

Abstract
Several factors are involved in the control of HIV transcription/replication, including epigenetic modifications at the promoter level. Analysis of the HIV long terminal repeat (LTR) methylation status in infected patients controlling viremia is scarce. Herein, we show a higher degree of DNA methylation in the 5′-LTR of long-term nonprogressor and elite controller (LTNP/EC) versus progressor patients and a positive correlation with time of infection, indicating a certain contribution of HIV LTR silencing in reducing the number of replicating viruses which may account for a delayed progression.
INTRODUCTION
The majority of patients with HIV infection have a progressive active viral transcription and replication, which results in loss of CD4+ T cells, appearance of clinical symptoms, and disease progression. In a few rare cases, however, this increase in viremia is not observed, and immunity is maintained in these individuals. These are asymptomatic patients who maintain low levels of viral replication in the absence of antiretroviral treatment. They are known as controllers, either elite controllers (control viremia to undetectable levels) or virus controllers (low levels of viremia) (6, 13). Most of these patients fall also within the category of long-term nonprogressors (LTNP) (3, 14), who also maintain low levels of viremia and high levels of CD4+ T cells for more than 10 years without receiving treatment. These individuals have become the center of many studies to identify mechanisms of spontaneous viral control.
Several factors take part in the control of viral transcription and replication: defective viral strains, robust host cell-mediated immune responses, and other host genetic factors that can affect viral replication (5, 6). However, many questions still remain concerning the cellular immune mechanisms of persistent control and the extent of this control being driven by genetic and molecular factors. Among these molecular factors, the epigenetic silencing of HIV transcription could play a role. The different levels of chromatin regulation, such as the formation of a restrictive chromatin state and nucleosome remodelling, as well as other epigenetic modifications, such as DNA methylation, could influence HIV replication by acting as an additional mechanism of viral restriction. Whereas the involvement of histones in the control of gene expression through the chromatin state modification is compelling (8, 9), the role of differential DNA methylation in HIV control is somehow controversial. Silencing of the provirus through CpG methylation has been described in other retroviruses, such as human T-cell leukemia virus 1 (HTLV-1) (16). In cell lines latently infected with HIV, the 5′-long terminal repeat (LTR) of the provirus has been found to be hypermethylated (10). Indeed, activation of viral expression from latently infected cells does appear to correlate with loss of methylation of the HIV-1 LTR DNA. Two recent studies have shown evidences in this direction, where the promoter region of HIV-1 was epigenetically regulated by DNA methylation (1, 12) and influenced the stability of HIV latency. Two regions or islands in the 5′-LTR promoter are susceptible to CpG methylation, determining the accessibility of regulatory transcription factors to initiate viral transcription, such as Sp1 and NF-κB in the first island. Also MBD2/HDAC2 binding sites have been identified in the second CpG island, where under cytosine methylation conditions recruitment of these two factors takes place and silencing of the provirus is maintained (12). One of these studies showed that a higher proportion of hypermethylated HIV promoters in memory CD4+ cells was present in aviremic patients receiving treatment than in viremic patients also under antiretroviral treatment. However, in another more recent report, this DNA hypermethylation of LTR was not observed in proviruses from resting CD4+ cells in aviremic patients undergoing highly active antiretroviral therapy (HAART) (2). One of the differences between the two studies performed by Blazkova was the time of infection in each patient. Since DNA methylation may be a late event that enhances silencing of already-latent viruses rather than contributing to entry into latency (7, 11), time may need to be considered a factor. To date, studies evaluating the involvement of this mechanism of silencing are few; most of the work has been done in vitro, with very little data obtained directly from infected patients, especially addressing latency. We conducted the present study to determine if this effect could also be observed in proviruses from LTNP/EC patients, and the level of DNA methylation in 5′-LTR could influence viral replication level in these patients.
RESULTS AND DISCUSSION
Our aim was to compare the methylation state of HIV promoter in 2 groups of infected patients with different phenotypes. We analyzed the 5′-LTR CpG methylation patterns in peripheral blood mononuclear cells (PBMCs) in patients who maintain undetectable or low levels of viremia (<50 or <2,000 RNA copies/ml, respectively) without treatment. These patients, who would fall into the category of long-term nonprogressors (LTNP) and/or elite controllers (EC), were compared with patients in whom HAART resulted in the suppression of plasma viremia (Table 1).
Table 1.
CpG methylation of the 5′-LTR of HIV-1 in patients with low or undetectable levels of viremiaa
| Group | ID | Type of patient | No. of yrs of infection | On HAART (no. of yrs) | No. of CD4+ T cells/μl (%) | VL (RNA/ml) | % LTR CpG methylation | % total DNA methylation | LTR methylation/total methylation ratio |
|---|---|---|---|---|---|---|---|---|---|
| LTNP/EC | A | LTNP | 21 | No | 1,047 (22) | 209 | 9.2 | 1.03 | 8.93 |
| B | LTNP | 13 | No | 630 (35) | 1,965 | 0 | 1.65 | 0 | |
| C | LTNP | 22 | No | 670 (28) | 847 | 8.5 | 0.86 | 9.88 | |
| D | LTNP | 24 | No | 765 (41) | 8,195 | 10.7 | 1.25 | 8.56 | |
| E | LTNP | 7 | No | 1,029 | 750 | 1.8 | |||
| F | EC | 22 | No | 570 (30) | <50 | 9 | 1.66 | 5.42 | |
| G | EC | 19 | No | 1,520 (38) | <50 | 1.43 | 1.08 | 1.32 | |
| H | EC | 17 | No | 1,162 (45) | <50 | 2.3 | 0.63 | 3.65 | |
| Median | 20 | 897* | 479.5 | 5.4* | 1.8 | 5.42* | |||
| Progressors | I | Progressor | 22 | Yes (2) | 578 (27) | <50 | 1 | 1.37 | 0.73 |
| J | Progressor | 21 | Yes (16) | 194 (16) | <50 | 1 | 0.88 | 1.14 | |
| K | Progressor | 8 | Yes (3) | 819 (43) | <50 | 0 | 1.23 | 0 | |
| L | Progressor | 21 | Yes (1) | 586 (36) | <50 | 0 | 1.87 | 0 | |
| M | Progressor | 10 | Yes (1) | 412 (15) | 174 | 0 | 2.6 | 0 | |
| Median | 21 | 2 | 578* | <50 | 0* | 1.37 | 0* |
VL, viral load; ID, patient identification;
, P < 0.05 in the Mann-Whitney test.
Total genomic DNA from each patient was isolated with a QIAamp DNA blood minikit (Qiagen Inc., Valencia, CA), and approximately 1 μg of this DNA was prepared for bisulfite treatment using an Epitect bisulfite kit (Qiagen Inc., Valencia, CA) by following the manufacturer's instructions. Bisulfite-treated DNA was amplified by a nested PCR using conditions and modified primers spanning the LTR region, described elsewhere (1). PCR products were cloned in the pSC-A-amp/kan vector system (StrataClone, Life Technologies). Ten clones from each sample were sequenced in an ABI 3100 genetic analyzer. Only clones with at least 95% conversion of cytosines outside CpG islands were used for the analysis of methylation in order to avoid technical bias with the bisulfite treatment, and the percentage of methylated CpGs was calculated as described elsewhere (4). From an initial group of 11 patients selected from the LTNP/EC cohort from Hospital Carlos III (15), only 8 patients could be analyzed. It is likely that the low percentage of HIV provirus (many LTNP/EC patients had undetectable proviral loads) hampered the analysis of methylation in some of the analyzed patients. Also, similar problems were encountered when analyzing progressors (HAART-treated patients) with low viral loads; of an initial group of 7 patients, only 5 could be analyzed. The Mann-Whitney test was used to analyze differences between groups. Medians of viral load were 479.5 RNA copies/ml (interquartile range [IQR] of <50 to 1,685.5 RNA copies/ml) for LTNP/EC and <50 RNA copies/ml (IQR of <50 to 111.5 RNA copies/ml) for progressors, and the CD4+ T cell count medians were 897 cells/μl for LTNP/EC and 578 cells/μl for progressor patients (Table 1). The 5′-LTR of HIV-1 in all progressor patients showed very low or no methylation at all (range of 0 to 1%) compared with that of the LTNP/EC group, where the percentage of methylated CpGs in the HIV promoter was higher. In this group, the median of CpG methylation was 5.4% (IQR of 1.52 to 9.15%) (P = 0.011) (Fig. 1). Despite the moderate DNA methylation levels in the HIV promoter observed in our LTNP/EC patients, we demonstrated a difference with progressors, indicating a certain silencing of HIV LTR in LTNP/EC. The observed DNA methylation level in our patients is different from the levels observed by Blazkova and colleagues in 2009 in aviremic patients (1) but more similar to the new study by the same authors (2), although comparison is difficult since analyzed cell types are not the same (PBMCs in our study versus memory CD4+ T cells in both of Blazkova et al.'s works). Most of the methylated CpGs found in our patients were within the Sp1 binding sites and second CpG island, which could reduce activation by Sp1 or impeding transcription by recruitment of methyl-CpG binding domain protein 2 (MBD2) (Fig. 2). The stability of gene expression contributing to these patients' viral control may correlate more closely with the density of DNA methylation, as opposed to a binary methylated/demethylated state.
Fig 1.
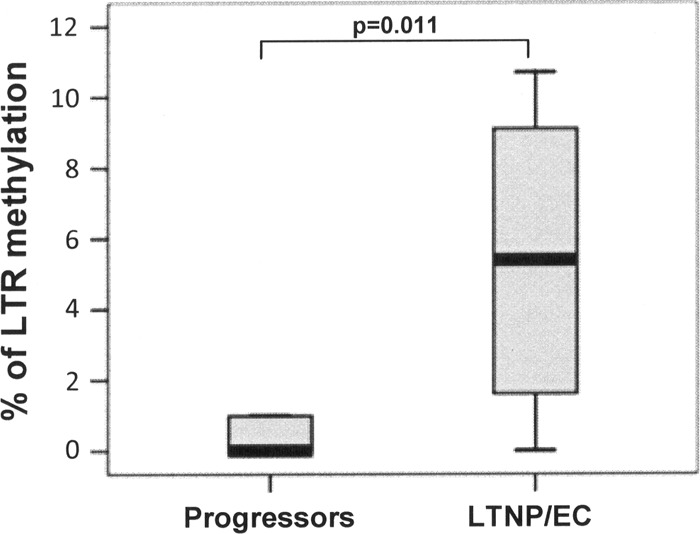
Percentage of CpG methylation (5-mCpG) in the 5′-LTR of HIV-1 in LTNP/EC (n = 8) and progressor (n = 5) patients.
Fig 2.

Level of HIV DNA methylation in the analyzed LTNP/EC individuals. (Top) 5′-LTR HIV region, approximate location of CpG sites susceptible of methylation (open circles) along with the regulatory regions within the LTR based on the HXB2 strain. NF-AT1/NRE/RBF-1 (nucleotides [nt] 237 to 338), NF-κB I and II (nt 350 to 373), Sp1 I, II, III (nt 377 to 407). (Bottom) LTNP/EC patients showing the highest degree of CpG methylation after bisulfite treatment. Closed and open circles represent methylated and nonmethylated CpG residues, respectively.
Next, the total DNA sample from each patient was also analyzed for quantification of global DNA methylation (5-mC). The MethylFlash methylated DNA quantification kit (Epigentek Inc., Farmingdale, NY) was used for this purpose. No differences were observed in the degree of global DNA methylation between the groups, which range from a median of 1.8% in LTNP/EC to 1.37% in progressors (P = 0.268). These values fell, in all patients except one, within the normal range of total DNA methylation (0.8 to 1.5%). Only patient M presented a slight hypermethylation (>2%). The ratio of LTR methylation with respect to total DNA methylation also showed differences between the groups (median ratio of 5.42 for LTNP/EC, 0 for progressors; P = 0.018). These results show that the higher methylation observed in LTNP/EC is not due to differences in the overall methylation of PBMCs, and hence their state of stimulation, but most likely tied to a local event related to HIV LTR.
Because of the low number of patients, we were unable to discern methylation patterns between LTNP and EC patients (median of 8.5% for LTNP versus median of 2.3% for EC; P = 0.786) or with their ratio of LTR methylation with respect to total DNA methylation (median of 9.88 for LTNP versus median of 3.6 for EC; P = 0.4). Also, no correlations between viral load and percentage of LTR methylation were observed when analyzing the total number of patients or within the LTNP/EC group. In the same manner, no correlation was observed with the degree of methylation and levels of CD4+ T cells. However, when analyzing the time of infection in all our patients (median of 20 years for LTNP/EC, 21 years for progressors), we observed a positive correlation with LTR CpG methylation (Spearman rank correlation rho of 0.661; P = 0.019). This correlation was primarily driven by the LTNP/EC group (rho = 0.802; P = 0.017). It seems that DNA methylation accumulates over time in these patients with controlled viremia over a long period of infection evolution. This DNA methylation of HIV LTR may be contributing to the reduction in the number of reactivable viral events over time and hence allowing a more efficient immune response with minimal activation, resulting in a delay or nonprogression of infection. Although the increase in methylation over time in our patients was observed only in the LTNP group, we should not exclude that this event may also be observed in patients on HAART with a long evolution under controlled viremia. In fact, this factor may help explain the different methylation ratios found in other studies in patients on HAART, although analyses with more progressor patients are needed to confirm this.
In the first of Blazkova et al.'s works, the presence of different populations of nonmethylated and methylated provirus within the same patient was reported when the observed LTR methylation was in an intermediate range (20 to 30%) (1), suggesting the presence of mixed virus populations. To explore this possibility in our patients, we further analyzed the sequence variability of HIV-1 promoters in the sequences obtained after bisulfite treatment. For this analysis, we used the neighbor-joining method to compare distances between sequences. The LTR sequences clearly segregated into two different clusters only in patients C and D (both LTNP); however, both clusters showed a mix of methylated and nonmethylated positions (Fig. 3). These 2 patients showed the highest methylation, but our results do not suggest the presence of two different proviral populations regarding promoter methylation.
Fig 3.

Phylogenetic tree of HIV-1 LTR sequences for each patient. Patients with LTR methylation are shown in black.
In summary, our data show that LTNP/EC patients carry some degree of HIV LTR methylation compared with progressor patients, who show no DNA methylation at all. Although levels observed in the LTNP/EC are moderate, the differences with progressors suggest that DNA methylation in the HIV 5′-LTR of LTNP/EC patients may participate along with other mechanisms in the control of viral replication. This methylation process may help in silencing some of the HIV proviruses, which in turn would favor a more efficient immunologic control. This greater response efficiency may result in a lower activation of the immune system and as a consequence in reduced disease progression.
ACKNOWLEDGMENTS
This work was supported by Fondo de Investigación Sanitaria (FIS) project PI09/00278 from the Instituto de Salud Carlos III.
B.R. designed research; J.A.P., T.P.-P., and B.S.-M. performed research; V.M., E.V., and C.G.-H. recruited and monitored study subjects; and J.A.P., C.T., and B.R. analyzed data and wrote the paper.
The authors declare no conflicts of interest.
Footnotes
Published ahead of print 12 September 2012
REFERENCES
- 1. Blazkova J, et al. 2009. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 5:e1000554 doi:10.1371/journal.ppat.1000554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blazkova J, et al. 2012. Paucity of HIV DNA methylation in latently infected, resting CD4+ T cells from infected individuals receiving antiretroviral therapy. J. Virol. 86:5390–5392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cao Y, Qin L, Zhang L, Safrit J, Ho DD. 1995. Virologic and immunologic characterization of long-term survivors of human immunodeficiency virus type 1 infection. N. Engl. J. Med. 332:201–208 [DOI] [PubMed] [Google Scholar]
- 4. Chávez L, Kauder S, Verdin E. 2011. In vivo, in vitro, and in silico analysis of methylation of the HIV-1 provirus. Methods 53:47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Colin L, Van Lint C. 2009. Molecular control of HIV-1 postintegration latency: implications for the development of new therapeutic strategies. Retrovirology 6:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deeks SG, Walker BD. 2007. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 27:406–416 [DOI] [PubMed] [Google Scholar]
- 7. Duverger A, et al. 2009. Determinants of the establishment of human immunodeficiency virus type 1 latency. J. Virol. 83:3078–3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Friedman J, et al. 2011. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 85:9078–9089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Imai K, Togami H, Okamoto T. 2010. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 285:16538–16545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ishida T, Hamamo A, Koiwa T, Watanabe T. 2006. 5′ Long terminal repeat (LTR)-selective methylation of latently infected HIV-1 provirus that is demethylated by reactivation signals. Retrovirology 3:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Karn J. 2011. The molecular biology of HIV latency: breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin. HIV AIDS 6:4–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kauder SE, Bosque A, Lindqvist A, Planelles V, Verdin E. 2009. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 5:e1000495 doi:10.1371/journal.ppat.1000495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lambotte O, et al. 2005. SEROCO-HEMOCO Study Group. HIV controllers: a homogeneous group of HIV-1-infected patients with spontaneous control of viral replication. Clin. Infect. Dis. 41:1053–1056 [DOI] [PubMed] [Google Scholar]
- 14. Pantaleo G, et al. 1995. Studies in subjects with long-term nonprogressive human immunodeficiency virus infection. N. Engl. J. Med. 332:209–216 [DOI] [PubMed] [Google Scholar]
- 15. Rodés B, et al. 2004. Differences in disease progression in a cohort of long-term non-progressors after more than 16 years of HIV-1 infection. AIDS 18:1109–1116 [DOI] [PubMed] [Google Scholar]
- 16. Taniguchi Y, et al. 2005. Silencing of human T-cell leukemia virus type I gene transcription by epigenetic mechanisms. Retrovirology 2:64. [DOI] [PMC free article] [PubMed] [Google Scholar]