

Abstract
The hemagglutinin (HA)-neuraminidase protein (HN) of paramyxoviruses carries out three discrete activities, each of which affects the ability of HN to promote viral fusion and entry: receptor binding, receptor cleaving (neuraminidase), and triggering of the fusion protein. Binding of HN to its sialic acid receptor on a target cell triggers its activation of the fusion protein (F), which then inserts into the target cell and mediates the membrane fusion that initiates infection. We provide new evidence for a fourth function of HN: stabilization of the F protein in its pretriggered state before activation. Influenza virus hemagglutinin protein (uncleaved HA) was used as a nonspecific binding protein to tether F-expressing cells to target cells, and heat was used to activate F, indicating that the prefusion state of F can be triggered to initiate structural rearrangement and fusion by temperature. HN expression along with uncleaved HA and F enhances the F activation if HN is permitted to engage the receptor. However, if HN is prevented from engaging the receptor by the use of a small compound, temperature-induced F activation is curtailed. The results indicate that HN helps stabilize the prefusion state of F, and analysis of a stalk domain mutant HN reveals that the stalk domain of HN mediates the F-stabilization effect.
INTRODUCTION
Fusion between enveloped viruses and the host cell is a key step in viral infectivity, and interfering with this process can lead to highly effective antivirals. Viral fusion is driven by specialized proteins that undergo an ordered series of conformational changes resulting in the close apposition of the viral and host membranes and the formation of a fusion pore (reviewed in reference 14). The first step of infection with most paramyxoviruses is binding of the receptor binding protein to cell surface receptors. In the case of human parainfluenza virus (HPIV), the receptor binding protein, hemagglutinin (HA)-neuraminidase (HN), binds to sialic acid-containing molecules. HN activates the viral fusion protein (F) to undergo the series of structural rearrangements that ultimately lead to direct fusion of the viral envelope with the plasma membrane of the cell (27, 29, 35, 63). For F activation to initiate, the F protein must have been properly cleaved from its F0 precursor to its active processed form (F1+F2) during synthesis in the infected cell. For HPIV (36, 37) and for most other paramyxoviruses (4, 8, 17, 20, 26), interaction of HN with its receptor is essential in order for F to promote membrane fusion during infection although there are a few exceptions, e.g., respiratory syncytial virus (RSV) (7, 59) and human metapneumovirus (hMPV) (60, 61). HN also plays a key role after the assembly of progeny virions since its sialic acid-cleaving (neuraminidase) activity facilitates the release of newly budded virions and thus the spread of infection.
HN is a tetrameric type II membrane protein consisting of a cytoplasmic domain, a membrane-spanning region, a stalk region, and a globular head. The stalk confers specificity for the homologous F in the fusion activation process (6, 12, 33, 50, 52, 62, 64, 65, 69, 71). Study of the crystal structures of the avian paramyxovirus Newcastle disease virus (NDV) HN (11, 72) and later the HPIV3 HN (28) and parainfluenza virus 5 (PIV5) HN (70) identified the locations of the primary binding/neuraminidase active-site residues on the globular head. For NDV our experimental data suggested two active sites on NDV HN, where site I exhibits both neuraminidase and receptor binding activity and site II possesses only the receptor binding function (38, 51). These two sites were also identified in the X-ray crystal structure of NDV HN (69, 72). Engagement of NDV's site I with the receptor leads to the activation of site II (47). For HPIV3 we showed that a second binding site on HN (site II), while functioning differently from that of NDV HN′s, contributes to HPIV3 HN′s F-triggering activity during the entry/fusion process (41, 47, 48, 51). The efficiency of F triggering by HN critically influences the degree of fusion mediated by F and thus the extent of viral entry (40, 52). The three functions of HN—binding, triggering, and cleaving (neuraminidase)—are balanced to determine the outcome of infection (50).
A real-time analysis of the sequence of events leading up to HN/F-mediated membrane fusion in live cells using bimolecular fluorescence complementation (BiFC) revealed that HN and F associate prior to receptor engagement (53). After receptor engagement, HN drives the formation of HN-F clusters at the site of fusion, and alterations in the HN-F interaction determine the fusogenicity of the glycoprotein pair (53). HN′s second sialic acid binding site directly modulates F activation (48, 53) and the physical interaction between F and HN. HN and F retain their association (53), and HN continues to act on F well beyond insertion of the fusion peptide of F protein into the target membrane. Thus, HN is responsible not only for initiating the activation of F but also for continuous stimulation of the process until the HN-receptor interaction is severed (45).
Dysregulated triggering of F by HN leading to premature activation of F is detrimental to infectivity in the natural host (15, 40). We have shown that small molecules can stimulate HN to prematurely trigger F distant from the target cell, thereby inactivating viruses (15). Therefore, for infectivity F must remain in an untriggered state until HN encounters its receptor.
Here, we report that, in addition to the three roles of receptor binding, cleaving, and F triggering, HN possesses a fourth function: maintenance of the F protein in a pretriggered state, thereby preventing untimely activation of the fusion machinery. HN, before it is engaged with its receptor, exerts a stabilizing effect on F. We employ a series of well-characterized HNs with mutations in either the globular head domain or in the stalk domain of HN to dissect this novel role and to show that the stalk domain of HN is essential for this F-protective function.
MATERIALS AND METHODS
Chemicals.
Zanamivir was prepared from Relenza Rotadisks (5 mg of zanamivir with lactose). A 50 mM stock solution was prepared by dissolving each 5-mg blister capsule in 285 μl of OptiMEM (Gibco). Stock solutions were stored at −20°C.
Antibodies.
Antibodies to the C-terminal heptad repeat (HRC) region of F were custom generated in rabbits by Invitrogen using a previously described HPIV HRC sequence (46).
Viruses.
Titers of HPIV3 virus stocks were assessed by plaque assay performed as described previously (40). Variant HPIV3 viruses were selected and grown as described previously (40).
Transient expression of glycoproteins.
HPIV3 HN and F cDNAs were digested with SacI or EcoRI and BamHI and ligated into digested pCAGGS and pEGFP (where EGFP is enhanced green fluorescent protein) mammalian expression vectors as previously described (38, 48–52). Constructs used in the assay for F protein triggering, HN with the N terminus of Venus fluorescent protein (HN-N-Venus), F with the C terminus of the cyan fluorescent protein (F-C-CFP), and HA with the C-terminus of Venus fluorescent protein (HA-C-Venus) were prepared as previously described (9). HA cDNA was codon optimized and synthesized by Epoch Biolabs and subcloned into the mammalian expression vector pCAGGS. Transfections were performed with Lipofectamine and Plus or Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions.
Cells.
293T (human kidney epithelial) cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) and antibiotics at 37°C and 5% CO2.
β-Gal complementation-based fusion assay.
We previously adapted a fusion assay based on alpha complementation of β-galactosidase (β-Gal) (34, 48). In this assay, receptor-bearing cells expressing the omega peptide of β-Gal are mixed with cells coexpressing envelope glycoproteins and the alpha peptide of β-Gal, and cell fusion leads to alpha-omega complementation. Fusion is stopped by lysing the cells, and, after addition of the substrate (Galacton-Star; Applied Biosystems), fusion is quantified on a Spectramax M5 microplate reader.
Measurement of the fusion between RBCs and envelope glycoprotein-expressing cells.
Monolayers of 293T cells transiently expressing viral glycoproteins were washed with OptiMEM and incubated for 1 h at 37°C in OptiMEM supplemented with 100 mg/ml cycloheximide (to prevent de novo protein synthesis) (39) and then incubated with 1% red blood cell (RBC) suspensions (pH 7.5) for 30 min at 4°C with or without zanamivir (2 mM). After the samples were rinsed to remove unbound RBCs, they were placed at 37°C for the time indicated in the figures with or without 2 mM zanamivir. The plates then were rocked, and the liquid phase was collected in V-bottomed tubes for measurement of released RBCs. The cells were then incubated at 4°C with 200 μl of RBC lysis solution, whereby the lysis of unfused RBCs with NH4Cl removes RBCs that have not fused with cells coexpressing envelope glycoproteins. The liquid phase was collected in V-bottom 96-well plates for measurement of bound RBCs. The cells were then lysed in 200 μl of 0.2% Triton X-100–phosphate-buffered saline (PBS) and transferred to flat-bottom 96-well plates for quantification of fused RBCs. The amount of RBCs in each of the above three compartments was determined by measuring the absorption at 405 nm.
Detection of fusion protein triggering.
293T cell monolayers, transiently transfected with uncleaved HA along with HN-N-Venus and F-C-CFP or with F-C-CFP alone, were incubated overnight in DMEM supplemented with 2 mM zanamivir to prevent fusion. The transfected cells were washed with OptiMEM and incubated for 1 h at 37°C in OptiMEM supplemented with 100 mg/ml cycloheximide (to prevent de novo protein synthesis) (39) only or additionally supplemented with 2 mM zanamivir or 2 mM zanamivir and a 3 mM concentration of the desired compound. The cells were then lysed in DH buffer (50 mM HEPES, 100 mM NaCl, 0.005 g/ml dodecyl maltoside, complete protease inhibitor cocktail [Roche]). HN and F were immunoprecipitated from postnuclear lysates with anti-GFP antibody-conjugated agarose beads (Santa Cruz) for 2 h at 4°C, washed, and resuspended in PBS and then incubated in the absence or presence of proteinase K (Sigma) (0.64 × 10−5 mg/ml [1×] or 1.28 × 10−5 mg/ml [2×]) for up to 90 min. Complete protease inhibitor cocktail (Roche) was added to stop proteinase K digestion after 90 min. The protein-bead complexes were supplemented with Laemmli sample buffer, heated to 99°C for 5 min, and then subjected to SDS-PAGE and Western blot analysis with polyclonal antibodies to the HRC region of F.
Peptide synthesis.
All peptides were produced by standard fluorenylmethoxycarbonyl (Fmoc) solid-phase methods. The cholesterol moiety was attached to the peptide via chemoselective reaction between the thiol group of an extra cysteine residue, added C-terminally to the sequence, and a bromoacetyl derivative of cholesterol, as previously described (21, 54, 57).
BiFC microscopy assay.
Bimolecular fluorescence complementation (BiFC) microscopy was performed as described previously (53). 293T cells were transiently transfected on biocoated Delta TPG dishes (Fisher Scientific) with the cDNA combinations indicated in the table and 10% red fluorescence protein (RFP) using Lipofectamine 2000 according to the manufacturer's instructions. The transfection ratio of the N-Venus/C-Venus/CFP constructs was 2:1. After 4 h of incubation at 37°C, the transfection mixture was replaced with complete medium (DMEM, 10% FBS, and 1% penicillin and streptomycin [Pen-Strep]) supplemented with 10 mM zanamivir. Fifteen hours later the medium was replaced with OptiMEM supplemented with 10 mM zanamivir and 100 ng/ml cycloheximide (Sigma) for 1 h at 37°C. Fluorescent images and the mean fluorometric ratio (calculated as the fluorescence intensity produced by BiFC divided by that of the RFP) were acquired using a confocal laser scanning microscope (Nikon TE-2000U Digital Eclipse C1si equipped with a spectral detector) using EZ-C1 acquisition and analysis software and a 60× (numerical aperture [NA], 1.4) oil objective. Venus, hybrid fluorescent complex, and RFP were excited at 488 nm (emission, 529 nm), 488 nm (emission, 513 nm), and 561 nm (emission, 610 nm), respectively. The same laser power and gain settings were used for all samples and for all replicate experiments.
Microscopy.
Equal volumes of purified wild-type HPIV3 and bovine serum albumin (BSA)-blocked 10-nm gold beads in 150 mM NaCl–10 mM HEPES, pH 7.5, were first mixed together on ice. Three microliters of the mixture was transferred onto glow-discharged, carbon-coated electron microscope grids and stained with nano-W (methylaminetungstate; NanoProbes, Yaphank, NY). Specimens for tomography were imaged using an FEI Company Tecnai G2 transmission electron microscope operating at 200 kV, coupled to a Gatan Ultrascan 1000 charge-coupled device. Typically, ∼57 images were collected for each tilt series in 2° increments from −55 to +55° by the FEI Explore 3D Automated Tomography Data Acquisition Software (ATDASW). Tilt series were imaged at a ×27,000 magnification with a pixel size of 0.4902 nm. Tomograms were then reconstructed using the fiducial-based method implemented in IMOD software (25).
Cell surface expression assay.
293T cell monolayers, transiently transfected with uncleaved HA along with HN-N-Venus and F-C-CFP or with F-C-CFP alone were incubated overnight in DMEM supplemented with neuraminidase (100 mU/well in six-well plates) to prevent fusion.
Cells were washed twice in phosphate-buffered saline (PBS) and incubated with anti-HPIV3 F rabbit polyclonal antibodies in 3% BSA and 0.1% sodium azide in PBS for 1 h. Samples were then washed twice in PBS and incubated with 1:100 of anti-mouse IgG(H+L) R-phycoerythrin conjugate (Caltag Laboratories). Cell surface expression of all samples was performed with fluorescence-activated cell sorting (FACS) (FACSCalibur; Becton, Dickinson).
RESULTS
The notion that HN may protect processed F from untimely activation until the time of receptor engagement derives from several of our recent findings (15, 32). We have shown that in the absence of HN, processed F is sensitive to degradation by added proteases (15), a measure of F activation (15, 45), whereas in the presence of an HN molecule that is not engaged to its receptor, F is protected from proteolysis (15). For F activation in the absence of HN, we substituted the influenza virus hemagglutinin (uncleaved HA) as a nonhomologous sialic acid binding protein (45, 58) in assays for F triggering. The HA is not cleaved when produced in these cells and is not exposed to low pH, both requirements for the HA to express its own membrane-fusing activities. Using heat to activate F, we showed that in the presence of uncleaved HA, at temperatures above 37°C, HPIV3 F mediates fusion with red blood cell (RBC) membranes (45).
HPIV3 F protein mediates fusion in the absence of HN under physiological conditions.
To determine whether F can also mediate cell-cell fusion at a physiological temperature (37°), we performed a quantitative cell fusion assay using the nonhomologous sialic acid binding protein uncleaved influenza virus HA, present solely for its tethering function. To compare fusion mediated by F with and without HN in this experiment, we used an HN molecule containing a mutation in the globular head (D216R), previously shown to eliminate neuraminidase activity while retaining binding and F-activation properties (41). The use of this HN for comparison allows the focused evaluation of the binding and fusion-promoting features of HN in the absence of neuraminidase activity, which would cause dissociation of the HN/F-bearing cells. In the cell-to-cell fusion assay (Fig. 1), fusion is assessed by syncytium formation, whose visualization is enhanced by coexpression of the red fluorescent protein (RFP). Quantitative fusion measurements were simultaneously carried out by β-galactosidase complementation (41). 293T cells were cotransfected with HPIV3 F and HN wild type (wt), HPIV3 F and HN D216R, HPIV3 F and uncleaved HA, F alone, uncleaved HA alone, or vector alone (Fig. 1). After an 18-h incubation with overlaid target cells, fusion was observed and quantified by the β-galactosidase complementation assay (Fig. 1G). When associated with receptor-engaged HN molecules (wt HN or D216R HN) (Fig. 1A and B), the HPIV3 F mediates levels of cell fusion similar to those seen when HPIV3 F is expressed in the presence of uncleaved influenza virus HA (Fig. 1C). When F is expressed without uncleaved HA to tether the cells (Fig. 1D), it presumably mediates some fusion since the cells are contacting each other. The results indicate that F alone can mediate some fusion over time at 37°.
Fig 1.
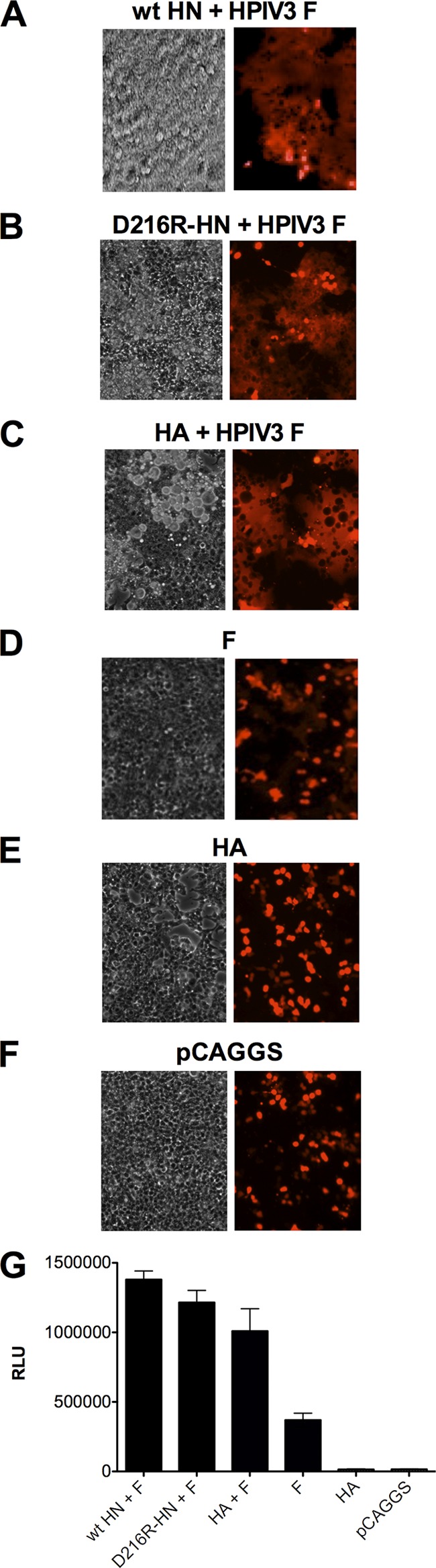
HPIV3 fusion protein mediates cell fusion at 37°C with uncleaved influenza virus hemagglutinin (HA) as a nonspecific binding protein. Cells expressing HPIV3 HN-wt and HPIV3 F (A), HPIV3 HN-D216R and HPIV3 F (B), uncleaved influenza virus HA and HPIV3 F (C), HPIV3 F (D), uncleaved influenza virus HA (E), or pCAGGS empty vector (F) were overlaid with receptor-bearing cells expressing red fluorescent protein (RFP) and incubated for 18 h at 37°C. Fusion under each condition was quantitated as described in Materials and Methods (G). Cell-cell fusion is observed on the left by syncytium formation (visible microscopy) and on the right by RFP redistribution (fluorescence microscopy). RLU, relative light units.
HN retains F in a preactivated state prior to receptor engagement.
HN and F associate prior to receptor engagement (53), and upon receptor engagement HN activates F to mediate fusion (45). A protective effect of HN on F thus can only be demonstrated in the absence of HN-receptor interaction. For this purpose zanamivir was used to prevent HN from engaging its sialic acid receptor (47, 49, 51), while having no effect on HA's receptor binding (16). We have shown that zanamivir blocks receptor engagement without itself stimulating HN to trigger F; in fact, exposure of HN to zanamivir prevents its ability to activate F (15, 45, 48, 50, 52). In this way the effect of nonreceptor-engaged HN can be assessed.
Cells expressing HPIV3 F with HN (wt or mutants) and uncleaved HA, HPIV3 F with uncleaved HA, F alone, or vector alone were incubated with an overlay of target cells in the presence of either medium alone or medium containing zanamivir. The use of uncleaved HA under these conditions allows the continued apposition of viral protein-expressing membranes and host cell membranes even in the presence of HN receptor dissociation caused by the addition of zanamivir. The level of cell-to-cell fusion (48) mediated by HPIV3 F was suppressed in the presence of HN when zanamivir was used to prevent HN′s receptor engagement (either wt HN, D216R HN, or D216N/P111S HN) at all time points (Fig. 2A), albeit less strongly at later times. An inhibitory peptide that blocks F-mediated fusion (54) abolished all cell-to-cell fusion mediated by HA/F, indicating that the observed fusion by the HA/F pair was attributable to F alone. Although continuous engagement of HN for the D216R HN was able to overcome the effect of peptide inhibition as we have shown (45), the combination of zanamivir with a fusion-inhibitory peptide (46) abolishes cell-to cell fusion even in the presence of the D216R HN. These data were supported by visual and fluorescence microscopy at 18 h (Fig. 2B), which confirmed that HN expression without receptor engagement prevents the cell-to-cell fusion otherwise mediated by the HA-F pair. The conclusion that HN inhibits fusion (rather than simply failing to activate fusion) is supported by the finding that F alone in the presence of uncleaved HA to tether adjacent cells mediates more fusion than in the presence of non-receptor-engaged HN.
Fig 2.
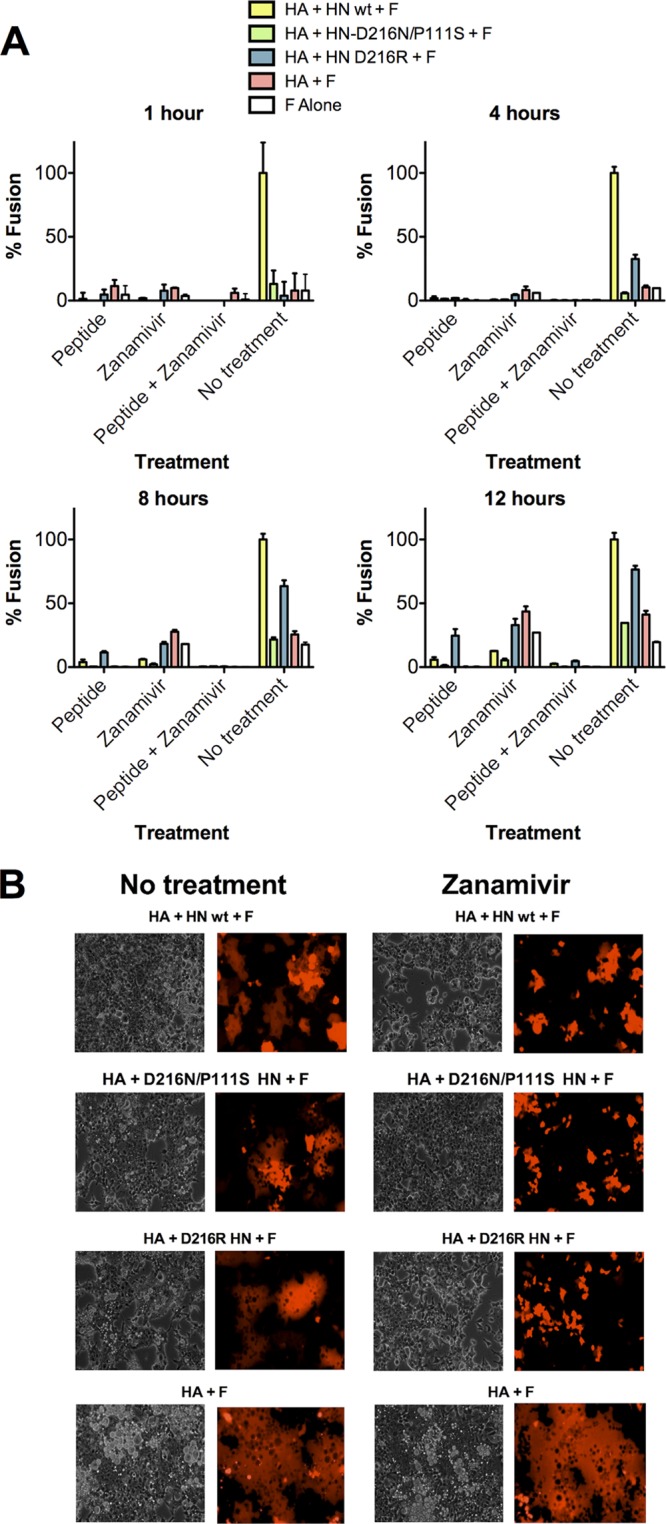
HN, before receptor engagement, prevents F activation. (A) Cell-to-cell fusion at 37°C mediated by HPIV3 F coexpressed with uncleaved influenza virus HA and HN wt, uncleaved influenza virus HA and HN P111S/D216N, uncleaved influenza virus HA and HN-D216R, uncleaved influenza virus HA, or alone at 1, 4, 8, and 12 h in the presence of treatments as indicated on the x axis. Fusion, quantitated as described in Materials and Methods, is shown as a percentage of the fusion mediated by the HA, HN wt, and HPIV3 F in the no-treatment group (± standard deviation). (B) Fusion was visualized at 18 h by visible and fluorescence microscopy for the no-treatment group and the zanamivir-treated group. The data reflect experiments repeated at least three times.
The HN stalk is critical for preventing F from undergoing heat activation.
A variant HPIV3 virus with a mutation in the stalk of HN (P111S) is poorly fusogenic and is inefficient at triggering F (45, 50, 52, 69). Virions bearing this HN molecule retain viability due to a mutation (D216N) in the globular head of HN that reduces neuraminidase activity, thereby allowing for prolonged receptor contact (49, 52). We have proposed that the impairment in F triggering mediated by this HN could protect F from indiscriminant triggering, resulting in more pretriggered F remaining available for fusion upon contact with target cells (50).
Here, we used this HN to assess the role of the stalk domain of HN in the F-stabilizing effect. First, we determined whether, in the presence of uncleaved HA to tether the glycoprotein-expressing and target membranes, the receptor-engaged P111S/D216N HN reduces or increases the energy required for F to undergo its conformational rearrangements toward fusion. Two other mutant HN molecules along with wt HN were also used to assess the importance of various regions of receptor-engaged HN for promotion of F triggering (Fig. 3). The HNs were evaluated for their ability to provide a stimulus for F's transition to fusion. The readout was fusion of F-expressing cells with target red blood cells (RBCs) at a range of temperatures, in the presence of influenza virus uncleaved HA to tether the membranes. In this way we determined whether each HN provided an advantage beyond the tethering function provided by HA. The D216R mutation in HN confers constitutive receptor engagement (41). The N551D mutation confers enhanced F triggering due to the action of HN′s sialic acid binding site II at the dimer interface (41, 48). Fig. 3 shows that the cells coexpress wt F and uncleaved influenza virus HA (present to serve the tethering function as in the experiments shown in Fig. 1 and 2) either alone or in the presence of wt HN, D216R HN, N551D HN, or P111S/D216N HN. After binding to receptor-bearing RBCs at 4°C, the cells were transferred to the indicated temperatures (on the x axis), and fusion was quantitated.
Fig 3.
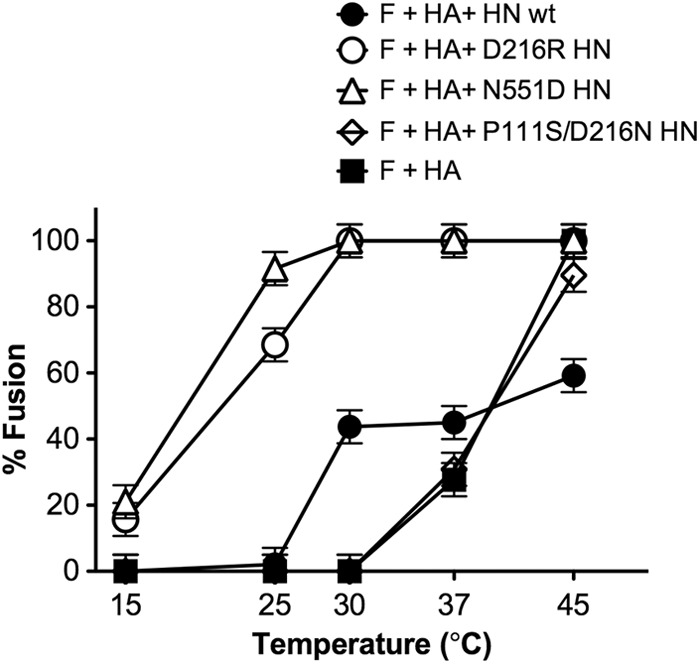
Temperature dependence of fusion mediated by F in the presence of HA alone or with D216R HN, N551D HN, or P111S/D216N HN. For the F-activation assay, monolayers of 293T cells coexpressing wt F and uncleaved influenza virus HA, alone or in the presence of wt HN, D216R HN, N551D HN, or P111S/D216N HN were allowed to bind to receptor-bearing RBCs at 4°C and then transferred to the indicated temperatures (x axis). After 60 min of incubation, fused RBCs were quantified, and results are expressed as a percentage of the total bound RBCs (y axis). The ordinate values are means of results (± standard deviations) of three experiments.
We found that temperature activation of F occurs at 37°C (with uncleaved HA serving to adhere the membranes and permit fusion), independent of HN. In the presence of HN molecules efficient at F triggering (N551D) or constitutively receptor bound (D216R), fusion occurs at lower temperatures. However, the stalk mutation-bearing HN molecule (P111S/D216N) fails to enhance F activation, with F-mediated fusion occurring at temperatures similar to those mediated solely by the nonspecific binding molecule HA. It appears here that in this period of time—1 h—the P111S/D216N HN offers no advantage over F alone, as long as the membranes are brought into apposition (by uncleaved HA in this case) so that F can act. This experiment also confirms that the presence of uncleaved HA on the glycoprotein-expressing cells does not affect the performance of HPIV3 HN molecules since the temperature curves for each HN are consistent with those we have published in the absence of HA (41, 42, 48), making the assay system suited for subsequent experiments. These results show that while F can be thermally activated if brought into apposition with its target cell, the specific characteristics of its partner HN molecule determine whether the efficiency of F triggering and fusion is enhanced.
We next assessed the effect of these HN molecules in the absence of receptor engagement (Fig. 4). We found that in the absence of HN receptor engagement (presence of zanamivir), the presence of an HN molecule significantly reduced HA-F fusion over 60 min at 37°C, compared to the no-HN samples (Fig. 4A). The stalk mutant P111S/D216N HN reduces fusion the most, to 20%. The two HNs with globular head mutations, D216R HN and N551D HN, also reduce HA-F fusion over 60 min at 37°C, though less than the P111S/D216N HN. Under the same conditions but with HN receptor engaged (Fig. 4E), the stalk mutant P111S/D216N HN still reduces fusion compared to the no-HN samples, while the other HN molecules promote fusion.
Fig 4.

The HN stalk domain is critical for preventing F from undergoing thermal activation. Monolayers of 293T cells coexpressing wt F and uncleaved influenza virus HA, either alone (no HN) or in the presence of the indicated HN molecules, were allowed to bind to receptor-bearing RBCs at 4°C and then transferred to 37°C for 60 min (A and E) or to 45°C for the indicated times (B, C, D, F, G, and H). Zanamivir to prevent HN′s receptor engagement was either present or absent, as indicated. After incubation, fused RBCs were quantified, and results are expressed as a percentage of the total bound RBCs (y axis). The ordinate values are means of results (± standard deviations) of at least three experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The effect of these differences is reduced at 45°C (Fig. 4B); however, the effect of the stalk mutant HN P111S/D216N remains evident. In the absence of HN-receptor interaction (presence of zanamivir), the presence of P111S/D216N HN significantly reduced HA-F fusion over 5 min compared to no-HN levels and appears to continue to reduce fusion after 15 min (Fig. 4C), although not to a statistically significant extent. By 30 min at 45°C (Fig. 4D), fusion has been maximally triggered by heat in all cases. Under the condition of receptor-engaged HN (no zanamivir), even at 45°C (Fig. 4F to H), the stalk mutant P111S/D216N HN provides protection against heat triggering of fusion, while the two other HN molecules promote additional fusion. Stabilization of F by P111S/D216N HN is evident only at the 5-min time point (Fig. 4F); at later time points the protective effect is supplanted by the overwhelming fusion triggered by heat.
The HN stalk domain protects F from the fusion-activating conformational change (retains F in a preactivated state) prior to receptor engagement.
We analyzed the state of F biochemically to determine whether the observed differences in cell-cell fusion in the presence or absence of non-receptor-engaged HN correlated with altered protein conformation (Fig. 5A). We have previously shown that F becomes protease sensitive after activation, and this biochemical assay can be used to detect conformational changes in the posttriggered protein (15, 45). This protease sensitivity assay provides a strong correlate of the initiation of the fusion activation process (if F is found to be sensitive) or of stabilization of, or failure to activate, F (if F is found to be resistant). We assessed the protease sensitivity of F when it was coexpressed with uncleaved HA alone or with HA and each HN—wt HN, D216N/P111S HN, or P111S HN—in the presence (R+) or absence (R−) of HN-receptor interaction (i.e., without or with zanamivir) (Fig. 5A). The D216N/P111S HN exists on a viable virus because the mutation at D216 eliminates receptor cleavage and permits more time for the defective triggering function to operate. The P111S HN meanwhile permits assessment of the effect of the single stalk mutation on HN′s effects on F whether HN is receptor engaged or not.
Fig 5.
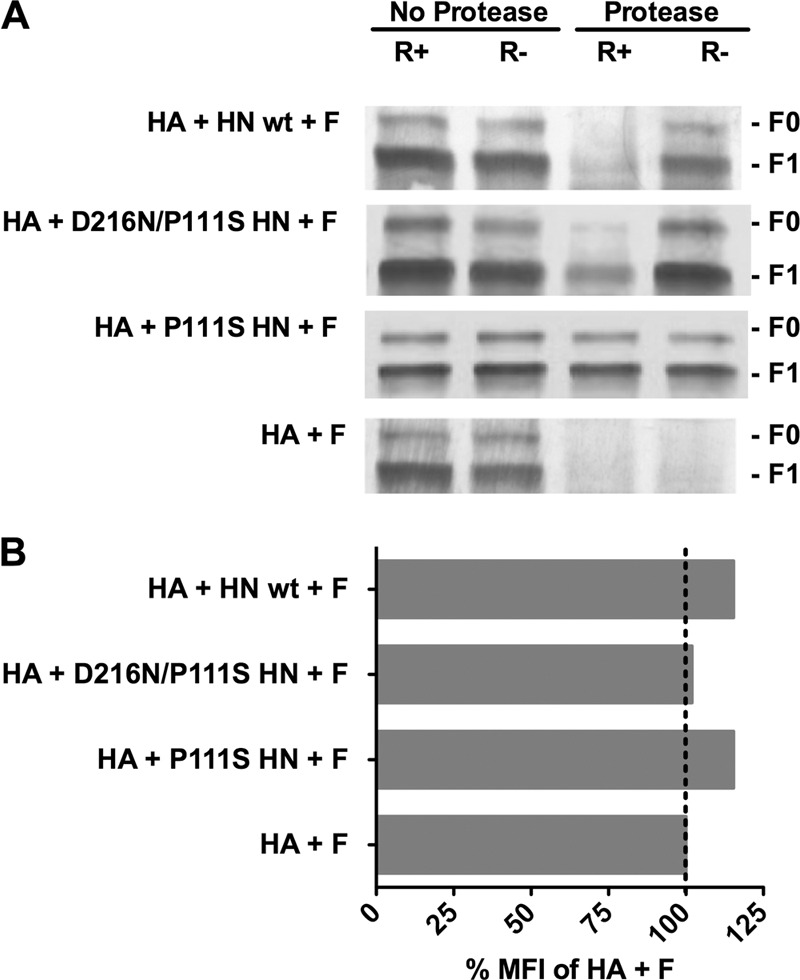
Structural rearrangements in F in are detected by altered protease sensitivity. (A) Monolayers of cells coexpressing F and uncleaved HA with the indicated HN or F and HA alone (bottom row) were incubated for 60 min at 37°C in medium either without zanamivir (receptor-engaged; R+) or with zanamivir to disengage HN from its receptor (R−). After the cells were lysed, the envelope glycoproteins were immunoprecipitated and then incubated in the presence or absence of protease K. The representative Western blot shows F proteolysis, detected by polyclonal anti-F HRC antibodies. F0 (precursor) protein and F1 are indicated. When each HN is present, but not engaging receptor (R−), F does not become protease sensitive. The stalk domain mutant HN (P111S HN) prevents protease digestion of F even when HN is receptor engaged (R+). (B) Flow cytometric analysis of cell surface F expression using rabbit polyclonal anti-F antibodies. The mean fluorescent intensity (MFI) of the cells expressing F with the indicated HN/HA pair is presented as a percentage of the mean fluorescent intensity of cells expressing F with HA alone.
When uncleaved HA and F are coexpressed without HN (Fig. 5A, bottom row), F is completely protease sensitive. In the presence of wt HN, F is stabilized and not protease sensitive when HN is not receptor engaged (R−), suggesting that F activation has not occurred and that HN stabilized F in a pretriggered state. F becomes protease-sensitive when wt HN is receptor-engaged (R+), suggesting that activation has initiated. For D216N/P111S HN, F is stabilized and not protease sensitive when HN is not receptor engaged and upon receptor engagement is protease sensitive. For P111S HN, an HN that we have shown to possess abrogated fusion promotion function (45, 50, 52), F is stabilized and not protease sensitive when HN is not receptor engaged but in this case is still protected from protease upon receptor engagement by HN (Fig. 5A, note the bands in both protease/R+ and protease/R− lanes for HA + P111S HN+ F). Surface expression levels of F in the presence of each receptor binding protein were analyzed by FACS (Fig. 5B) and found to be similar regardless of the coexpressed HN.
The results shown in Fig. 4 and 5 imply that the HN proteins bearing the P111S mutation can interact with F. We previously described the use of the technique of bimolecular fluorescence complementation (BiFC) to measure the HN-F interaction. The BiFC strategy is based on the reconstitution of an active fluorescent complex when fragments of fluorescent proteins, fused to putative interacting pairs, associate (18, 19, 24, 31). We have shown that this strategy effectively measures the interaction between HN-F pairs by quantitating the relative mean fluorescence produced upon HN-F interaction (53). To evaluate HN-F interaction in the presence of the P111S mutation, we again employed BiFC. The cells were transfected with the indicated BiFC-tagged HN and F molecules (HN-N-Venus and F-C-CFP) along with red fluorescent protein (RFP). In this experiment fluorescent emission at 513 nm indicates HN-F interaction since only when HN and F associate is the fluorescent probe reconstituted. RFP was used as reference for the BiFC mean fluorometric ratio calculation (24), so that the higher the ratio, the stronger is the interaction. We compared the P111S mutated HN with wt HN for the HN-F interaction under identical conditions in the absence of receptor engagement. In the experiment reported in Table 1, cells coexpressing the indicated HN or uncleaved HA-N-Venus and F-C-CFP were treated overnight with zanamivir. One hour before observation, the cells were treated with cycloheximide to block de novo protein synthesis (and thereby limit our observation to premade viral glycoproteins) and resupplemented with zanamivir to prevent HN from engaging receptor. Table 1 lists the mean fluorimetric ratio for each indicated HN-F pair. For comparison with a noninteractive protein pair, we show the value for uncleaved HA and F (53). Diffuse, evenly distributed fluorescence was visualized across the cell surfaces showing HN-F interaction prior to receptor binding (data not shown). The single mutant P111S HN, which completely fails to promote fusion, interacts with F before HN-receptor interaction, as demonstrated by BiFC (53) (Table 1), and F is not protease sensitive even when HN is receptor bound, remaining in its pretriggered state.
Table 1.
Interaction of HPIV3 HN and F measured by BiFC
| Protein | Mean fluorometric ratioa |
|---|---|
| wt HN | 1.35 |
| D216N/P111S HN | 2.25 |
| P111S HN | 1.974 |
| HA | 0.862 |
Values are based on the interaction of HA or HN protein and F protein. 293T cells were cotransfected with constructs encoding F-C-CFP and HN-N-Venus as indicated in the text; receptor engagement was prevented by addition of 10 mM zanamivir. These experiments were subjected to immunoblot analysis to assess expression across all samples. Proteins were detected by goat anti-GFP horseradish peroxidase-conjugated antibody. The mean fluorometric ratio resulting from protein-protein interaction was measured in an average of at least 10 fields per experiment, and the values are averages of results from three separate experiments, showing equal expression across all samples.
F protein adopts a postfusion state on virion surfaces in the absence of HN.
The data shown in Fig. 2, 4, and 5 indicate that HPIV3 HN, prior to receptor engagement, increases the stability of the prefusion state of F. In support of this notion, a previous structural investigation of intact PIV5 virions showed that in areas of the surface where F was observed to be present in distinct spikes without HN, the ectodomain structure was consistent with the postfusion state (32). The electron cryo-micrographs revealed, in addition to the areas of distinct individual spikes, areas of a continuous layer. Since the spikes were found to correspond to the postfusion state, the authors concluded that the prefusion state of the F protein requires stabilization, most probably by the association with hemagglutinin-neuraminidase (32).
We analyzed HPIV3 viral particles using negative-stain electron tomography data (Fig. 6) and observed clearly different organizations of the prefusion HN/F pair (unactivated F) and the postfusion state of F on viral membranes. Surface density on a subpopulation of small viral particles (Fig. 6A and E) shows clusters of glycoprotein spikes that bear clear resemblance to reported crystal structures for postfusion HPIV3 F trimers (67) and to the moderate-resolution cryo-negative stain reconstruction reported for PIV5 F (32) (Fig. 6G). This suggests that for HPIV3 as well, in the absence of HN, F on the surface of the virus may readily adopt its postfusion state.
Fig 6.

Negative-stain electron microscope tomography of HPIV3 virus particles. (A) A subpopulation of small particles (V, virus interior) bear predominantly F proteins in postfusion conformation (white arc and carat), with some F in prefusion form (black carat). (B and C) The majority of typical particles in our preparations show a double-layered surface (white box), with little postfusion F observed. (D) Close-up of the upper region of the particle shown in panel A. (E) Close-up of the lower region of the particle in panel A. (F) Average radial density plots show clear differences between regions of primarily F in postfusion conformation (red) and the more typical double-layer coat on most particles (black). (G) Models and dimensions for coupled HN/F and postfusion F based upon reported crystal structures (28, 42, 67–69) (yellow, HN; cyan, prefusion F; red, postfusion F) and a cryo-negative stain electron microscopy reconstruction for postfusion F (red density) (32). au, arbitrary units.
In contrast, the majority of viral particles, which are generally larger than the small, primarily postfusion F-bearing particles, exhibit surfaces with a fairly continuous coat of spikes with two layers of alternating density (Fig. 6B and C, boxed regions), similar to the configuration of HN and F shown in yellow and cyan in Fig. 6G. Based upon comparisons to the reported prefusion F structure (68) and the HPIV3 model we generated (42), we interpret these patterns to reflect staggered HN-F-HN-F-HN-F-HN spikes. The radial density along the perimeter of the two types of particles was measured (Fig. 6F), and two peaks at ∼13 nm and at ∼16 nm are clearly distinguishable. Based on the dimensions as shown in Fig. 6G, we interpret these peaks as corresponding to the prefusion F and the postfusion F, respectively. The peak corresponding to HN is clearly separate at ∼22 nm. These data support the role of HN in stabilizing the prefusion F on the surface of HPIV3.
DISCUSSION
We have recently shown that small-molecule- or receptor mimic-mediated activation of the paramyxovirus fusion protein (for HPIV3 or Nipah virus) before the virus reaches proximity to the appropriate target cell, leads to inactivation of viral particles (15, 56). For HPIV3, we have directly shown that the mechanism of small molecule-mediated viral inactivation occurs via the corruption of the viral fusion machinery, inducing HN to prematurely trigger F (15). Dysregulation of the fusion process in lung tissue by a virus bearing a highly fusogenic HN/F pair leads to production of noninfective particles (40). Under selective pressure, this fusogenic virus evolved to gain mutations that rendered the HN/F fusion pair less readily activated and the F more stable at higher temperatures (42). For Sendai virus it has been shown that incubation at high temperatures led to inactivation of viral fusion without affecting HN′s receptor binding avidity, consistent with triggering and inactivation of F (66). These findings from several different paramyxoviruses indicate that stability of the prefusion state of F is important for infectivity and that overactive fusion activation is detrimental (8, 40, 58).
Our results contrast to some extent with a previous report in which coexpression of PIV5 HN and F did not provide resistance to the heat-induced triggering of F (9). In the study by Connolly et al., cells coexpressing F and HN were heated at temperatures up to 60°C and analyzed for altered binding to conformation-sensitive monoclonal antibodies (MAb), and the presence of HN was not seen to affect the change in F's reactivity to MAbs (9). It is not clear whether HN was receptor engaged in the experiments; unless the cells in which HN and F were coexpressed were treated to deplete receptors, it is likely that HN was receptor engaged. Our results show that only non-receptor-engaged HN exerts a stabilizing role, which is consistent with the requirement for F to remain in its nontriggered state prior to receptor engagement. Furthermore, in the Connolly report, the postfusion state is detected at very high temperatures approaching 60°C, which is much higher than the physiologically relevant temperatures at which we observe a stabilizing effect of HN on F.
We have used alteration in F protein's sensitivity to degradation by added protease as an indication of conformational change in F and interpret the reduced protease sensitivity in the presence of HN to indicate that HN prevents that conformational change. We have considered the alternative interpretation that HN-F interaction shields the protease digestion sites and that protease sensitivity would thus reflect a change in HN-F interaction rather than a progression of F through the steps of fusion activation. Recent results argue against this interpretation. We have shown that unprocessed F (F0)—while it cannot be fully activated to the postfusion state since it lacks an accessible fusion peptide—can undergo conformational changes consistent with early stages of activation and that this partially extended state of F can be captured and detected using membrane-anchored peptides (45, 54). The F0 expressed alone is not protease sensitive, but activation to an extended intermediate, which occurs only in the presence of receptor-engaged HN, causes F to become protease sensitive, indicating that the protease sensitivity depends on activation of F, not on altered HN-F interactions that expose protease sites. Protease-sensitive F represents an F that has initiated the conformational changes of activation; i.e., it is not necessarily a postfusion state F but rather one that has initiated activation (45).
Our results support a key role for the stalk domain of HN in determining physical interaction with F and in the protection of F from untimely activation. We have previously shown that specific properties of the globular head of HN influence the actions of the stalk and that stalk residues modulate the F-triggering property of receptor-engaged HN. Here, we analyzed the protective effect of HN on F when HN is not receptor engaged. Mutant HN molecules allowed us to distinguish between effects of the head and the stalk and to dissect specific contributions of each region. We took advantage of the fusion machinery of a specific virus that bears an HN with two mutations: D216N, which decreases receptor-cleaving activity, and P111S, which causes a defect in F triggering (45, 49, 50, 52, 69). The P111S mutation in the stalk of HN renders the HN molecule more stabilizing of F's prefusion state (Fig. 3, 4, and 5). Interestingly, the stalk mutation in the HN of this virus arose when the selective pressure of growth in cultured cells was applied to the virus bearing only the D216N HN mutation. It seems feasible that the stalk mutation emerged to compensate for the diminished receptor-cleaving activity, generating a fusion machinery that is less active and more consistent with viability. The finding that this HN exerts an enhanced stabilizing effect on F adds a new element to the balance of features of the HN/F pair that determine viability (8, 40, 50, 53). Enhancement of the F-protective role may, for example, counteract the effect of an avidly receptor binding HN head.
The stalk region of other paramyxovirus receptor binding proteins has also been recognized as critical for interaction with F during the entry process (5, 6, 8, 30, 33, 69). Several reports have suggested that a weaker interaction between the receptor binding protein and the fusion protein leads to increased fusion for those paramyxoviruses that use proteinaceous receptors (2, 3, 10, 44). For sialic acid binding viruses, it has been suggested that the converse is true: weaker receptor binding protein interaction with the fusion protein leads to decreased fusion (23, 33). For both groups of viruses, either sialic acid- or proteinaceous receptor-requiring, mutations in conserved residues in the receptor binding protein stalks led to alterations in fusion promotion. For all the paramyxoviruses studied that utilize a receptor binding protein for F activation, the proline at the position corresponding to P111 in HPIV3 is required for proper fusion activation (1, 6, 52, 69). For measles virus, experimental constraint of the flexibility of the tetrameric stalk of the binding protein (H) in the region of the corresponding proline (P108) abrogates fusion promotion, supporting the notion that this region of paramyxoviruses is key for F activation and may require a conformational change in the stalk tetramer (1, 6, 69). It is possible that the P111S mutation in the HPIV3 HN stabilizes the stalk, thereby decreasing the rearrangements associated with F triggering and enhancing the protective role of HN′s stalk domain in blocking F activation before receptor engagement.
Taken together, our data suggest a modified model for HN-F interaction during the viral life cycle to include an F-stabilizing role for HN prior to receptor engagement. HN and F are associated prior to receptor binding (53), and during this time HN guards F against premature activation (15, 40), with the stalk of HN serving a primary role in the efficacy of this protection. Upon receptor engagement, HN drives the formation of HN-F clusters at the site of fusion (53). HN and F remain associated as fusion activation proceeds (53), and HN continues to act on F well beyond insertion of the fusion peptide into the target (45); disengagement of HN from the receptor during the onset of fusion, even after insertion of the fusion peptide into the target, halts the process. While receptor engagement is key for the activation step, the specific HN heads are interchangeable, and it is the HN stalk that is responsible for the triggering function (55). Thus, HN is essential for distinct purposes before receptor engagement, when it acts to stabilize F in its pretriggered state, and after receptor engagement, when its interactions with F become stimulatory and persist throughout the activation of fusion. In contrast to the prevailing dichotomy (13, 22, 29, 43) which holds that the paramyxovirus receptor binding protein either restrains F from triggering until receptor is engaged and then releases F to allow it to fuse or engages F only upon receptor binding and then activates it to fuse, we propose that both mechanisms are at play. The switch of HN from protector to activator is determined by receptor engagement. If the F-stabilizing role of HN is critical for the survival of viruses and production of infectious particles in the natural host, these findings represent a new paradigm for understanding paramyxovirus entry.
ACKNOWLEDGMENTS
We are grateful to Ashton Kutcher and Jonathan Ledecky for their support, without which the microscopy critical to this work would not have been possible, to Dan and Nancy Paduano for their essential support of innovative research projects, and to the Friedman Family Foundation for our laboratories at Weill Cornell Medical College (WCMC). The work was supported by NIH grant R01AI31971 to A.M., NIH grants 3R01AI031971-19S1 (Research Supplement to Promote Diversity in Health-Related Research Program) and R01-GM099989 to K.L. and by WCMC-Qatar Biomedical Research Program Grant 5723081000 to A.M. M.P. is the Friedman Family Research Scholar in Pediatric Infectious Diseases.
Footnotes
Published ahead of print 20 September 2012
REFERENCES
- 1. Ader N, et al. 2012. Structural rearrangements of the central region of the morbillivirus attachment protein stalk domain trigger f protein refolding for membrane fusion. J. Biol. Chem. 287:16324–16334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aguilar HC, et al. 2007. Polybasic KKR motif in the cytoplasmic tail of Nipah virus fusion protein modulates membrane fusion by inside-out signaling. J. Virol. 81:4520–4532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aguilar HC, et al. 2006. N-glycans on Nipah virus fusion protein protect against neutralization but reduce membrane fusion and viral entry. J. Virol. 80:4878–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bagai S, Lamb R. 1995. Quantitative measurement of paramyxovirus fusion: differences in requirements of glycoproteins between simian virus 5 and human parainfluenza virus 3 or Newcastle disease virus. J. Virol. 69:6712–6719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bishop KA, et al. 2008. Residues in the stalk domain of the Hendra virus g glycoprotein modulate conformational changes associated with receptor binding. J. Virol. 82:11398–11409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bose S, et al. 2011. Structure and mutagenesis of the parainfluenza virus 5 hemagglutinin-neuraminidase stalk domain reveals a four-helix bundle and the role of the stalk in fusion promotion. J. Virol. 85:12855–12866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chaiwatpongsakorn S, Epand RF, Collins PL, Epand RM, Peeples ME. 2011. Soluble respiratory syncytial virus fusion protein in the fully cleaved, pretriggered state is triggered by exposure to low-molarity buffer. J. Virol. 85:3968–3977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chang A, Dutch RE. 2012. Paramyxovirus fusion and entry: multiple paths to a common end. Viruses 4:613–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Connolly SA, Leser GP, Jardetzky TS, Lamb RA. 2009. Bimolecular complementation of paramyxovirus fusion and hemagglutinin-neuraminidase proteins enhances fusion: implications for the mechanism of fusion triggering. J. Virol. 83:10857–10868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Corey EA, Iorio RM. 2007. Mutations in the stalk of the measles virus hemagglutinin protein decrease fusion but do not interfere with virus-specific interaction with the homologous fusion protein. J. Virol. 81:9900–9910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crennell S, Takimoto T, Portner A, Taylor G. 2000. Crystal structure of the multifunctional paramyxovirus hemagglutinin-neuraminidase. Nat. Struct. Biol. 7:1068–1074 [DOI] [PubMed] [Google Scholar]
- 12. Deng R, Wang Z, Mirza A, Iorio R. 1995. Localization of a domain on the paramyxovirus attachment protein required for the promotion of cellular fusion by its homologous fusion protein spike. Virology 209:457–469 [DOI] [PubMed] [Google Scholar]
- 13. Dutch RE. 2010. Entry and fusion of emerging paramyxoviruses. PLoS Pathog. 6:e1000881 doi:10.1371/journal.ppat.1000881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eckert DM, Kim PS. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70:777–810 [DOI] [PubMed] [Google Scholar]
- 15. Farzan SF, et al. 2011. Premature activation of the paramyxovirus fusion protein before target cell attachment with corruption of the viral fusion machinery. J. Biol. Chem. 286:37945–37954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Greengard O, Poltoratskaia N, Leikina E, Zimmerberg J, Moscona A. 2000. The anti-influenza virus agent 4-GU-DANA (Zanamivir) inhibits cell fusion mediated by human parainfluenza virus and influenza virus HA. J. Virol. 74:11108–11114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Horvath CM, Paterson RG, Shaughnessy MA, Wood R, Lamb RA. 1992. Biological activity of paramyxovirus fusion proteins: factors influencing formation of syncytia. J. Virol. 66:4564–4569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hu CD, Chinenov Y, Kerppola TK. 2002. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9:789–798 [DOI] [PubMed] [Google Scholar]
- 19. Hu CD, Kerppola TK. 2003. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 21:539–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu X, Ray R, Compans RW. 1992. Functional interactions between the fusion protein and hemagglutinin-neuraminidase of human parainfluenza viruses. J. Virol. 66:1528–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ingallinella P, et al. 2009. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. U. S. A. 106:5801–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iorio RM, Mahon PJ. 2008. Paramyxoviruses: different receptors—different mechanisms of fusion. Trends Microbiol. 16:135–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iorio RM, Melanson VR, Mahon PJ. 2009. Glycoprotein interactions in paramyxovirus fusion. Future Virol. 4:335–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kerppola TK. 2006. Visualization of molecular interactions by fluorescence complementation. Nat. Rev. Mol. Cell Biol. 7:449–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kremer J, Mastronarde D, McIntosh J. 1996. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116:71–76 [DOI] [PubMed] [Google Scholar]
- 26. Lamb R. 1993. Paramyxovirus fusion: a hypothesis for changes. Virology 197:1–11 [DOI] [PubMed] [Google Scholar]
- 27. Lamb RA, Paterson RG, Jardetzky TS. 2006. Paramyxovirus membrane fusion: lessons from the F and HN atomic structures. Virology 344:30–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lawrence MC, et al. 2004. Structure of the haemagglutinin-neuraminidase from human parainfluenza virus type III. J. Mol. Biol. 335:1343–1357 [DOI] [PubMed] [Google Scholar]
- 29. Lee B, Ataman ZA. 2011. Modes of paramyxovirus fusion: a Henipavirus perspective. Trends Microbiol. 19:389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee JK, et al. 2008. Functional interaction between paramyxovirus fusion and attachment proteins. J. Biol. Chem. 283:16561–16572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin HP, Vincenz C, Eliceiri KW, Kerppola TK, Ogle BM. 2010. Bimolecular fluorescence complementation analysis of eukaryotic fusion products. Biol. Cell 102:525–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ludwig K, et al. 2008. Electron cryomicroscopy reveals different F1+F2 protein states in intact parainfluenza virions. J. Virol. 82:3775–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Melanson VR, Iorio RM. 2006. Addition of N-glycans in the stalk of the Newcastle disease virus HN protein blocks its interaction with the F protein and prevents fusion. J. Virol. 80:623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moosmann P, Rusconi S. 1996. Alpha complementation of LacZ in mammalian cells. Nucleic Acids Res. 24:1171–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moscona A. 2005. Entry of parainfluenza virus into cells as a target for interrupting childhood respiratory disease. J. Clin. Invest. 115:1688–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moscona A, Peluso R. 1991. Properties of the human parainfluenza virus type 3 RNA polymerase/replicase in vitro: consensus with other negative-stranded RNA viruses. J. Virol. 65:4470–4474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moscona A, Peluso RW. 1993. Relative affinity of the human parainfluenza virus 3 hemagglutinin-neuraminidase for sialic acid correlates with virus-induced fusion activity. J. Virol. 67:6463–6468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Murrell M, Porotto M, Weber T, Greengard O, Moscona A. 2003. Mutations in human parainfluenza virus type 3 HN causing increased receptor binding activity and resistance to the transition state sialic acid analog 4-GU-DANA (zanamivir). J. Virol. 77:309–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Obrig TG, Culp WJ, McKeehan WL, Hardesty B. 1971. The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J. Biol. Chem. 246:174–181 [PubMed] [Google Scholar]
- 40. Palermo L, et al. 2009. Human parainfluenza virus infection of the airway epithelium: the viral hemagglutinin-neuraminidase regulates fusion protein activation and modulates infectivity. J. Virol. 83:6900–6908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Palermo LM, Porotto M, Greengard O, Moscona A. 2007. Fusion promotion by a paramyxovirus hemagglutinin-neuraminidase protein: pH modulation of receptor avidity of binding sites I and II. J. Virol. 81:9152–9161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Palmer SG, et al. 2012. Adaptation of human parainfluenza virus to airway epithelium reveals fusion properties required for growth in host tissue. mBio 3(3):e00137–12 doi:10.1128/mBio.00137-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Plemper RK, Brindley MA, Iorio RM. 2011. Structural and mechanistic studies of measles virus illuminate paramyxovirus entry. PLoS Pathog. 7:e1002058 doi:10.1371/journal.ppat.1002058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Plemper RK, Hammond AL, Gerlier D, Fielding AK, Cattaneo R. 2002. Strength of envelope protein interaction modulates cytopathicity of measles virus. J. Virol. 76:5051–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Porotto M, et al. 2011. Spring-loaded model revisited: Paramyxovirus fusion requires engagement of a receptor binding protein beyond initial triggering of the fusion protein. J. Virol. 85:12867–12880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Porotto M, et al. 2006. Inhibition of Hendra virus membrane fusion. J. Virol. 80:9837–9849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Porotto M, et al. 2006. Paramyxovirus receptor-binding molecules: engagement of one site on the hemagglutinin-neuraminidase protein modulates activity at the second site. J. Virol. 80:1204–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Porotto M, Fornabaio M, Kellogg GE, Moscona A. 2007. A second receptor binding site on human parainfluenza virus type 3 hemagglutinin-neuraminidase contributes to activation of the fusion mechanism. J. Virol. 81:3216–3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Porotto M, Greengard O, Poltoratskaia N, Horga M-A, Moscona A. 2001. Human parainfluenza virus type 3 HN-receptor interaction: the effect of 4-GU-DANA on a neuraminidase-deficient variant. J. Virol. 75:7481–7488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Porotto M, Murrell M, Greengard O, Doctor L, Moscona A. 2005. Influence of the human parainfluenza virus 3 attachment protein's neuraminidase activity on its capacity to activate the fusion protein. J. Virol. 79:2383–2392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Porotto M, et al. 2004. Inhibition of parainfluenza type 3 and Newcastle disease virus hemagglutinin-neuraminidase receptor binding: effect of receptor avidity and steric hindrance at the inhibitor binding sites. J. Virol. 78:13911–13919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Porotto M, Murrell M, Greengard O, Moscona A. 2003. Triggering of human parainfluenza virus 3 fusion protein (F) by the hemagglutinin-neuraminidase (HN): an HN mutation diminishing the rate of F activation and fusion. J. Virol. 77:3647–3654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Porotto M, Palmer SG, Palermo LM, Moscona A. 2012. Mechanism of fusion triggering by human parainfluenza virus type III: communication between viral glycoproteins during entry. J. Biol. Chem. 287:778–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Porotto M, et al. 2010. Inhibition of Nipah virus infection in vivo: targeting an early stage of paramyxovirus fusion activation during viral entry. PLoS Pathog. 6:e1001168 doi:10.1371/journal.ppat.1001168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Porotto M, et al. 2012. The second receptor binding site of the globular head of the Newcastle disease virus hemagglutinin-neuraminidase activates the stalk of multiple paramyxovirus receptor binding proteins to trigger fusion. J. Virol. 86:5730–5741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Porotto M, Yi F, Moscona A, LaVan DA. 2011. Synthetic protocells interact with viral nanomachinery and inactivate pathogenic human virus. PLoS One 6:e16874 doi:10.1371/journal.pone.0016874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Porotto M, et al. 2010. Viral entry inhibitors targeted to the membrane site of action. J. Virol. 84:6760–6768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Russell CJ, Kantor KL, Jardetzky TS, Lamb RA. 2003. A dual-functional paramyxovirus F protein regulatory switch segment: activation and membrane fusion. J. Cell Biol. 163:363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. San-Juan-Vergara H, et al. 2012. Cholesterol-rich microdomains as docking platforms for respiratory syncytial virus in normal human bronchial epithelial cells. J. Virol. 86:1832–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schildgen V, et al. 2011. Human metapneumovirus: lessons learned over the first decade. Clin. Microbiol. Rev. 24:734–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schowalter RM, Chang A, Robach JG, Buchholz UJ, Dutch RE. 2009. Low-pH triggering of human metapneumovirus fusion: essential residues and importance in entry. J. Virol. 83:1511–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sergel T, McGinnes LW, Peeples ME, Morrison TG. 1993. The attachment function of the Newcastle disease virus hemagglutinin-neuraminidase protein can be separated from fusion promotion by mutation. Virology 193:717–726 [DOI] [PubMed] [Google Scholar]
- 63. Smith EC, Popa A, Chang A, Masante C, Dutch RE. 2009. Viral entry mechanisms: the increasing diversity of paramyxovirus entry. FEBS J. 276:7217–7227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stone-Hulslander J, Morrison TG. 1999. Mutational analysis of heptad repeats in the membrane-proximal region of Newcastle disease virus HN protein. J. Virol. 73:3630–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tanabayashi K, Compans R. 1996. Functional interactions of paramyxovirus glycoproteins: identification of a domain in Sendai virus HN which promotes cell fusion. J. Virol. 70:6112–6118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wharton SA, Skehel JJ, Wiley DC. 2000. Temperature dependence of fusion by Sendai virus. Virology 271:71–78 [DOI] [PubMed] [Google Scholar]
- 67. Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. U. S. A. 102:9288–9293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yuan P, et al. 2011. Structure of the Newcastle disease virus hemagglutinin-neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc. Natl. Acad. Sci. U. S. A. 108:14920–14925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yuan P, et al. 2005. Structural studies of the parainfluenza virus 5 hemagglutinin-neuraminidase tetramer in complex with its receptor, sialyllactose. Structure 13:803–815 [DOI] [PubMed] [Google Scholar]
- 71. Yuasa T, et al. 1995. A cell fusion-inhibiting monoclonal antibody binds to the presumed stalk domain of the human parainfluenza type 2 virus hemagglutinin-neuraminidase protein. Virology 206:1117–1125 [DOI] [PubMed] [Google Scholar]
- 72. Zaitsev V, et al. 2004. Second sialic acid binding site in Newcastle disease virus hemagglutinin-neuraminidase: implications for fusion. J. Virol. 78:3733–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]