

Abstract
It is widely appreciated that memory processing engages a wide range of molecular signaling cascades in neurons, but how these cascades are temporally and spatially integrated is not well understood. To explore this important question, we used Aplysia californica as a model system. We simultaneously examined the timing and subcellular location of two signaling molecules, MAPK (ERK1/2) and protein kinase A (PKA), both of which are critical for the formation of enduring memory for sensitization. We also explored their interaction during the formation of enduring synaptic facilitation, a cellular correlate of memory, at tail sensory-to-motor neuron synapses. We find that repeated tail nerve shock (TNS, an analog of sensitizing training) immediately and persistently activates MAPK in both sensory neuron somata and synaptic neuropil. In contrast, we observe immediate PKA activation only in the synaptic neuropil. It is followed by PKA activation in both compartments 1 h after TNS. Interestingly, blocking MAPK activation during, but not after, TNS impairs PKA activation in synaptic neuropil without affecting the delayed PKA activation in sensory neuron somata. Finally, by applying inhibitors restricted to the synaptic compartment, we show that synaptic MAPK activation during TNS is required for the induction of intermediate-term synaptic facilitation, which leads to the persistent synaptic PKA activation required to maintain this facilitation. Collectively, our results elucidate how MAPK and PKA signaling cascades are spatiotemporally integrated in a single neuron to support synaptic plasticity underlying memory formation.
During signal transduction, single molecules often generate different cellular effects, depending on their temporal dynamics, spatial distribution, and interacting partners (1). In considering the wide range of molecules implicated in memory processing, the question of how multiple signaling cascades are integrated in time and space to contribute to memory formation and its underlying synaptic plasticity remains a fundamental issue.
We have begun to explore this general question in Aplysia californica, a model system well suited for mechanistic analyses of simple forms of learning. We focused on two molecules, MAPK (ERK1/2) and protein kinase A (PKA), both known to be engaged in many forms of memory and synaptic plasticity (2–4). Recent studies, however, suggest the timing, cellular location, and cross-talk between these kinases are critical in determining their ultimate effects (5–10). Thus, in addition to knowing that MAPK and PKA are required, it also is important to understand their spatiotemporal dynamics and their interactions during memory formation.
Aplysia provides unique advantages for analyzing these questions. In Aplysia, memory for sensitization induced by tail shock (TS) is supported in large measure by synaptic facilitation at identified tail sensory-to-motor neuron (SN-MN) synapses (11). As an analog of behavioral training, tail nerve shock (TNS) also induces synaptic facilitation (12–14). A single TNS induces short-term facilitation (STF) lasting <30 min, whereas repeated spaced TNS induces intermediate-term (ITF) and long-term facilitation (LTF) lasting hours and days, respectively. TS/TNS triggers the release of serotonin (5-HT) around SN soma and SN-MN synapses, which activates a series of signaling cascades, including MAPK and cAMP/PKA (11, 12). MAPK activation is required for the formation of ITF and LTF, but not for STF, whereas cAMP/PKA is required for all three (15–18). Finally, although signaling in the synaptic compartment is critical for all forms of synaptic facilitation, it has not yet been established that MAPK and PKA can indeed be activated and exert their function locally at the SN-MN synapse. Nor is it known how they interact with each other during synaptic facilitation.
In the present paper, we simultaneously examined MAPK and PKA activation in two subcellular compartments (SN soma and synaptic neuropil) at two time points (immediately and 1 h) after TNS. We found that MAPK was activated immediately and persistently in both compartments after repeated TNS. In contrast, although immediate and persistent PKA activation by repeated TNS also occurred in synaptic neuropil, we observed only delayed PKA activation in SN soma. Interestingly, MAPK activation during, but not after, TNS was essential for synaptic, but not somatic, PKA activation. Synaptic integration of these two signaling cascades in turn led to ITF. These results provide unique insights into both the spatial and temporal features of these two critical molecular cascades, and suggest a model of how they interact to regulate synaptic plasticity underlying memory formation.
Results
Spatiotemporal Profile of MAPK Activation.
As a first step in the analysis of spatiotemporal integration of the MAPK and PKA signaling cascades contributing to synaptic facilitation, we examined whether TNS activates MAPK and PKA with overlapping spatiotemporal profiles. We chose TNS for this study for two reasons: (i) it induces the same key features of sensitization as behavioral TSs, including 5-HT release, enhanced neuronal excitability, and synaptic facilitation, while providing additional advantages of repeatability and temporal precision (12, 19); and (ii) it provides the experimental advantage of allowing the use of reversible inhibitors during training. We harvested both the pleural sensory cluster, which is enriched in somata of SNs (SN soma), and neuropil underneath the pedal MN cluster, which is relatively enriched with SN-MN synapses (synaptic neuropil) (12), at two different time points: immediately or 1 h after TNS. As a verification of our tissue samples (Fig. S1A), both SN soma and synaptic neuropil were enriched in the mRNA of sensorin, an SN-specific neuropeptide (20). In addition, synaptic neuropil was enriched with vesicle-associated membrane protein, a synaptic protein marker. MAPK activation in each sample was analyzed by Western blotting and revealed by an increase in its phosphorylation. In parallel, PKA activation in the same samples was measured by modified PepTag PKA assay (Fig. S1B).
We found that five temporally spaced TNSs induced significant MAPK activation in SN soma that was observed immediately after the last TNS (178.8 ± 19.7%, n = 7, P = 0.034) and lasted for at least 1 h (178.9 ± 12.2%, n = 11, P < 0.001) (Fig. 1A1), consistent with previous findings that repeated TS or 5-HT induces persistent MAPK activation in SN soma (15). In addition, we observed immediate and persistent MAPK activation in synaptic neuropil (immediate: 160.5 ± 9.8%, n = 10, P = 0.006; 1 h: 169.2 ± 16.4%, n = 13, P = 0.002). There was no significant difference in the level of MAPK activation observed between either different time points or compartments. In contrast, MAPK activation was not observed in either compartment, either immediately (SN soma: 110.9 ± 29.6%, n = 8, P = 0.969; synaptic neuropil: 109.0 ± 16.1%, n = 8, P = 0.891) or at 1 h (SN soma: 109.7 ± 12.1%, n = 6, P = 0.656; synaptic neuropil: 89.7 ± 5.6%, n = 6, P = 0.202) after a single TNS (Fig. 1A2). (However, Philips et al. (10) show that a single shock does activate MAPK in a specific narrow temporal window at 45 min.) These findings support the hypothesis that MAPK signaling is required for long-lasting facilitation after repeated-trial training, but not for STF induced by a single training trial (15).
Fig. 1.

Spatiotemporal profiles of MAPK activation by TNS. (A1) MAPK activation is observed immediately (imm) and at 1 h (1hr) after repeated TNS in both SN soma and synaptic neuropil (Syn. Np). (A2) A single TNS does not give rise to immediate or 1-h MAPK activation in either compartment. (B) The presence of U0126 during, but not after, repeated TNS blocks 1-h MAPK activation, suggesting that persistent MEK activation is not required for sustained MAPK activation by TNS. (C) Somatic U0126 significantly blocks MAPK activation in SN soma without affecting MAPK activation in synaptic neuropil, and vice versa, suggesting independent activation of somatic and synaptic MAPK.
Persistent MAPK activation might be mediated by sustained activation of upstream signaling (21). To test this possibility, we applied U0126, an inhibitor of MEK (the upstream activator of MAPK), or its inactive analog, U0124, either 30 min before and during repeated TNS, or immediately after TNS until harvesting. The presence of U0126 during training abolished the MAPK activation normally seen 1 h following TNS in both SN soma (U0124: 226.0 ± 54.7%, n = 7, P = 0.016; U0126: 117.9 ± 8.5%, n = 6, P = 0.065) and synaptic neuropil (U0124: 149.0 ± 17.9%, n = 7, P = 0.031; U0126: 104.4 ± 5.9%, n = 7, P = 0.826) (P < 0.05 U0124 vs. U0126). However, application of U0126 after TNS had no effect (SN soma: 267.6 ± 38.7%, n = 6, P = 0.011; synaptic neuropil: 192.7 ± 26.2%, n = 6, P = 0.014; P < 0.05 U0126 during vs. post TNS) (Fig. 1B) (a 2 × 2 ANOVA [treatment: F(2,34) = 8.0, P = 0.001; compartment: F(1,34) = 4.7, P = 0.037; treatment × compartment, F(2,34) = 0.81, P = 0.454] followed by Bonferroni’s post hoc test). These data indicate that MAPK is activated by MEK during TNS, but is not actively maintained through upstream MEK activity at least out to 1 h post training.
The detection of increased MAPK activity in both SN soma and synaptic neuropil by TNS might be the result of MAPK activation by local signaling in each compartment and/or translocation of active MAPK between the compartments. To distinguish between these two possibilities, we used a two-chamber dish (22), which permits restriction of U0126 to one compartment or the other. We found that blocking TNS-induced MAPK activation in the somatic compartment did not affect MAPK activation in synaptic neuropil (somatic U0126—SN soma: 107.4 ± 6.7%, n = 7, P = 0.677; synaptic neuropil: 182.2 ± 19.5%, n = 7, P = 0.005), and the reverse also was observed (synaptic U0126—SN soma: 154.7 ± 16.6%, n = 7, P = 0.027; synaptic neuropil: 104.5 ± 5.7%, n = 7, P = 0.490) (Fig. 1C), indicating that MAPK was activated locally in each compartment.
Spatiotemporal Profile of PKA Activation.
In the same samples in which we analyzed the spatiotemporal recruitment of MAPK by TNS, we also measured PKA activation. As shown in Fig. 2A, repeated TNS induced significant PKA activation in synaptic neuropil that was observed immediately after the last TNS (136.4 ± 8.7%, n = 12, P = 0.004), and persisted for at least 1 h (124.2 ± 9.1%, n = 14, P = 0.038). In contrast, there was no significant change in PKA activity in SN soma immediately after the last TNS (91.0 ± 5.8%, n = 9, P = 0.282), whereas there was a delayed increase in somatic PKA activity at 1 h (112.6 ± 5.4%, n = 13, P = 0.035).
Fig. 2.
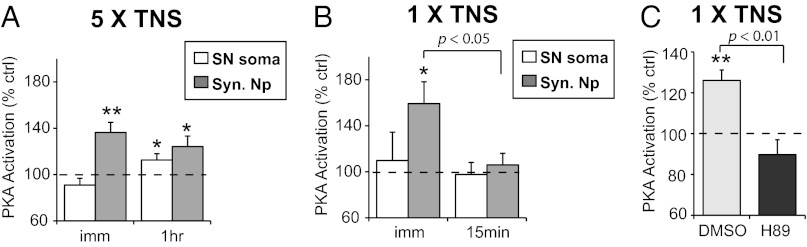
Spatiotemporal profiles of PKA activation by TNS. (A) PKA activation is observed immediately and at 1 h after repeated TNS in synaptic neuropil (Syn. NP), whereas in SN soma PKA activation is delayed, observed at 1 h post TNS. (B) A single TNS induces transient PKA activation in synaptic neuropil that returns to baseline by 15 min. (C) The transient PKA activation in synaptic neuropil by a single TNS is blocked by H89, a PKA inhibitor.
Consistent with the notion that PKA is engaged in both short-term and long-lasting plasticity (16–18), we also observed transient PKA activation after a single TNS. Interestingly, this activation occurred specifically in synaptic neuropil (159.3 ± 19.0%, n = 8, P = 0.022), but not in SN soma (109.8 ± 24.6%, n = 7, P = 0.965), and returned to baseline in synaptic neuropil within 15 min (106.0 ± 10.1%, n = 8, P = 0.211) (Fig. 2B). In addition, in the presence of H89, a PKA inhibitor, the PKA activation in the synaptic compartment after a single TNS was blocked (vehicle: 126.0 ± 5.1%, n = 8, P < 0.001; H89: 89.7 ± 7.3%, n = 8, P = 0.167; P = 0.001 between groups) (Fig. 2C).
Interaction Between MAPK and PKA Activation.
The analysis of the individual spatiotemporal profiles of MAPK and PKA activation now permitted the identification of potential functional interactions between these signaling cascades. Specifically, we examined their interactions following repeated TNS, because (i) both kinases are implicated in long-lasting synaptic facilitation induced by repeated training (15, 16, 18), and (ii) they are activated with overlapping spatiotemporal profiles by this pattern of training (Figs. 1 and 2).
We first asked whether MAPK activation induced by TNS regulates PKA activation. We applied U0126 during repeated TNS to block MAPK activation, and examined how it might affect PKA activation. Interestingly, U0126 completely abolished PKA activation in synaptic neuropil observed at both immediate (vehicle: 129.0 ± 7.8%, n = 11, P = 0.021; U0126: 95.4 ± 4.4%, n = 11, P = 0.126) and 1-h (vehicle: 126.4 ± 9.7%, n = 10, P = 0.018; U0126: 102.2 ± 5.8%, n = 10, P = 0.749) time points (P < 0.05 vehicle vs. U0126) after repeated TNS (Fig. 3A) (a 2 × 2 ANOVA [drug: F(1,39) = 7.3, P = 0.010; time: F(1,39) = 0, P = 1; drug × time, F(1,39) = 0.59, P = 0.447) followed by Bonferroni’s post hoc test]. Surprisingly, this effect was specific to the synaptic neuropil and did not affect the increase in somatic PKA activity observed at 1 h post TNS (vehicle: 135.2 ± 15.7%, n = 11, P = 0.045; U0126: 127.4 ± 11.2%, n = 9, P = 0.040; P = 0.690 between groups) (Fig. 3A). Although both somatic and synaptic regions were uniformly exposed to U0126, we considered the possibility that MAPK activation may not have been disrupted in the SN soma. This, however, was not the case as we confirmed that this treatment resulted in a uniform disruption of MAPK activation (Fig. S2A). Collectively, these data suggest that the increases in PKA activity in different compartments after TNS are regulated by distinct molecular processes.
Fig. 3.

Interaction between MAPK and PKA activation induced by repeated TNS. (A) U0126 during repeated TNS blocks PKA activation in synaptic neuropil, but has no effect on PKA activation in SN soma. (B) The presence of H89, a PKA inhibitor, does not affect MAPK activation by repeated TNS in either compartment.
Several lines of evidence show that our results are not due to nonspecific effects of U0126. First, a whole-bath application of U0126 had differential effects on PKA activation in different compartments. Second, similar to our earlier finding that post-TNS application of U0126 had no effect on 1-h MAPK activation (Fig. 1B), it also did not affect 1-h PKA activation in synaptic neuropil (vehicle: 140.8 ± 12.0%, n = 8, P = 0.023; U0126: 137.3 ± 12.1%, n = 8, P = 0.026; P = 0.838 between groups) (Fig. S2B). Third, application of U0126 alone did not affect basal PKA activity either in SN soma (103.5 ± 7.5%, n = 7, P = 0.959) or in synaptic neuropil (107.6 ± 4.1%, n = 7, P = 0.081) (Fig. S2C). Finally, the presence of U0126 during a single TNS did not affect the transient PKA activation in synaptic neuropil (vehicle: 130.1 ± 4.5%, n = 6, P = 0.033; U0126: 124.2 ± 4.2%, n = 6, P = 0.026; P = 0.356 between groups) (Fig. S2D).
In a reciprocal fashion, we examined the requirement of PKA for MAPK activation induced by repeated TNS. We used H89 to block PKA activity during and after repeated TNS, and asked how it affected MAPK activation. We found that H89 did not affect MAPK activation by repeated TNS either in SN soma (immediate—vehicle: 180.1 ± 25.8%, n = 10, P = 0.006; H89: 180.6 ± 21.6%, n = 10, P = 0.004; 1 h: vehicle: 183.4 ± 30.0%, n = 6, P = 0.015; H89: 184.9 ± 22.4%, n = 8, P = 0.001), or in synaptic neuropil (immediate—vehicle: 144.2 ± 13.2%, n = 10, P = 0.010; H89: 129.0 ± 11.0%, n = 10, P = 0.035; 1 h: vehicle: 154.6 ± 25.8%, n = 6, P = 0.011; H89: 157.5 ± 15.2%, n = 8, P = 0.007). There is no significant difference in the level of MAPK activation observed in the presence of vehicle versus H89 at either time point or in either compartment (Fig. 3B), although, as shown above, H89 blocked TNS-induced PKA activation (Fig. 2C).
Collectively, these findings reveal a unique functional interaction between MAPK and PKA activation induced by repeated TNS: MAPK activation during repeated TNS is essential for persistent PKA activation after repeated TNS. This interaction occurs exclusively in synaptic neuropil, but not in SN soma.
Functional Significance of Synaptic Integration of MAPK to PKA Signaling.
Our molecular analysis revealed a unique integration of MAPK to PKA signaling specifically restricted to the synaptic neuropil after repeated TNS. Thus, next we asked how this integration functionally contributed to synaptic facilitation. To answer this question, we examined ITF, a phase of synaptic facilitation that lasts 1–2 h after repeated TNS. Importantly, Sherff and Carew (22) have shown that ITF can be induced by local application of repeated pulses of 5-HT in the synaptic compartment, and it requires local translation at SN-MN synapses but not transcription in SN soma. Thus, this form of ITF provides a good target for examining the role of synaptic signaling cascades in plasticity. Furthermore, both MAPK and PKA previously were shown to play important roles in ITF/intermediate-term memory induced by repeated-trial training (15, 16). However, the specific requirement of MAPK and PKA locally at the synapses in the induction and maintenance of ITF has not yet been examined.
We recorded from pairs of SNs and MNs, and applied inhibitors restricted to the synaptic compartment of a two-chamber dish (22) (Fig. 4A). Based on previous studies (15, 16) and our molecular analysis (Figs. 1–3), we hypothesize that synaptic MAPK activation during TNS is required for the induction of ITF, which leads to downstream persistent synaptic PKA activation after TNS that supports the maintenance of ITF. Indeed, we found that U0126 applied during TNS in the synaptic compartment completely blocked ITF (vehicle: 176.9 ± 18.9%, n = 6, P = 0.010; U0126: 85.0 ± 5.2%, n = 5, P = 0.044; P < 0.01 between groups) without affecting STF (vehicle: 282.5 ± 17.4%, n = 7, P < 0.001; U0126: 290.4 ± 32.9%, n = 5, P = 0.004; not significant [NS] between groups) (Fig. 4B) (a 2 × 2 ANOVA [drug: F(1,22) = 4.99, P = 0.038; time: F(1,22) = 54.2, P < 0.001; drug × time, F(1,22) = 4.66, P = 0.044] followed by Bonferroni’s post hoc test). Furthermore, to examine the requirement of synaptic PKA activation in ITF maintenance, after establishing ITF (ITF-1 tested at 20–35 min post TNS), we applied either H89 or its vehicle only to the synaptic compartment; a second ITF test (ITF-2) was delivered 35 min later. Although ITF was stable in the control preparations (ITF-1: 172.3 ± 16.7%, n = 7, P = 0.005; ITF-2: 158.6 ± 18.3%, n = 6, P = 0.024; NS between the two tests), H89 blocked the maintenance of ITF, causing it to return promptly to baseline levels (first ITF: 157.9 ± 12.0%, n = 7, P = 0.003; second ITF: 99.8 ± 7.2%, n = 6, P = 0.983; P < 0.01 between the two tests) (Fig. 4C) (a 2 × 2 ANOVA [drug: F(1,25) = 6.24, P = 0.021; time: F(1,25) = 5.93, P = 0.023; drug × time, F(1,25) = 2.39, P = 0.136] followed by Bonferroni’s post hoc test). Finally, blocking synaptic PKA activation with H89 during a single TNS significantly reduced STF (vehicle: 357.8 ± 61.5%, n = 7, P = 0.006; H89: 190.4 ± 27.2%, n = 7, P = 0.016; P = 0.037 between groups) (Fig. 4D). Thus, these findings suggest that synaptic PKA activation is required for STF and the maintenance of ITF.
Fig. 4.

Requirement of synaptic MAPK and PKA activation for synaptic facilitation. (A) A diagram depicting intracellular recording from paired SNs and MNs in a two-chamber dish with application of inhibitors (shaded region) restricted to the synaptic compartment. (B) U0126 in the synaptic compartment during repeated TNS blocks ITF induction, without affecting STF. (C) H89 in the synaptic compartment after repeated TNS blocks ITF maintenance. (D) Blocking synaptic PKA activation by H89 significantly attenuats STF induced by a single TNS. Representative traces of synaptic facilitation are shown for each experiment. Data are presented as mean percent ± SEM of baseline EPSP.
Discussion
Our findings are summarized in the model shown in Fig. 5. Collectively, our data suggest the view that following repeated TNS, MAPK is immediately and persistently activated in both somatic and synaptic compartments. In parallel, PKA is immediately and persistently activated in the synaptic compartment. However, in the somatic compartment PKA activation is delayed. Importantly, we have identified a specific form of spatiotemporal integration of these two cascades: activation of MAPK at SN-MN synapses during TNS is essential for the persistent increase in synaptic PKA activity after TNS, the combined effects of which lead to the formation of ITF. The form of training we have used (repeated TNS) also gives rise to LTF, which is both transcription and translation dependent (11). Given the previously identified role of MAPK and PKA in transcriptional regulation (23–25), our model is consistent with the view that somatic MAPK and PKA activation may converge on transcriptional machinery to support LTF formation. In contrast, a single TNS induces only transient synaptic PKA activation without coincident MAPK activation, resulting only in STF.
Fig. 5.

Model depicting the spatiotemporal integration of MAPK and PKA signaling cascades in mediating synaptic facilitation. Repeated TNS triggers the release of endogenous 5-HT, which binds to 5-HT receptors on the membranes of SN soma and SN-MN synapses to activate adenylate cyclase (AC)-dependent intracellular signaling cascades, including MAPK and PKA. At the synapse, transient PKA activation leads to STF (red arrows). In addition, synaptic MAPK activation resulting from repeated TNS is required to sustain synaptic PKA activation, resulting in ITF (green arrows). Repeated TNS also induced persistent somatic MAPK activation and delayed somatic PKA activation, which we posit converge on regulation of transcriptional machinery that leads to LTF (blue arrows).
Spatiotemporal Regulation of MAPK and PKA Signaling in Memory Processing.
MAPK and PKA activation is critical for synaptic plasticity and memory formation in Aplysia, as well as many other systems (2–4). Although these kinases are thought to function in both somatic and synaptic compartments, previous studies often measured their activation in SN soma as a proxy for their activation at synapses (15, 26). However, given these distinct molecular environments, it is likely that these molecules are coupled with different upstream and downstream elements in different compartments.
In the present study, in addition to confirming previous findings of MAPK activation in SN soma by repeated TNS, we found comparable MAPK activation in synaptic neuropil. Importantly, inhibiting synaptic MAPK activation (without affecting somatic MAPK activation) during TNS blocked the induction of ITF. Collectively, these findings highlight the importance of synaptic MAPK signaling in plasticity and support the hypothesis that MAPK may regulate local translation and synaptic adhesion proteins during memory formation (27, 28). Furthermore, we showed that in both compartments, TNS-induced MAPK activation persisted for at least 1 h. In addition, post-TNS application of U0126 had no effect on 1-h MAPK activation, suggesting that persistent MAPK activation did not rely on persistent upstream MEK signaling. One intriguing possibility is that repeated TNS may lead to inhibition of protein phosphatases, which critically control the duration of MAPK activation and can be regulated by plasticity-related stimuli (29–31).
Interestingly, we found differential profiles of PKA activation in SN soma and synaptic neuropil. Immediately after a single or repeated TNS, PKA was activated in synaptic neuropil, but not in SN soma, whereas at 1 h after repeated TNS, PKA activity was elevated in both compartments. A previous study from our laboratory showed that PKA was rapidly activated in SN soma by exogenous 5-HT application (26). Thus, if provided a sufficient amount of 5-HT, PKA also may be activated in SN soma. However, our findings suggest that TNS, which, similar to TS, triggers comparable endogenous 5-HT release onto SN soma and SN-MN synapses (12, 19), gives rise to preferential activation of PKA in synaptic neuropil compared with SN soma. Although the mechanisms underlying this phenomenon are as yet unknown, our preliminary analysis reveals that PKA catalytic subunit is highly enriched in neuropil, which may contribute to its preferential activation in this compartment.
To our surprise, we found that although PKA activity was increased in both SN soma and synaptic neuropil at 1 h after repeated TNS, MAPK activation was required only for synaptic, but not for somatic, PKA activation. We hypothesize that this finding is a result of the differential distribution of the regulatory machinery of PKA in the two compartments. It is known that there are two types of PKA, differing in their regulatory (R) subunits. RI is mostly soluble in the soma, whereas RII is enriched at nerve terminals (32). Repeated or prolonged 5-HT induces degradation of RI, relieving its inhibition of the catalytic subunit, thus giving rise to increased PKA activity (18, 33). Similar mechanisms may underlie the delayed increase in somatic PKA activity after repeated TNS. In contrast, 5-HT does not lead to RII degradation (33), suggesting that a different mechanism may be engaged to prolong synaptic PKA activation. One critical regulator of the duration of synaptic PKA activation is phosphodiesterase-4 (PDE4), which can be phosphorylated by MAPK, in turn reducing its activity (34). Inhibition of PDE4 can rescue memory deficits produced by blocking hippocampal MAPK activation (35). Furthermore, RII binding to microtubules via microtubule-associated protein 2 (MAP2) is important for maintaining synaptic PKA activation (36). MAPK phosphorylation of MAP2 regulates its interaction with microtubules (37). Similar mechanisms may account for MAPK regulation of persistent synaptic PKA activation by repeated TNS. Overall, our findings expand the original view that PKA acts primarily (if not exclusively) as a freely diffusible molecule. Rather, our results agree with recent findings that PKA signaling cascades can be spatially organized in microdomains, which potentially increases their signaling specificity and diversity (1, 6, 38).
Finally, synaptic plasticity at SN-MN synapses may be induced in both an activity-dependent and activity-independent manner (16). In our synaptic experiments, most SNs did not fire action potentials during TNSs. Thus, our molecular data mostly represent activity-independent signaling events. In support of this notion, we did not find MAPK activation either immediately or at 1 h following a single TNS, which is reliably observed following a single trial of activity-dependent training (39). However, we cannot exclude the possibility of a modest contribution of activity-dependent signaling to our molecular data.
Molecular Routing of Signaling Cascades.
It commonly has been observed across a range of systems that synaptic plasticity and memory formation may be induced by different patterns of training, which often may engage different molecular signaling cascades and share common mechanisms. These observations raise two interesting questions: (i) How do different training patterns funnel through a common downstream signaling molecule? (ii) How do signals from a single molecule diverge to subserve different forms of plasticity? Recent studies from Aplysia begin to provide some clues to these questions.
In Aplysia, ITF may be induced either following repeated 5-HT/TNS or by a single pulse of 5-HT combined with KCl depolarization (KCl + 5-HT). Importantly, the induction of both forms of ITF requires MAPK activation (15, 39), whereas the maintenance of each form differentially requires either PKA or PKC, respectively (16). Recently, we found that in response to repeated 5-HT, MAPK was activated under a specific set of conditions involving coactivation of two G proteins (ApRas and ApRap), but only when ApRas activation exceeded that of ApRap. This relationship was reversed for MAPK activation by KCl + 5-HT (40). Furthermore, MAPK activated by KCl + 5-HT was required for membrane translocation of PKC, a critical step for PKC activation (39). Finally, in the present paper, we demonstrate a requirement of MAPK activation for persistent synaptic PKA activation, which results in ITF formation by repeated TNS. Taking these studies together, one plausible explanation is that the differential engagement of ApRas and ApRap in response to different patterns of training routes MAPK signaling to distinct downstream cascades (PKA vs. PKC) to mediate the expression of specific forms of ITF.
From a broader scientific perspective, dynamically changing the spatiotemporal properties of a molecule and its interactions with other molecules can significantly enhance the range of molecular options available for the induction of diverse forms of behavioral and synaptic plasticity. Thus, our current analysis of spatiotemporal integration of MAPK and PKA signaling in mediating synaptic facilitation may provide valuable insights for understanding this additional dimension of molecular mechanisms underlying memory processing.
Materials and Methods
Animals and Physiological Procedures.
Wild-caught adult A. californica (150–350 g) (Marinus, Long Beach, CA, or Charles Hollahan, Santa Barbara, CA) were maintained in an aquarium for at least 3 d before experimentation. They were anesthetized by injection of isotonic MgCl2 (∼120 mL/100 g of body weight). Pleural–pedal ganglia were removed with the tail nerve (TN) attached and pinned in Sylgard-coated dishes containing a 1:1 mixture of MgCl2 and artificial seawater (ASW; 460 mM NaCl/55 mM MgCl2/11 mM CaCl2/10 mM KCl/10 mM Tris, pH 7.6). The TN was cut immediately above the caudal bifurcated point. Ganglia were desheathed and incubated in ASW for at least 1 h before training. The TN was drawn into a suction electrode. Each train of TNS lasted 2 s, with 5-ms 30-V pulses at 40 Hz. For repeated TNS, a total of five trains were applied with 15-min intertrial intervals. Ganglia from one side of the animal received TNS, whereas those from the other side were mock treated as control. Although the somata of most serotonergic neurons are located in the cerebral ganglia, local serotonergic fibers in pleural–pedal ganglia were able to release 5-HT, with magnitude and duration similar to those of the intact circuit (19). For experiments in which the SN soma and synapse were differentially exposed to drugs, the ganglia were pinned in a two-chamber dish as previously described (22). The pedal ganglion, containing the MN cluster and SN-MN synapses, was pinned on one side of a plastic barrier (synaptic compartment). The pleural ganglion, containing the SN somata, was pinned on the other side (somatic compartment).
Parallel Measurement of PKA and MAPK Activation.
Either 1 min (immediately) or 1 h after the last TNS, the dish was quickly transferred into ice-cold 1:1 MgCl2:ASW. SN soma and synaptic neuropil were quickly excised, homogenized, and immediately frozen on dry ice/ethanol mixture. Subsequently, part of the homogenate was subjected to a modified PepTag PKA assay (Promega) for measuring PKA activity in the sample, whereas the remainder was resolved on 4–12% Bis-Tris gels (Invitrogen) for Western blotting of phospho- and total-MAPK, as previously described (15), and protein quantification with Sypro Ruby Gel Stain (Bio-Rad) (SI Materials and Methods).
Pharmacology.
To block MAPK activation, 20-µM U0126 (Calbiochem or Tocris) was perfused into the dish and present either from 30 min before and throughout TNS, or from immediately after TNS until harvesting. Either the vehicle (0.05% DMSO) or the inactive analog U0124 (20 µM, Calbiochem or Tocris) was used as control. Because there was no significant difference between these control groups, the data were pooled. To inhibit PKA activity, H89 (Calbiochem) was added to a static bath to a final concentration of 20 µM and immediately mixed into ASW by pipetting. H89 or its vehicle (0.2% DMSO) was present from 50 min before TNS until harvesting, with mixing every 20–30 min. H89 also may inhibit several other kinases (41, 42). However, several studies from our laboratory and others show that H89’s effects in Aplysia are mimicked by other functional blockers of PKA signaling.
Synaptic Facilitation.
Electrophysiological responses were recorded intracellularly from tail SNs and MNs that make monosynaptic connections in pleural–pedal ganglia pinned in a two-chamber dish, as previously described (22). TNS induced action potentials in a small subset of SNs. Those were excluded in our analysis because the current study focused on activity-independent facilitation. Three pretests (intertrial interval = 15–20 min) were made by eliciting a single SN action potential (with a 5-ms depolarizing current pulse) and recording the evoked baseline MN excitatory postsynaptic potential (EPSP) amplitude. To examine the requirement of synaptic MAPK activation in ITF induction, the perfusion was stopped after the last pretest, and 20 µM U0126 was applied to the synaptic compartment. Twenty minutes later, another pretest was made to ensure the drug did not affect basal transmission [also (15)]. After another 10 min, repeated TNS was delivered. STF was tested at 5 min after the first TNS. The same synapse was tested again for ITF at 35 min after the last TNS. To examine the requirement of synaptic PKA in ITF maintenance, repeated TNS was delivered after the pretests. The first ITF was tested at 20–35 min post TNS. Afterward, the perfusion was stopped, and 20 µM H89 was applied to the synaptic compartment. A second ITF was tested 35 min later. To examine the requirement of synaptic PKA in STF, 20 µM H89 was applied to the synaptic compartment after the pretests. Forty minutes later, another pretest was made to ensure the treatment did not affect basal transmission. After another 10 min, a single TNS was delivered, and STF was tested at 5 min afterward. To further ensure H89 did not affect basal transmission, in a separate group, the preparation was treated as described previously but no TNS was given. Baseline EPSP amplitude was determined by the average of the pretests.
Data Analysis.
Statistical analyses were performed using ANOVA followed by Bonferroni post hoc tests for selected groups. For paired comparisons, Student t tests (paired tests for within-animal comparisons and unpaired tests for between-animal comparisons) were used. Data are presented as mean percent ± SEM of mock-treated controls that did not receive TNS, unless otherwise specified. All P values are reported for two-tail analysis. Statistical significance is defined as *P < 0.05, **P < 0.01.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) Grant R01 MH 041083 (to T.J.C.), National Science Foundation Grant IOB-0444762 (to T.J.C), and NIH Grant R01 MH 081151 (to T.J.C. and Kelsey C. Martin).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1209956109/-/DCSupplemental.
References
- 1.Kholodenko BN, Hancock JF, Kolch W. Signalling ballet in space and time. Nat Rev Mol Cell Biol. 2010;11(6):414–426. doi: 10.1038/nrm2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abel T, Nguyen PV. Regulation of hippocampus-dependent memory by cyclic AMP-dependent protein kinase. Prog Brain Res. 2008;169:97–115. doi: 10.1016/S0079-6123(07)00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma SK, Carew TJ. The roles of MAPK cascades in synaptic plasticity and memory in Aplysia: Facilitatory effects and inhibitory constraints. Learn Mem. 2004;11(4):373–378. doi: 10.1101/lm.81104. [DOI] [PubMed] [Google Scholar]
- 4.Adams JP, Sweatt JD. Molecular psychology: Roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol. 2002;42:135–163. doi: 10.1146/annurev.pharmtox.42.082701.145401. [DOI] [PubMed] [Google Scholar]
- 5.Gerits N, Kostenko S, Shiryaev A, Johannessen M, Moens U. Relations between the mitogen-activated protein kinase and the cAMP-dependent protein kinase pathways: Comradeship and hostility. Cell Signal. 2008;20(9):1592–1607. doi: 10.1016/j.cellsig.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 6.Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci. 2010;35(2):91–100. doi: 10.1016/j.tibs.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 7.Pagani MR, Oishi K, Gelb BD, Zhong Y. The phosphatase SHP2 regulates the spacing effect for long-term memory induction. Cell. 2009;139(1):186–198. doi: 10.1016/j.cell.2009.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pouysségur J, Volmat V, Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem Pharmacol. 2002;64(5-6):755–763. doi: 10.1016/s0006-2952(02)01135-8. [DOI] [PubMed] [Google Scholar]
- 9.Shalin SC, Hernandez CM, Dougherty MK, Morrison DK, Sweatt JD. Kinase suppressor of Ras1 compartmentalizes hippocampal signal transduction and subserves synaptic plasticity and memory formation. Neuron. 2006;50(5):765–779. doi: 10.1016/j.neuron.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 10.Philips GT, Tzvetkova EI, Carew TJ. Transient mitogen-activated protein kinase activation is confined to a narrow temporal window required for the induction of two-trial long-term memory in Aplysia. J Neurosci. 2007;27(50):13701–13705. doi: 10.1523/JNEUROSCI.4262-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kandel ER. The molecular biology of memory storage: A dialog between genes and synapses. Biosci Rep. 2004;24(4-5):475–522. doi: 10.1007/s10540-005-2742-7. [DOI] [PubMed] [Google Scholar]
- 12.Marinesco S, Carew TJ. Serotonin release evoked by tail nerve stimulation in the CNS of aplysia: Characterization and relationship to heterosynaptic plasticity. J Neurosci. 2002;22(6):2299–2312. doi: 10.1523/JNEUROSCI.22-06-02299.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma SK, Sherff CM, Stough S, Hsuan V, Carew TJ. A tropomyosin-related kinase B ligand is required for ERK activation, long-term synaptic facilitation, and long-term memory in aplysia. Proc Natl Acad Sci USA. 2006;103(38):14206–14210. doi: 10.1073/pnas.0603412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang F, Goldsmith JR, Byrne JH. Neural analogue of long-term sensitization training produces long-term (24 and 48 h) facilitation of the sensory-to-motor neuron connection in Aplysia. J Neurophysiol. 1994;72(2):778–784. doi: 10.1152/jn.1994.72.2.778. [DOI] [PubMed] [Google Scholar]
- 15.Sharma SK, et al. Differential role of mitogen-activated protein kinase in three distinct phases of memory for sensitization in Aplysia. J Neurosci. 2003;23(9):3899–3907. doi: 10.1523/JNEUROSCI.23-09-03899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sutton MA, Carew TJ. Parallel molecular pathways mediate expression of distinct forms of intermediate-term facilitation at tail sensory-motor synapses in Aplysia. Neuron. 2000;26(1):219–231. doi: 10.1016/s0896-6273(00)81152-6. [DOI] [PubMed] [Google Scholar]
- 17.Ghirardi M, et al. Roles of PKA and PKC in facilitation of evoked and spontaneous transmitter release at depressed and nondepressed synapses in Aplysia sensory neurons. Neuron. 1992;9(3):479–489. doi: 10.1016/0896-6273(92)90185-g. [DOI] [PubMed] [Google Scholar]
- 18.Chain DG, et al. Mechanisms for generating the autonomous cAMP-dependent protein kinase required for long-term facilitation in Aplysia. Neuron. 1999;22(1):147–156. doi: 10.1016/s0896-6273(00)80686-8. [DOI] [PubMed] [Google Scholar]
- 19.Philips GT, Sherff CM, Menges SA, Carew TJ. The tail-elicited tail withdrawal reflex of Aplysia is mediated centrally at tail sensory-motor synapses and exhibits sensitization across multiple temporal domains. Learn Mem. 2011;18(4):272–282. doi: 10.1101/lm.2125311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schacher S, Wu F, Panyko JD, Sun ZY, Wang D. Expression and branch-specific export of mRNA are regulated by synapse formation and interaction with specific postsynaptic targets. J Neurosci. 1999;19(15):6338–6347. doi: 10.1523/JNEUROSCI.19-15-06338.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu GY, Deisseroth K, Tsien RW. Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat Neurosci. 2001;4(2):151–158. doi: 10.1038/83976. [DOI] [PubMed] [Google Scholar]
- 22.Sherff CM, Carew TJ. Parallel somatic and synaptic processing in the induction of intermediate-term and long-term synaptic facilitation in Aplysia. Proc Natl Acad Sci USA. 2004;101(19):7463–7468. doi: 10.1073/pnas.0402163101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin KC, et al. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 1997;18(6):899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- 24.Kaang BK, Kandel ER, Grant SG. Activation of cAMP-responsive genes by stimuli that produce long-term facilitation in Aplysia sensory neurons. Neuron. 1993;10(3):427–435. doi: 10.1016/0896-6273(93)90331-k. [DOI] [PubMed] [Google Scholar]
- 25.Michael D, et al. Repeated pulses of serotonin required for long-term facilitation activate mitogen-activated protein kinase in sensory neurons of Aplysia. Proc Natl Acad Sci USA. 1998;95(4):1864–1869. doi: 10.1073/pnas.95.4.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Müller U, Carew TJ. Serotonin induces temporally and mechanistically distinct phases of persistent PKA activity in Aplysia sensory neurons. Neuron. 1998;21(6):1423–1434. doi: 10.1016/s0896-6273(00)80660-1. [DOI] [PubMed] [Google Scholar]
- 27.Kelleher RJ, 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004;116(3):467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 28.Bailey CH, et al. Mutation in the phosphorylation sites of MAP kinase blocks learning-related internalization of apCAM in Aplysia sensory neurons. Neuron. 1997;18(6):913–924. doi: 10.1016/s0896-6273(00)80331-1. [DOI] [PubMed] [Google Scholar]
- 29.Sindreu C, Palmiter RD, Storm DR. Zinc transporter ZnT-3 regulates presynaptic Erk1/2 signaling and hippocampus-dependent memory. Proc Natl Acad Sci USA. 2011;108(8):3366–3370. doi: 10.1073/pnas.1019166108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci. 2003;6(1):34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- 31.Sharma SK, Bagnall MW, Sutton MA, Carew TJ. Inhibition of calcineurin facilitates the induction of memory for sensitization in Aplysia: Requirement of mitogen-activated protein kinase. Proc Natl Acad Sci USA. 2003;100(8):4861–4866. doi: 10.1073/pnas.0830994100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J, Hu JY, Schacher S, Schwartz JH. The two regulatory subunits of aplysia cAMP-dependent protein kinase mediate distinct functions in producing synaptic plasticity. J Neurosci. 2004;24(10):2465–2474. doi: 10.1523/JNEUROSCI.4331-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurosu T, Hernández AI, Schwartz JH. Serotonin induces selective cleavage of the PKA RI subunit but not RII subunit in Aplysia neurons. Biochem Biophys Res Commun. 2007;359(3):563–567. doi: 10.1016/j.bbrc.2007.05.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoffmann R, Baillie GS, MacKenzie SJ, Yarwood SJ, Houslay MD. The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J. 1999;18(4):893–903. doi: 10.1093/emboj/18.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang HT, et al. Inhibition of the phosphodiesterase 4 (PDE4) enzyme reverses memory deficits produced by infusion of the MEK inhibitor U0126 into the CA1 subregion of the rat hippocampus. Neuropsychopharmacology. 2004;29(8):1432–1439. doi: 10.1038/sj.npp.1300440. [DOI] [PubMed] [Google Scholar]
- 36.Zhong H, et al. Subcellular dynamics of type II PKA in neurons. Neuron. 2009;62(3):363–374. doi: 10.1016/j.neuron.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vaillant AR, et al. Signaling mechanisms underlying reversible, activity-dependent dendrite formation. Neuron. 2002;34(6):985–998. doi: 10.1016/s0896-6273(02)00717-1. [DOI] [PubMed] [Google Scholar]
- 38.Gervasi N, Tchénio P, Preat T. PKA dynamics in a Drosophila learning center: Coincidence detection by rutabaga adenylyl cyclase and spatial regulation by dunce phosphodiesterase. Neuron. 2010;65(4):516–529. doi: 10.1016/j.neuron.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 39.Shobe JL, et al. Temporal phases of activity-dependent plasticity and memory are mediated by compartmentalized routing of MAPK signaling in aplysia sensory neurons. Neuron. 2009;61(1):113–125. doi: 10.1016/j.neuron.2008.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ye X, Shobe JL, Sharma SK, Marina A, Carew TJ. Small G proteins exhibit pattern sensitivity in MAPK activation during the induction of memory and synaptic facilitation in Aplysia. Proc Natl Acad Sci USA. 2008;105(51):20511–20516. doi: 10.1073/pnas.0808110105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351(Pt 1):95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lochner A, Moolman JA. The many faces of H89: A review. Cardiovasc Drug Rev. 2006;24(3-4):261–274. doi: 10.1111/j.1527-3466.2006.00261.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.