA from KSHV virions (virional RNA). (B) Concentration of virional RNA extracted from induced supernatant of BCBL-1 cells. (C) OD260/OD280 ratio of virional RNA extracted from BCBL-1 KSHV virions. The concentration and purity of BCBL-1 virional RNA were measured by a NanoDrop 1000 spectrophotometer. (D) Four known virional transcripts (PAN RNA, ORF54, ORF58, and ORF59, carried by KSHV) were detected by RT-PCR. BCBL-1 virional RNA refers to virional RNA extracted from BCBL-1 KSHV virions that were first reverse transcribed and then used as the template in RT-PCR. RNase+BCBL-1 virional RNA refers to virional RNA extracted from BCBL-1 KSHV virions that was first treated with RNase A before reverse transcription and then used as the template in RT-PCR. BCBL-1 RNA refers to RNA extracted from BCBL-1 cells first reverse transcribed and then used as the template in RT-PCR (positive control). H2O indicates that there was no template in RT-PCR. DL2000 refers to DL2000 DNA marker. The products of RT-PCR were loaded onto a 1.5% agarose gel for electrophoresis. (E) RNAs extracted from sucrose gradient-purified KSHV virions from the supernatant of reactivated Vero/Bac36 cells (Vero/Bac36 KSHV virion), TRExBCBL-1-Rta cells (TRExBCBL-1-Rta KSHV virion), or BCBL-1 cells (BCBL-1 KSHV virion) or RNA extracted from BCBL-1 cell pellet (BCBL-1-pellet) collected at the same time with supernatant were measured by an Agilent 2100 Bioanalyzer. RNA size was referenced to the RNA ladder analyzed at the same time. All of the procedures were performed in accordance with the manufacturer's instructions. Vertical arrows indicate the small RNA peak in each RNA sample. Data shown are the means ± SD from three independent experiments.
Virional RNAs were then extracted from the supernatants of reactivated Vero/Bac36, TRExBCBL-1-Rta, and BCBL-1 cells, and their sizes were measured using an Agilent 2100 Bioanalyzer. Each virional RNA contained a peak at around 25 nt (Fig. 2E), which was similar to that found in the BCBL-1 cell pellet RNA (Fig. 2E). This result indicated that small RNA was present in virional RNAs extracted from all three cell lines that harbor KSHV.
Virus miRNAs are detected in small RNAs extracted from KSHV virions.
KSHV has its own viral miRNAs (7, 14, 23, 34, 39, 44), so we determined whether virus miRNAs were present in KSHV virional small RNAs. Four highly expressed viral miRNAs in KSHV-positive cells were chosen as detection targets. Indeed, four KSHV miRNAs were readily detected in the virional RNA using both Bulge-Loop RT-PCR (Fig. 3A) and probe-specific TaqMan RT-PCR (Fig. 3B). These miRNA signals were generated from RNA rather than any contaminated viral genomic DNA, because no miRNA signal was generated when virional RNA without reverse transcription was directly used as the template for probe-specific TaqMan RT-PCR (Fig. 3B, −RT). These miRNA signals were more likely to be generated from RNA packaged inside the virus particles, because pure miRNAs bound externally on the virus particles were sensitive to RNase treatment (Fig. 3B, +RNase+RT). In addition to virional RNA extracted from the mature C type KSHV particles (mature KSHV virions), these four virus miRNAs were also detected in the total RNA extracted from both A type and B type KSHV particles (Fig. 3C). Overall, these data demonstrate that at least four viral miRNAs were present in all three types of KSHV particles.
Fig 3.

Viral miRNAs are detected in KSHV virional RNA. (A) Four viral miRNAs are detected in BCBL-1 virional RNA by Bulge-Loop RT-PCR. Four viral miRNAs were detected using corresponding Bulge-Loop RT-PCR primers. RNA samples from the indicated cells were reverse transcribed as the template for RT-PCR. DL2000 refers to the DL2000 DNA marker (TaKaRa Bio Inc., Shiga, Japan). BCBL-1 refers to the RNA extracted from BCBL-1 cells used as the template for Bulge-Loop RT-PCR after reverse transcription (positive control). 293T refers to RNA extracted from HEK293T cells used as the template for Bulge-Loop RT-PCR after reverse transcription (negative control). BCBL-1 virion refers to virional RNA extracted from BCBL-1 KSHV virion used as the template for Bulge-Loop RT-PCR after reverse transcription. TRExBCBL-1-Rta refers to RNA extracted from latent TRExBCBL-1-Rta cells used as the template for Bulge-Loop RT-PCR after reverse transcription (positive control). H2O indicates that there was no template in Bulge-Loop RT-PCR. The products of Bulge-Loop RT-PCR for four viral miRNAs or U6 snRNA from the indicated samples were loaded onto a 1% agarose gel for electrophoresis. (B) Four viral miRNAs are detected in BCBL-1 virional RNA by TaqMan RT-PCR. Equal amounts of virional RNA were used in each treatment. −RT refers to virional RNA used as the template directly without reverse transcription. +RNase+RT refers to virional RNA treated with 100 U/ml RNase A at 37°C for 2 h before reverse transcription. (C) Four viral miRNAs are detected in all three types of KSHV particles by Bulge-Loop RT-PCR. miR-K12-3-5p, miR-K12-4-3p, miR-K12-10a, and miR-K12-10b are detected in RNAs extracted from all three types of KSHV particles by Bulge-Loop RT-PCR. Relative expression levels of four viral miRNAs were set using a relative quantification method (2−ΔΔCT). Data shown are the means ± SEM from three independent experiments.
Cellular miRNAs are also detected in small RNAs extracted from KSHV virions.
We then asked whether KSHV virions contained any cellular miRNAs. We used a deep sequencing approach to address this question. We performed HiSeq deep sequencing with small RNAs (15 to 35 nt) extracted from KSHV virions that were generated from Vero/Bac36 cells, TRExBCBL-1-Rta cells, and BCBL-1 cells. Annotated HiSeq deep sequencing data confirmed the presence of the four viral miRNAs (see Table S1 in the supplemental material) that were detected by Bulge-Loop RT-PCR and probe-specific TaqMan RT-PCR (Fig. 3A to C), while it also uncovered that additional virus miRNAs were present within KSHV virions (see Table S1).
In addition to the virus-encoded miRNAs, the HiSeq deep sequencing data also showed that more than 100 cellular miRNAs were present in the KSHV virions that were generated from all three KSHV-positive cell lines. Many of the top 50 miRNAs were repeatedly detected in small RNAs extracted from the KSHV virions that were purified from different cellular backgrounds (see Table S2 in the supplemental material).
Viral preparations have no detectable miRNA-containing vesicles.
Several extracellular vesicles have been reported to contain miRNAs (reviewed in reference 27). Although there is no obvious structures of exosomes in our purified virus samples (Fig. 1C), we wanted to make sure the miRNAs detected in our viral preparations were not artifacts. We first measured the floating density of all three types of KSHV particles in sucrose gradients. The floating density of KSHV virions in sucrose gradients (Fig. 4A) is much larger than that of other vesicles reported to contain miRNAs (see Table S3 in the supplemental material). However, to fully eliminate the possibility that miRNAs are packaged in vesicles other than KSHV particles during viral production, we detected the presence of typical markers for those miRNA-containing extracellular vesicles by immunoblot analysis. As shown in Fig. 4B, neither CD63 nor CD81 could be detected in our purified virus sample while viral tegument protein ORF45 was readily detected, which strongly suggests that our viral preps have no contamination of miRNA-containing vesicles reported previously.
Fig 4.
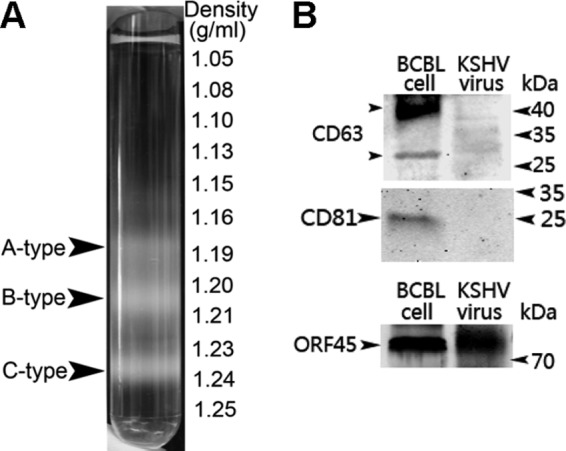
Purified KSHV particles are not contaminated with miRNA-containing extracellular vesicles. (A) The floating density of all three types of KSHV particles in 30 to 60% noncontinuous sucrose gradient centrifugation. Induced supernatant was first centrifuged at 29,000 × g for 2 h as described in Materials and Methods. The pellet was then centrifuged at 50,000 × g for 4 h after being loaded onto 30 to 60% noncontinuous sucrose gradient. The floating densities of all three types of KSHV particles (A type, ∼1.18 g/ml; B type, ∼1.20 to 1.21 g/ml; C type, ∼1.23 to 1.24 g/ml) are much larger than that of typical exosomes (∼1.15 g/ml). (B) Immunoblot analysis for miRNA-containing vesicle markers CD63 and CD81 and viral tegument protein ORF45 with purified KSHV particles. KSHV in the CD63 and CD81 panel refers to KSHV particles purified in 500 ml induced supernatant from BCBL-1 cells. KSHV in the ORF45 panel refers to KSHV particles purified in 100 ml induced supernatant from BCBL-1 cells. Supernatant from induced BCBL-1 cells was centrifuged at 29,000 × g for 2 h as described in Materials and Methods. The pellet was loaded onto the gel before sucrose gradient ultracentrifugation. BCBL cell refers to the protein sample prepared with BCBL-1 cells.
Virion-associated miRNAs colocalize with KSHV virion.
To further confirm that miRNAs are packaged in KSHV particles other than vesicles like exosomes, we performed in situ hybridization-electron microscopy (ISH-EM) to directly detect representative viral and cellular miRNAs packaged in KSHV virions using 5′ biotin-labeled locked nucleic acid (LNA) scramble sequence or probes specific for KSHV miR-K12-4-3p and cellular hsa-miR-21. Under our conditions for miRNA in situ hybridization, we could not detect any gold particles with the scramble probe (Fig. 5A to C), while gold particles were repeatedly observed to colocalize with KSHV virions using probes specific for representative viral (Fig. 5D to F) and cellular (Fig. 5G to I) miRNAs. Since external RNAs were readily degraded by RNase treatment (Fig. 3B), the result indicated that both miR-K12-4-3p (Fig. 5D to F) and hsa-miR-21(Fig. 5G to I) were present inside KSHV virions. Overall, these data demonstrate that both KSHV virion-associated viral and cellular miRNAs were present inside the KSHV virion.
Fig 5.

Virion-associated miRNAs colocalize with KSHV virions. Both virally encoded miR-K12-4-3p and cellular hsa-miR-21 colocalize with KSHV virions (C type KSHV particles) under ISH-EM. (A to C) Scramble probe was used in ISH-EM. (D to F) Biotin-labeled LNA probe for KSHV-encoded miR-K12-4-3p was used in ISH-EM. (G to I) Biotin-labeled LNA probe for cellular hsa-miR-21 was used in ISH-EM. Representative results are shown from three independent experiments. White arrows indicate the positions of 10-nm gold particles. The scale bar for each image represents 50 nm. Detailed procedures and parameters for ISH-EM are stated in Materials and Methods.
usRNAs are present in KSHV virional small RNAs.
We tested whether other classes of small RNAs were present in KSHV virional small RNAs in addition to miRNAs. Annotated HiSeq deep sequencing data indicate that no endogenous short interfering RNAs (endo-siRNAs) or Piwi interacting RNAs (piRNAs) were present in the KSHV virional small RNAs extracted from Vero/Bac36, TRExBCBL-1-Rta, or BCBL-1 cells. This is consistent with the notion that KSHV does not carry any viral siRNAs or piRNAs. In addition, no viral miRNA off-target RNA (moRNA) sequence (23, 44) could be recovered from any virional small RNAs. Analysis of the length of distribution of miRNAs identified a peak of 17 to 19 nt for some miRNAs, in addition to the typical peak of 21 to 23 nt of common miRNAs (Fig. 6A to E). This result suggested that these 17- to 19-nt RNAs were not transient degradation products of miRNAs, and they were probably the previously reported unusual small RNAs (usRNAs) derived from miRNAs (20). We also found a group of highly abundant small RNA sequences with a peak of 18 nt that mapped to U2 small nuclear RNAs (snRNA) (Fig. 6F). Their reads account for approximately 1.08, 24.50, 29.96, and 81.46% of total reads of Vero/Bac36 KSHV virional RNA, TRExBCBL-1-Rta KSHV virional RNA, BCBL-1 KSHV virional RNA, and BCBL-1 pellet RNA, respectively. Their high abundance dismisses the notion that they are transient degradation products of U2 snRNAs during viral production. These sequences did not contain position-specific motifs that were similar to the usRNAs that have been reported previously (20), suggesting that these U2 snRNA-derived usRNAs were generated by different mechanism(s). Taken together, these data indicate that besides miRNAs, miRNA-derived and non-miRNA-derived usRNAs are also present in KSHV virional small RNAs.
Fig 6.

usRNAs are detected in KSHV virional small RNAs. The upper panels show the length distribution of sequences for miR-K12-10b-3p (A), miR-K12-5-3p (B), hsa-miR-148a (C), hsa-miR-21 (D), hsa-miR-30e (E), and U2 snRNA-derived usRNA (F) recovered from HiSeq deep sequencing data of virional small RNAs from Vero/Bac36 cells, TRExBCBL-1-Rta cells, and BCBL-1 cells and small RNA from BCBL-1 pellets. Lower panels show the sequence of each miRNA and corresponding miRNA-derived usRNA or the top three sequences of U2 snRNA-derived usRNA recovered from HiSeq deep sequencing data. Blue rectangles show the peak of usRNA derived from miRNA.
Selectivity of miRNAs detected in KSHV virion.
To test whether miRNAs were selectively packaged in KSHV virions, we initially used probe-specific TaqMan RT-PCR to compare the miRNAs in these three different types of KSHV particles. Although the amount of miRNAs varied in the three types of KSHV particles, the trend for the expression level of the four viral miRNAs goes up and down similarly in the A type and B type KSHV particles (Fig. 7A), suggesting that the A type and B type KSHV particles contained a similar ratio of these four viral miRNAs. The trend of expression levels four the viral miRNAs was different in KSHV virions from that found in the A type and B type virus particles (Fig. 7A), suggesting that the relative ratio of four viral miRNAs in the virion was different from that in the other two types of KSHV particles. miRNAs were significantly enriched inside the KSHV virions compared to U6 snRNA (Fig. 7B), suggesting that miRNAs were preferentially packaged into KSHV particles compared to other small RNA species. Differential expression analysis of miRNAs using scatter plot showed that one group of miRNAs was enriched in KSHV virions, whereas two other groups of virional miRNAs were either equally expressed or downregulated (Fig. 7C; also see Table S4 in the supplemental material). These data demonstrate that the expression levels between intracellular and extracellular miRNAs are different and the relative ratio of miRNAs in KSHV virions is different from that in A and B types of KSHV particles.
Fig 7.

Selectivity of miRNAs detected in KSHV virions. (A) Comparison of relative expression level of four viral miRNAs in A, B, and C types of KSHV particles. The lowest expressed miR-K12-4-3p in A type virus particles was set as the reference. (B) The ratio of extracellular to intracellular abundance of four viral miRNAs or U6 snRNA. (C) Differential expression analysis between virional miRNAs and corresponding miRNAs detected in BCBL-1 cell pellets harvested at the same time with induced supernatant is shown by scatter plot. miRNA reads in each sample are normalized to obtain the read number of transcripts per million. Fold change was calculated as log2(normalized read number per million in BCBL-1 virion divided by normalized read number per million in the BCBL-1 pellet). Fold changes of >1 (P < 0.05) are defined as upregulated. Fold changes of <−1 (P < 0.05) are defined as downregulated. Fold changes between −1 and 1 are defined as equally expressed. Representative miRNAs in KSHV virions are indicated in the figure, and the full list is shown in Table S4 in the supplemental material. Data shown in panels A and B are means ± SD from three independent experiments.
Quantification of miRNAs in KSHV virion.
The total copy numbers of these four virional miRNAs in the supernatant of reactivated BCBL-1 cells were then quantified using their corresponding miRNA mimics as the standards. The copy numbers of these four viral miRNAs in KSHV virions ranged from 1010 to 1011 per liter in the induced supernatant of BCBL-1 cells (Fig. 8A). Compared to the uninduced supernatants of BCBL-1 cells, the induced supernatants contained greater than one or two orders of magnitude more miRNAs (Fig. 8A), suggesting that miRNAs were transported into the supernatant by the newly released viral progeny. The average copy numbers of four selected viral miRNAs per KSHV virion then were calculated as described in Materials and Methods. The average copy numbers of four selected viral miRNAs per KSHV virion range from about 700 to 7,000 more (Fig. 8B), implying that they have functions.
Fig 8.

Quantification of miRNAs in KSHV virions. (A) Quantification of viral miRNAs in induced or uninduced supernatant of reactivated BCBL-1 cells by probe-specific TaqMan RT-PCR. Copy number of miRNAs was analyzed by probe-specific TaqMan RT-PCR using corresponding miRNA mimics as the standards. Tenfold serial dilutions of miRNA mimics (with known copy numbers of miRNA) were transcribed as the template in RT-PCR for generating standard curves. The copy numbers of each viral miRNA were calculated and referenced to the standard curve. (B) The average copy number of four viral miRNAs per KSHV virion. The average copy number of viral miRNA per KSHV virion was determined with the following formula: average copy number of viral miRNA per KSHV virion equals total miRNA copy number in KSHV virions divided by total viral genome copy number in KSHV virions. The number above each column represents the average copy number for the corresponding viral miRNA. Data shown are the means ± SD from six independent experiments.
KSHV virional miRNAs are biologically functional.
We then determined whether these virional miRNAs were biologically relevant. To address this question, we first determined whether virional miRNAs were transported into host cells during KSHV de novo viral infection. Using probe-specific TaqMan RT-PCR, we found that the selected four virional miRNAs were only detected in RNA extracted from HEK293T cells that were de novo infected with KSHV but not from mock-infected HEK293T cells (Fig. 9A). The data suggest that these viral miRNAs were present in target cells during de novo viral infection. All four viral miRNAs were detected in HEK293T cells as early as 10 min after viral infection (Fig. 9A), strongly suggesting that these viral miRNAs were transported into cells together with KSHV virions and that they were not newly transcribed.
Fig 9.

Virional miRNAs are biologically functional during de novo viral infection. (A) Virional miRNAs were brought into host cells during viral infection. Equal volumes of supernatant containing KSHV treated with or without UV were incubated with HEK293T cells at 4°C for 1 h and then transferred immediately to 37°C for 10 min with brief shaking. After incubation, HEK293T cells with or without incubation at 37°C were extensively wash with 1× PBS five times, RNA was subsequently extracted using TRIzol reagent, and viral miRNAs were detected by TaqMan RT-PCR. Products of TaqMan RT-PCR for miRNAs or U6 snRNA from the indicated samples were loaded onto 1% agarose gels for electrophoresis. (B) Fluorescent pictures for HEK293T cells pretransfected with miR-K12-4-3p sensor, miR-K12-5-5p sensor, or hsa-miR-21 sensor and then mock infected with culture media or infected with KSHV or UV-inactivated KSHV, respectively. Images were taken 24 h after viral infection. (C) Relative firefly Luciferase activity in HEK293T cells shown in panel B. The estimated MOI used in all three experiments was 5. Data shown are the means ± SD from three independent experiments. ***, P < 0.001; **, P < 0.01; n.s., not significant; Student's t test was used to determine significance.
We then assessed the biological relevance of these virional miRNAs using KSHV virions or UV-inactivated KSHV virions. The numbers of GFP-positive cells were greatly decreased for HEK293T cells infected with UV-treated rKSHV.219 (Fig. 9B), indicating that KSHV was transcriptionally inactivated by UV treatment. The activity of the reporter gene targeted by virional miRNA miR-K12-4-3p in HEK293T cells infected with KSHV or UV-treated KSHV was significantly decreased compared to that in HEK293T cells that had been mock infected with culture media of latent KSHV-positive cells (Fig. 9C), while the activity of reporter genes targeted by nonvirional miRNA, miR-K12-5-5p (see Table S1 in the supplemental material), was not significantly changed (Fig. 9C), suggesting that these two virional miRNAs were biologically functional during viral infection. Overall, these results indicate that virional miRNAs are brought into host cells and that they are biologically relevant during de novo viral infection.
DISCUSSION
Viruses have long been considered to contain only one type of nucleic acid as their genetic material. The genetic content has usually been recognized as the main discriminator during viral classification (26, 38). Many viral classification systems have been set up based on the viral genome content and other viral characteristics, e.g., the famous Baltimore classification system, which is based on viral genetic contents and their strategy of replication. The discovery of many RNA species in a group of DNA virus particles has redefined this consensus (3, 6, 9, 10, 13, 15, 36, 38, 40, 48). These discoveries have opened up a new avenue of DNA virus biology, particularly herpesvirus biology. However, the functions of these virional RNAs are largely unknown in the context of viral infection. The exact composition of the RNAs present in DNA virus particles also remains undefined. Based on the diversity of RNA molecules discovered in those DNA virus particles and their ability to possess their own viral miRNAs, we hypothesized that virus miRNAs could also be present in those DNA virus particles.
In this study, we utilized KSHV as a model herpesvirus and showed that KSHV particles contained small regulatory RNA molecules based on indirect evidence by Bulge-Loop RT-PCR, probe-specific TaqMan RT-PCR, and HiSeq deep sequencing (Fig. 3 and 6; also see Table S1 and S2 in the supplemental material) and by direct evidence through ISH-EM probing virional miRNAs (Fig. 5). These small regulatory RNAs are KSHV miRNAs, cellular miRNAs, miRNA-derived usRNAs, and U2 snRNA-derived usRNAs. miRNAs were enriched in KSHV virions compared to other non-miRNA RNA (Fig. 7B). One group of poorly expressed miRNAs in the host cells was enriched in the KSHV virions (Fig. 7C; also see Table S4). All of these data suggest that some miRNAs are preferentially packaged into KSHV virions during virus production. The different ratio of viral miRNAs in mature KSHV virions compared to that in A and B types of KSHV particles suggest that those virional miRNAs have a role in KSHV virion maturation. Virional miRNAs were able to be transported into host cells, and they were competent to dampen gene expression during de novo viral infection (Fig. 9C).
Most recently, Amen and Griffiths showed that in the presence of actinomycin D, viral miRNAs were able to be detected in target cells at 60 min after herpes B virus infection (2). Although they had not examined the purity of their condensed virus preparation, which could be contaminated with exosomes or other miRNA-containing entities, their data suggested that viral miRNAs are present in the herpes B virus particle, a DNA virus particle. The assembly process of virus particles is shared by most, if not all, herpesviruses (29), so the acquisition of miRNAs may be a common phenomenon in herpesviruses. As almost all of the herpesviruses, polyomaviruses, and adenoviruses tested so far have been demonstrated to encode their own viral miRNAs (http://www.mirbase.org/cgi-bin/browse.pl), it is likely that these DNA virus particles also contain miRNAs encoded by viruses.
The high abundance of U2 snRNA-derived usRNAs (Fig. 6F) suggests that they are not randomly degraded fragments, especially taking into consideration that those RNA fragments were surrounded by RNases released by lysed cells at the time of virus collection. These highly abundant U2 snRNA-derived usRNAs may play an essential role in KSHV de novo viral infection, or they may help KSHV assemble virus particles during the late lytic replication phase. The fact that no KSHV-encoded moRNAs are detected in our deep sequencing data suggests that viral moRNAs previously found in KSHV-positive cells at early times postinduction are more likely to be degraded intermediates.
Colloidal gold-LNA-miRNA probe-based ISH-EM provides a way to detect the precise location of miRNAs at the nanometer scale. ISH-EM may be useful for dissecting the functions and localizations of miRNAs when combined with other technologies, such as immunogold staining.
Many questions remain. The underlying mechanism(s) of virional miRNA and usRNA packaging and selectivity is not known. Pure miRNAs and usRNAs are negatively charged and sensitive to RNase (Fig. 3B). It is conceivable that they are associated with other partners, such as RNA-binding proteins, forming more stable ribonucleoprotein (RNP) complexes. Studies from host noncoding RNA 7SL present in HIV-1 virions showed that the package of 7SL is dependent on a cis element in 7SL and the interactions between 7SL, HIV-1 Gag, and host cell signal recognition particle (SRP) (16). It is possible that KSHV utilizes a similar strategy to package its virional miRNAs and usRNAs. However, mass spectrometry analysis of KSHV virion proteins (4, 49) showed that the KSHV virion does not contain any homologues to the RNA-binding proteins that have been reported in the virions of herpes simplex virus (HSV) (41). Meanwhile, those virional miRNAs and usRNAs do not share an obvious homologous domain. Thus, it remains unclear whether any unknown viral or host RNA-binding proteins are responsible for the packaging of virional miRNAs. One possibility is that some known virion protein with unknown RNA binding function is responsible for virional miRNA packaging. Another explanation is that there is some RNA binding protein in the KSHV virion, but its expression level is very low and could not be detected by mass spectrometry. Additionally, the RNA binding protein(s) that is responsible for KSHV virional miRNA packaging may exert its RNA sorting function during the process of virus assembly but does not stay inside KSHV virions after mature KSHV virions are formed.
The localization of virional miRNAs and usRNAs in KSHV virions is also unknown at present. They may localize in the tegument and/or inside the capsid. Our ISH-EM data suggest that KSHV virional miRNAs mainly localize in the tegument of KSHV virions (Fig. 5). This is consistent with the observation that mature miRNAs mainly localize in the cytoplasmic compartment of the cells (19). However, we also observed that in some cases the virional hsa-miR-21 localizes inside the viral capsid (Fig. 5H and data not shown). Several miRNAs, including hsa-miR-21, have been shown to have a nucleolar location in muscle cells (35). It is unclear whether hsa-miR-21 also localize in the nucleolus in KSHV-positive cells. If so, hsa-miR-21 may be acquired by KSHV during capsid formation in the nuclear compartment. Future studies with higher resolution methods, such as electron tomography (ET) or other biochemical methods, are needed to determine the exact localization of virional miRNAs in KSHV virions and whether the location will affect their function.
We noticed that there were miRNAs released in the culture medium of latently infected KSHV-positive cells (Fig. 8A, uninduced). The relative centrifugation force used for KSHV purification is much smaller than that commonly used for exosome purification. We did not observe any obvious exosomes in our sample under EM, so these miRNAs may come from entities other than exosomes. It will be interesting to know how these miRNAs are generated, in what form they exist in the culture medium, and what their functions are.
Another interesting issue is how these U2 snRNA-derived usRNAs are generated. Although we named them U2 snRNA-derived usRNAs, we do not have any evidence to support whether they are processed from a U2 snRNA precursor or are transcribed from its own promoter. The primary sequence of this usRNA is very similar to that of hsa-miR-1246. It will be interesting to know whether miRNA machineries are required for the production of U2 snRNA-derived usRNA, since there has been no report of miRNAs derived from snRNAs yet.
In addition, the in vivo role of these virional miRNAs and usRNAs remains unclear. Some miRNAs have been shown to be upregulated at early time points after KSHV de novo viral infection (17, 37, 43, 47). Those miRNAs include virional hsa-miR-21 and hsa-miR-146a. hsa-miR-146a might contribute to KS development by promoting premature release of KSHV-infected endothelial progenitors into circulation through repressing the expression of CXCR4 (37). However, most virional miRNAs have not been studied in the context of KSHV infection. There are several possibilities regarding the roles of virional miRNAs and usRNAs. (i) No specific function, i.e., small RNAs are simply randomly packaged inside virions or they are simply the consequence of viral production. (ii) No specific function, i.e., small RNAs are simply packaged into the virus particles during capsidation or envelopment, because they are capable of interacting with capsid proteins or tegument proteins. (iii) Virional miRNAs and/or usRNAs are required for virus particles maturation, i.e., RNA species, including virional miRNAs, usRNAs, mRNAs, and noncoding RNAs, are structural elements of KSHV particles, like some RNAs found in retroviruses (30, 46). (iv) Virional miRNAs and usRNAs may be used to help the virus evade the host immune system, which might facilitate latency during de novo viral infection.
Studies from Epstein-Barr virus (EBV) showed that virional RNAs in EBV virions can help EBV set up the viral latency, induce cytokines synthesis, and evade the host T-cell response to newly infected B cells (15). KSHV virional miRNAs and usRNAs may have similar functions due to the intimate relations between these two gammaherpesviruses. Based on the abundance of some virional miRNAs and U2 snRNA-derived usRNA and the potential roles of miRNAs in the regulation of gene expression, it is believed that virional miRNAs and usRNAs play critical roles in the KSHV life cycle, especially in preparation of an environment (such as viral host immune system evasion and antiapoptosis) for virus during de novo viral infection.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funds from the National Basic Research Program of China (2011CB504805 and 2012CB519002), the Natural Science Foundation of China (30770098 and 30970154), and the 100 Talent Program of the Chinese Academy of Sciences to K.L.
We thank all members of the Lan laboratory for stimulating discussions. We are also grateful to Hongyu Deng, Sheng Shen, and Lili Wang at the Institute of Biophysics, Chinese Academy of Sciences, for their technical suggestions on virional miRNA detection by ISH-EM and Shufeng Sun at the Institute of Biophysics, Chinese Academy of Sciences, for on-site assistance with EM.
Footnotes
Published ahead of print 12 September 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Abend JR, Uldrick T, Ziegelbauer JM. 2010. Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi's sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J. Virol. 84:12139–12151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amen MA, Griffiths A. 2011. Identification and expression analysis of herpes B virus-encoded small RNAs. J. Virol. 85:7296–7311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2a. Audic S, Claverie JM. 1997. The significance of digital gene expression profiles. Genome Res. 7:986–995 [DOI] [PubMed] [Google Scholar]
- 3. Bechtel J, Grundhoff A, Ganem D. 2005. RNAs in the virion of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:10138–10146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bechtel JT, Winant RC, Ganem D. 2005. Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:4952–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bellare P, Ganem D. 2009. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 6:570–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bresnahan WA, Shenk T. 2000. A subset of viral transcripts packaged within human cytomegalovirus particles. Science 288:2373–2376 [DOI] [PubMed] [Google Scholar]
- 7. Cai X, et al. 2005. Kaposi's sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. U. S. A. 102:5570–5575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chandran B. 2010. Early events in Kaposi's sarcoma-associated herpesvirus infection of target cells. J. Virol. 84:2188–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chung SW, Arnott JA, Yang Y, Wong PM. 2003. Presence of prepackaged mRNA in virions of DNA adenovirus. J. Biol. Chem. 278:50635–50640 [DOI] [PubMed] [Google Scholar]
- 10. Cliffe AR, Nash AA, Dutia BM. 2009. Selective uptake of small RNA molecules in the virion of murine gammaherpesvirus 68. J. Virol. 83:2321–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cullen BR. 2010. Five questions about viruses and microRNAs. PLoS Pathog. 6:e1000787 doi:10.1371/journal.ppat.1000787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gottwein E, Cullen BR. 2010. A human herpesvirus microRNA inhibits p21 expression and attenuates p21-mediated cell cycle arrest. J. Virol. 84:5229–5237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Greijer AE, Dekkers CA, Middeldorp JM. 2000. Human cytomegalovirus virions differentially incorporate viral and host cell RNA during the assembly process. J. Virol. 74:9078–9082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grundhoff A, Sullivan CS, Ganem D. 2006. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA 12:733–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jochum S, Ruiss R, Moosmann A, Hammerschmidt W, Zeidler R. 2012. RNAs in Epstein-Barr virions control early steps of infection. Proc. Natl. Acad. Sci. U. S. A. 109:E1396–E1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Keene SE, Telesnitsky A. 2012. Cis-acting determinants of 7SL RNA packaging by HIV-1. J. Virol. 86:7934–7942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lagos D, et al. 2010. miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat. Cell Biol. 12:513–519 [DOI] [PubMed] [Google Scholar]
- 18. Lei X, et al. 2010. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat. Cell Biol. 12:193–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leung AK, Sharp PA. 2006. Function and localization of microRNAs in mammalian cells. Cold Spring Harb. Symp. Quant. Biol. 71:29–38 [DOI] [PubMed] [Google Scholar]
- 20. Li Z, et al. 2009. Characterization of viral and human RNAs smaller than canonical MicroRNAs. J. Virol. 83:12751–12758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liang D, et al. 2011. A human herpesvirus miRNA attenuates interferon signaling and contributes to maintenance of viral latency by targeting IKKepsilon. Cell Res. 21:793–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin X, et al. 2011. miR-K12-7-5p encoded by Kaposi's sarcoma-associated herpesvirus stabilizes the latent state by targeting viral ORF50/RTA. PLoS One 6:e16224 doi:10.1371/journal.pone.0016224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin YT, et al. 2010. Small RNA profiling reveals antisense transcription throughout the KSHV genome and novel small RNAs. RNA 16:1540–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu CC, et al. 2010. MicroRNAs encoded by Kaposi's sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep. 11:784–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu F, Stedman W, Yousef M, Renne R, Lieberman PM. 2010. Epigenetic regulation of Kaposi's sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J. Virol. 84:2697–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lwoff A, Horne R, Tournier P. 1962. A system of viruses. Cold Spring Harb. Symp. Quant. Biol. 27:51–55 [DOI] [PubMed] [Google Scholar]
- 27. Meckes DG, Jr, Raab-Traub N. 2011. Microvesicles and viral infection. J. Virol. 85:12844–12854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mesri EA, Cesarman E, Boshoff C. 2010. Kaposi's sarcoma and its associated herpesvirus. Nat. Rev. Cancer 10:707–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234 [DOI] [PubMed] [Google Scholar]
- 30. Muriaux D, Mirro J, Harvin D, Rein A. 2001. RNA is a structural element in retrovirus particles. Proc. Natl. Acad. Sci. U. S. A. 98:5246–5251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O. 2009. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 5:376–385 [DOI] [PubMed] [Google Scholar]
- 32. Nakamura H, et al. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J. Virol. 77:4205–4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nealon K, et al. 2001. Lytic replication of Kaposi's sarcoma-associated herpesvirus results in the formation of multiple capsid species: isolation and molecular characterization of A, B, and C capsids from a gammaherpesvirus. J. Virol. 75:2866–2878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pfeffer S, et al. 2005. Identification of microRNAs of the herpesvirus family. Nat. Methods 2:269–276 [DOI] [PubMed] [Google Scholar]
- 35. Politz JC, Hogan EM, Pederson T. 2009. MicroRNAs with a nucleolar location. RNA 15:1705–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Prichard MN, et al. 1998. Identification of persistent RNA-DNA hybrid structures within the origin of replication of human cytomegalovirus. J. Virol. 72:6997–7004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Punj V, et al. 2010. Kaposi's sarcoma-associated herpesvirus-encoded viral FLICE inhibitory protein (vFLIP) K13 suppresses CXCR4 expression by upregulating miR-146a. Oncogene 29:1835–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roizman B. 2000. Redefining virology. Science 288:2327–2328 [DOI] [PubMed] [Google Scholar]
- 39. Samols MA, Hu J, Skalsky RL, Renne R. 2005. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:9301–9305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sciortino MT, Suzuki M, Taddeo B, Roizman B. 2001. RNAs extracted from herpes simplex virus 1 virions: apparent selectivity of viral but not cellular RNAs packaged in virions. J. Virol. 75:8105–8116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sciortino MT, Taddeo B, Poon AP, Mastino A, Roizman B. 2002. Of the three tegument proteins that package mRNA in herpes simplex virions, one (VP22) transports the mRNA to uninfected cells for expression prior to viral infection. Proc. Natl. Acad. Sci. U. S. A. 99:8318–8323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Suffert G, et al. 2011. Kaposi's sarcoma herpesvirus microRNAs target caspase 3 and regulate apoptosis. PLoS Pathog. 7:e1002405 doi:10.1371/journal.ppat.1002405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tsai YH, et al. 2009. The M type K15 protein of Kaposi's sarcoma-associated herpesvirus regulates microRNA expression via its SH2-binding motif to induce cell migration and invasion. J. Virol. 83:622–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Umbach JL, Cullen BR. 2010. In-depth analysis of Kaposi's sarcoma-associated herpesvirus microRNA expression provides insights into the mammalian microRNA-processing machinery. J. Virol. 84:695–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vieira J, O'Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240 [DOI] [PubMed] [Google Scholar]
- 46. Wang SW, Aldovini A. 2002. RNA incorporation is critical for retroviral particle integrity after cell membrane assembly of Gag complexes. J. Virol. 76:11853–11865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu YH, et al. 2011. The manipulation of miRNA-gene regulatory networks by KSHV induces endothelial cell motility. Blood 118:2896–2905 [DOI] [PubMed] [Google Scholar]
- 48. Xing L, Tikoo SK. 2004. Viral RNAs detected in virions of porcine adenovirus type 3. Virology 321:372–382 [DOI] [PubMed] [Google Scholar]
- 49. Zhu FX, Chong JM, Wu L, Yuan Y. 2005. Virion proteins of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:800–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.