

Abstract
Many biological processes generate force, and proteins have evolved to resist and respond to tension along different force axes. Single-molecule force spectroscopy allows for molecular insight into the behavior of proteins under force and the mechanism of protein folding in general. Here, we have used src SH3 to investigate the effect of different pulling axes under the low-force regime afforded by an optical trap. We find that this small cooperatively folded protein shows an anisotropic response to force; the protein is more mechanically resistant to force applied along a longitudinal axis compared to force applied perpendicular to the terminal β strand. In the longitudinal axis, we observe an unusual biphasic behavior revealing a force-induced switch in the unfolding mechanism suggesting the existence of two parallel unfolding pathways. A site-specific variant can selectively affect one of these pathways. Thus, even this simple two-state protein demonstrates a complex mechanical unfolding trajectory, accessing multiple unfolding pathways under the low-force regime of the optical trap; the specific unfolding pathway depends on the perturbation axis and the applied force.
Many cellular processes such as protein folding/unfolding, protein degradation, and nucleic acid splicing are mechanical processes in which force plays a significant physiological role (1, 2). The advent of single-molecule force spectroscopy has allowed the observation of these events at an unprecedented resolution and yielded significant mechanistic details (3, 4). This single-molecule force spectroscopy approach has also provided insight into our basic understanding of how proteins fold and unfold (5–7). In mechanical unfolding experiments, the applied force privileges a particular direction in space along which a well-defined reaction coordinate—namely, the end-to-end extension of the molecule—emerges as a natural metric of the extent of the reaction (1, 8, 9). The location of the transition state along this defined reaction coordinate can be determined directly from such experiments. This information, in combination with recent theoretical approaches (10), can provide detailed information on the protein folding free energy landscape (11).
Several studies have investigated the factors influencing the mechanical response of proteins under force; most of these studies are under the high-force regime of the atomic force microscopy (AFM). The term “mechanical stability” refers to a protein’s average unfolding force at a given loading rate. Because most of these AFM studies are carried out under high loading rates, they are far from equilibrium; the mechanical stability reflects the kinetic likelihood to unfold and is therefore not a measure of stability in the thermodynamic sense. These experimental studies have led to the suggestion that native-state topology governs mechanical stability (2, 12, 13). In general, β-sheet proteins appear to be mechanically stronger than α-helical proteins (12). The network of hydrogen bonds between adjacent β strands can act as a clamp conferring mechanical resistance (14). In addition, side-chain interactions and long-range contacts outside the mechanical clamp region have also been shown to play a role in determining the response of a protein under force (15–18).
A combination of AFM experiments, protein engineering, and molecular dynamics (MD) simulations have revealed a lot about mechanical unfolding pathways (14, 19–22). Such studies have shown that structurally similar proteins can access different mechanical unfolding pathways (19, 23). Studies on the I27 domain of titin, in which an unfolding intermediate becomes populated at high loading rates, highlight the complexity of mechanical unfolding (19, 20, 24). These experiments also suggest that the mechanical unfolding pathways accessed at these high forces differ from those observed in the zero force denaturant-induced unfolding pathways (16, 24, 25).
The geometry of force application (i.e., the axis of the applied force with respect to protein topology) also plays a role in the mechanical response of proteins. Coarse-grained simulations suggest that proteins follow different unfolding trajectories along different pulling geometries (9, 26). AFM experiments and MD simulations have shown that the average unfolding force varies with pulling geometry (13, 27–29).
To date, however, almost all experiments analyzing a protein’s mechanical response under different pulling geometries have relied on AFM spectroscopy with high loading rates, and unfolding occurs at relatively high forces. Using optical tweezers, however, one can apply forces at low, likely physiological, loading rates and obtain mechanistic information much closer to equilibrium. Such low-force unfolding trajectories are often assumed to mirror the unfolding in the absence of force. In these experiments, the pulling geometry is easily defined by the placement of unique cysteine residues that serve as the anchor points. This approach was used to apply force on different regions of T4 lysozyme, in a study evaluating interdomain cooperativity (30). Surprisingly, the data suggested that the pulling geometry did not affect the unfolding trajectory. Here, we have investigated the role of pulling geometry on the unfolding trajectory using the optical trap methodology on the src SH3 domain.
The src SH3 domain is a simple two-state folding protein composed predominantly of β strands that has been extensively characterized in solution (31, 32). We investigate the response of src SH3 to mechanical force under two different pulling axes (Fig. 1A). One axis is oriented longitudinally relative to the terminal β strand, and the other axis is oriented orthogonal to this strand. The longitudinal force (parallel to the terminal β strand) is expected to result in “shearing” of the β strands, while the orthogonal force (perpendicular to the terminal β strand) would be expected to “unzip” the strands. Because, in the latter case, the hydrogen bonds are ruptured successively rather than concurrently, the unzipping geometry is expected to disrupt a β sheet at lower average unfolding forces (33).
Fig. 1.

(A) The structure of the src SH3 domain (Protein Data Bank ID code 1SRL); the two pulling geometries apply a shearing (A7C/N59C) (black) and an unzipping (R19C/N59C) (red) force relative to the β strand between the N and the C termini. (B) Schematic representation of the experimental setup used to apply force on proteins using optical tweezers.
Our results demonstrate that in these near equilibrium conditions there exists a significant anisotropy in the mechanical unfolding behavior with the protein having a greater mechanical stability along the longitudinal force axis. The force dependence of the unfolding rates along this longitudinal geometry shows a unique biphasic behavior. The change in the distance to the transition state (the change in the rate constants as a function of force) between these force regimes cannot be explained by a sequential barrier mechanism. Rather, our data suggest that in this specific geometry the protein can access two parallel unfolding pathways. Thus, we find that at low forces the pulling geometry affects both the mechanical stability and the unfolding trajectory, and that even at these low forces, the protein can access different unfolding trajectories than in the absence of force.
Results and Discussion
Defining the Pulling Axes.
The force axis is defined by the placement of unique cysteine residues in the protein that are used to attach molecular handles to apply force. Two double-cysteine variants of the src SH3 domain were generated (Fig. 1A). The variant A7C/N59C results in a longitudinal force axis with respect to the N-terminal β strand (shearing), while R19/N59C creates a perpendicular (unzipping) axis. All cysteine substitutions were placed in solvent-exposed positions where mutations have been shown to have a negligible effect on the stability of the protein (32).
Bulk kinetic and equilibrium denaturation studies were carried out under the same solvent conditions as those used in the single-molecule mechanical studies (pH, ionic strength, etc.). The proteins unfold reversibly and cooperatively with a ΔGunf [using the two-state assumption and a linear extrapolation (34, 35)] of 2.20 ± 0.05 kcal/mol and 2.40 ± 0.10 kcal/mol for A7C/N59C and R19C/N59C src SH3, respectively, close to that for cysteine-free wild-type src SH3 under similar conditions (2.25 ± 0.07 kcal/mol), suggesting that the cysteine mutations do not severely perturb the protein. The unfolding/refolding kinetics confirm the two-state nature of the process (Fig. S1 and Table S1). Because both the stability and unfolding kinetics are not significantly affected by the placement of the cysteine residues, any dramatic changes observed in the mechanical unfolding behavior can be attributed to the pulling axes and not the mutations themselves. It should be noted that the kinetic and thermodynamic parameters determined here are different from the previously reported values for wild-type src SH3 (32), which were carried out under different buffer conditions (Tris vs. phosphate).
The optical tweezers setup used in these studies is diagramed in Fig. 1B (8, 36). Briefly, a single src SH3 protein is tethered between two polystyrene beads using dsDNA “handles” attached via thiol chemistry (6, 37). One bead is held in place on a pipette tip by suction, and a dual beam counter-propagating optical trap manipulates the other bead. The force acting on the trapped bead, and thus on the tethered single molecule, as well as the relative extension of the tether are measured.
Anisotropic Response to Force–Force Ramp Studies.
Both variants were initially characterized by force ramp experiments at a constant loading rate (8 pN/s). Here, the unfolding event is seen as a “rip” in the force versus trap position trace (Fig. 2A). The A7C/N59C shearing axis unfolds at a significantly higher average force than the R19C/N59C unzipping geometry (Fu = 35.0 ± 0.5 pN, n = 175 vs. Fu = 14.0 ± 0.3 pN, n = 180) (Fig. 2B). The difference in mechanical resistance is consistent with previous AFM studies (13, 27, 28) and highlights the role of protein topology in conferring mechanical stability.
Fig. 2.

(A) Typical unfolding/refolding traces obtained in a force-ramp experiment for A7C/N59C src SH3 (black/gray) and R19C/N59C src SH3 (red/pink). A single protein molecule is stretched (loading rate 8 pN/ sec) until the unfolding event is seen as a “rip” (indicated by arrows). The protein is then allowed to refold by moving the trap to low forces. (B) Histogram of unfolding forces for A7C/N59C src SH3 (black, Fu = 35 ± 0.5 pN) and R19C/N59C src SH3 (red, Fu = 14 ± 0.3 pN).
For both variants, the change in contour length associated with unfolding is consistent with that expected for complete unfolding of the molecule (Fig. S2). Because these are nonequilibrium experiments, the unfolding force, Fu, depends on the kinetics of unfolding. To evaluate the free energies involved in these transitions, we utilized Crooks fluctuation theorem (CFT) to extrapolate free energies from measurements of the nonequilibrium work in both the forward and reverse directions (30, 38, 39), which yielded a ΔGunf = 2.7 ± 0.3 kcal/mol for the R19C/N59C variant. Reliable estimates were not obtained for the A7C/N59C variant, as the unfolding and refolding work distributions did not overlap. The similarity between the thermodynamic stability determined by CFT and that in bulk experiments indicates that the DNA handles do not grossly perturb the system.
The Force Dependence of the Unfolding Rates ( ) Is Biphasic for the Shearing Axis.
) Is Biphasic for the Shearing Axis.
What change in the energy landscape can explain the difference in the mechanical stability (average unfolding forces) between the two pulling geometries? There are two possibilities: (i) The pulling geometry does not change the transition state, but simply alters the relative position of the transition state along the reaction coordinate, or (ii) the pulling geometry alters the structure of the transition state, which in turn could affect both the energy and position of the rate-limiting barrier.
To further explore these potential scenarios, we carried out force-jump/force-quench experiments to obtain the unfolding and refolding rates as a function of force. The kinetics were recorded for seven individual tethers, and the data shown were obtained from one such tether. Each tether was analyzed separately, and there was very little variation in rates between the tethers. For the R19C/N59C variant (the unzipping direction), ln ku increases linearly across the range of measured forces (Fig. 3A); the data were fit to a simple Bell’s model: k(F) = kmk0 exp(FX‡/kBT), where km represents the contribution of experimental parameters such as the bead size, trap stiffness, and handle length to the observed rate constant; k0 is the intrinsic rate constant of the molecule in the absence of force; F is the applied force; X‡ is the distance to the transition state ( and
and  ); kB is the Boltzmann’s constant; and T is the temperature (40). The slope of F vs. ln ku for R19C/N59C src SH3 yields a distance to the transition state,
); kB is the Boltzmann’s constant; and T is the temperature (40). The slope of F vs. ln ku for R19C/N59C src SH3 yields a distance to the transition state, 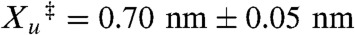 . The linear nature of the plot is consistent with a single pathway model in which force continuously tilts the free energy landscape, thereby lowering the height of the barrier (41). The relatively short distance to the transition state is typical of globular proteins and reflects the brittle nature of unfolding.
. The linear nature of the plot is consistent with a single pathway model in which force continuously tilts the free energy landscape, thereby lowering the height of the barrier (41). The relatively short distance to the transition state is typical of globular proteins and reflects the brittle nature of unfolding.
Fig. 3.

The logarithm of the (A) unfolding and (B) refolding rates of A7C/N59C src SH3 (black) and R19C/N59C src SH3 (red) obtained from force-jump/force-quench experiments were plotted as a function of force to obtain the unfolding and refolding distances to the transition state according to Bell’s model. Typical unfolding and refolding traces are shown in the inset.
The A7C/N59C variant does not show this simple linear behavior, but rather exhibits biphasic dependence (Fig. 3A). ln ku shows a weaker dependence on force in the 15–25 pN range as compared to that above 25 pN. The biphasic behavior is well captured by fitting the data to the sum of two Bell terms. These fits yield significantly different distances to the transition state for the two force regimes, 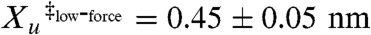 and
and  . Thus, under this shearing force, src SH3 demonstrates anti-Hammond behavior where the transition state moves away from the native state with increasing force (42). While no such behavior was observed for the unzipping axis, curvature outside of the accessible force range cannot be ruled out. It should be noted that under the force-ramp conditions (Fig. 2), the average unfolding force is high (around 35 pN), so we do not expect the biphasic behavior to have much effect on the observed unfolding force distribution under these conditions. In fact, using the approach outlined by Dudko et al. (43), the force ramp data adequately recapitulates the measured force-dependent unfolding rates (Fig. S3).
. Thus, under this shearing force, src SH3 demonstrates anti-Hammond behavior where the transition state moves away from the native state with increasing force (42). While no such behavior was observed for the unzipping axis, curvature outside of the accessible force range cannot be ruled out. It should be noted that under the force-ramp conditions (Fig. 2), the average unfolding force is high (around 35 pN), so we do not expect the biphasic behavior to have much effect on the observed unfolding force distribution under these conditions. In fact, using the approach outlined by Dudko et al. (43), the force ramp data adequately recapitulates the measured force-dependent unfolding rates (Fig. S3).
Refolding Is Insensitive to the Geometry of the Applied Force.
The refolding rates and their force dependence are similar in both the shearing and unzipping geometries (Fig. 3B), suggesting that the refolding mechanisms may be similar. The rates were fit to the Bell’s model [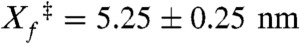 (A7C/N59C) and 4.30 ± 0.20 nm (R19C/N59C)]. In the low-force regime, the sum of the measured distances to the unfolding and refolding transition states equal the total extension change for both pulling axes, consistent with a two-state model (44).
(A7C/N59C) and 4.30 ± 0.20 nm (R19C/N59C)]. In the low-force regime, the sum of the measured distances to the unfolding and refolding transition states equal the total extension change for both pulling axes, consistent with a two-state model (44).
The Force-Induced Switch Suggests Multiple Unfolding Pathways.
Three possible models could explain the biphasic dependence of ln ku on the unfolding force: (i) sequential barriers along a single reaction pathway, (ii) interplay of force and local curvature on a multidimensional free energy landscape, and (iii) parallel unfolding pathways.
(i) Sequential unfolding barriers can produce biphasic behavior when there is a change in the rate-limiting step. This change, however, will result in biphasic behavior only when the barrier furthest from the native state, the “outer” barrier, is the highest at low force (Fig. 4A) (45, 46). Force alters the landscape by favoring more extended states, and the effect scales with the distance along the reaction coordinate. Thus, if the inner barrier is the rate-limiting step at low force, it will continue to be the highest at all forces (Fig. 4B). If, however, the outer barrier is higher at low force, at some force, there will be a switch and the inner barrier will become the rate-limiting step. In this sequential barrier scenario, the distance to the transition state,  , should be higher at low forces (outer barrier is rate limiting) compared to high forces (inner barrier is rate limiting). Such sequential barriers have been shown in receptor–ligand unbinding reactions (45, 46), and a similar sequential model with the formation of an intermediate at high force (Fig. 4C) was demonstrated in AFM studies of the titin I27 domain (19). Our force-jump data on src SH3, however, follow the opposite trend: The value of
, should be higher at low forces (outer barrier is rate limiting) compared to high forces (inner barrier is rate limiting). Such sequential barriers have been shown in receptor–ligand unbinding reactions (45, 46), and a similar sequential model with the formation of an intermediate at high force (Fig. 4C) was demonstrated in AFM studies of the titin I27 domain (19). Our force-jump data on src SH3, however, follow the opposite trend: The value of  increases from low forces to high forces. Thus, sequential barriers are not the cause of the observed biphasic behavior.
increases from low forces to high forces. Thus, sequential barriers are not the cause of the observed biphasic behavior.
Fig. 4.

Schematic representations of the features of the energy landscape that can account for a biphasic force dependence of ln ku. Sequential barrier scenario in which the (A) outer barrier or the (B) inner barrier is rate limiting. (C) Population of an intermediate at high force. (D) Parallel pathway scenario in which the two unfolding pathways (black and red) are accessible at different forces.
(ii) The change in the force dependence of the rate constants could also result from the interplay of force and the curvature in the landscape (10, 41). For example, the Hammond effect reflects the movement of the transition state closer to the native state with increasing perturbant (47). However, this effect cannot account for our observation, as the change we observe for src SH3 reflects anti-Hammond behavior, in that the transition states move away from the native state with increasing force (42).
All of the one-dimensional descriptions of the reaction discussed above involve the crossing of an activation barrier tilted under the applied force, with a single variable (the end-to-end extension) capturing the molecule’s many degrees of freedom. To account for our data, we must take into account the multidimensional nature of the energy landscape. Suzuki and Dudko have proposed a model in which the multidimensionality of the free energy landscape can give rise to different trends in the force dependence of the unfolding rates (48). In principle, this could result in unlimited scenarios for the force dependence of unfolding rates. For example, if at lower forces the unfolding pathway is not aligned with the direction of force and at higher forces the pathway gets reoriented more parallel to the direction of force, we would expect anti-Hammond behavior.
(iii) The third scenario posits that the anti-Hammond behavior arises from a multidimensional landscape where the protein can access two parallel trajectories, one dominating at low force and one dominating at high force (Fig. 4D). Such anti-Hammond behavior has been used as evidence for the presence of parallel pathways in bulk experiments (49, 50). In this scenario, each pathway will have its own transition state whose location will vary along the reaction coordinate. Although, in principle, both pathways are always accessible for unfolding, the pathway with the lowest barrier will dominate at a particular force. We believe that our data are most easily explained via parallel unfolding pathways.
Unfolding in the Shearing Geometry Accesses Parallel Pathways.
To further probe the mechanical response of src SH3, we constructed two site-specific mutations (I34A and S47A) in the shearing and unzipping geometries. Both of these variants have been studied in bulk (32). Force–ramp studies show no appreciable differences in the average unfolding forces for the variants (Table S2). Next, we evaluated the effects of the mutations by investigating the force dependence of unfolding (Fig. 5). All four variants show similar behavior to the parent proteins. In the unzipping geometry, both variants demonstrate a linear force dependence with similar slopes or distances to the transitions state (Fig. 5A and Table S2). In the shearing direction, both mutations also retain the biphasic anti-Hammond behavior observed in the parent protein (Fig. 5B).
Fig. 5.

Force versus ln ku plots for the I34A (blue) and S47A (green) variants of src SH3 along the (A) unzipping and (B) shearing geometries. The plots for the parent proteins, R19C/N59C and A7C/N59C, are shown in red and black, respectively. The location of the mutations is shown in the inset.
The I34A mutation shows similar effects in both pulling directions—unfolding and refolding are slowed, with a greater effect on folding than unfolding (Fig. 5, Table 1, and Fig. S4). In the shearing direction, the effects on both the low-force and high-force regimes are the same (Fig. 5B and Table 1). Thus, I34 appears to be equally involved in the transition state structure under both pulling geometries.
Table 1.
Comparison of the unfolding/folding rates in the presence and absence of force for the src SH3 variants
| Mutation | Bulk zero-force unfolding (folding) 2.5 M (0.55 M) GdmCl | Mechanical R19C/N59C unfolding (folding) 15 pN (5.5 pN) | Mechanical A7C/N59C low-force unfolding (folding) 20 pN (5.5 pN) | Mechanical A7C/N59C high-force unfolding (folding) 35 pN (5.5 pN) |
| I34A | A7C/N59C ↓1.5x (↓6x) | ↓1.5x (↓4x) | ↓1.5x (↓4x) | |
| R19C/N59C ↓2.5x (↓5x) | ↓2x (↓7x) | |||
| S47A | A7C/N59C ↑1.5x (↓2.5x) | ↑3.5x (↓1.5x) | no change (↓1.5x) | |
| R19C/N59C ↓1.5x (↓4x) | ↑3x (N.D.) |
N.D., not determined.
In the shearing geometry, S47A differentially affects the two unfolding regimes: It increases the unfolding rate approximately 3.5-fold in the low-force regime, but does not appear to affect the high-force regime (Fig. 5B, Table 1, and Fig. S4). Thus, this mutation appears to uncouple the two regimes, and the simplest interpretation is the presence of two structurally and energetically independent transitions. It is also formally possible that the protein traverses a single barrier over a multidimensional landscape and that the mutation alters the curvature such that in the accessible force range, the high-force regimes still overlap. In the unzipping geometry, the S47A mutation behaves similarly to the low-force shearing regime and increases the unfolding rate approximately 3-fold (Fig. 5A and Table 1).
In sum, these mutations implicate at least two different trajectories for the mechanical unfolding of src SH3 under the low-force regime of the optical trap and suggest the existence of parallel unfolding pathways that are preferentially accessed at different forces. While the presence of anti-Hammond behavior and the existence of parallel force-dependent unfolding trajectories have been suggested before (26, 51), to our knowledge, a transition has not been directly observed previously.
Comparison of Mechanical and Bulk Zero-Force Unfolding Pathways.
The different trajectories accessed under these low forces leads us to ask which, if any, of the observed mechanical pathways represents the intrinsic unfolding mechanism of src SH3 in the absence of force. Because the previous bulk studies were carried out under different buffer conditions, we repeated the bulk folding and refolding kinetics under our conditions in the presence of the engineered cysteine residues (Fig. 6 and Table S1). A simple linear extrapolation of the F vs. ln k plots to zero force does not match the ensemble data for either pulling geometries, as it results in an unfolding rate at zero force that is slower, and a refolding rate at zero force that is faster than in the bulk solution studies. While at first this may seem paradoxical, there are several issues that must be considered. The first is the effect of the spring constant of the system. For both folding and unfolding, a positive spring constant results in a decrease in the rate constants (52). The second issue is that as the rate constants approach zero force, there will be curvature in the slopes of the line (i.e., the change in the rate constants as a function of force) (53). This curvature occurs due to the change in the distance between the folded and unfolded states (the end-to-end extension of the molecule) as a function of force. Because the end-to-end extension of the molecule is measured along the applied force vector, it will approach zero at zero force, even though the radius of gyration is nonzero. Because the change in extension is so small at these low forces (less than 4 pN), we cannot directly observe this curvature. Furthermore, because the end-to-end extension is different for the two geometries, the extent of curvature will be different for A7C/N59C and R19C/N59C. Therefore, our data cannot make any direct comparison between the unfolding rates observed in bulk and in single-molecule experiments. We can, however, compare the effect of mutations (Table 1).
Fig. 6.

Ensemble kinetic chevron plots for src SH3 variants along both pulling geometries. The parent proteins, A7C/N59C and R19C/N59C, are shown in black and red, respectively. The I34A and S47A variants are shown in blue and green, respectively, for the A7C/N59C (solid) and R19C/N59C (dotted) geometries. The stopped-flow experiments were performed at 25 °C under the same buffer conditions as those used in the optical tweezers (10 mM Tris pH 7.0, 250 mM NaCl).
The mutations have relatively small destabilizing effects (approximately 0.5–1 kcal/mol) in the background of a marginally stable protein; therefore, we compare the change in folding/unfolding rates at a measured denaturant value instead of calculating a quantitative phi-value (Table 1). The I34A mutation decreases the folding and unfolding rates to the same extent as observed in single-molecule experiments. Thus, I34 appears to be similarly involved in the transition state in bulk and in both pulling geometries. The effects of the S47A mutation in the absence of force are complex; it induces apparent curvature in the unfolding limb at high [GdmCl] in the wild-type and A7C/N59C background (Fig. S5). At low [GdmCl], the mutation increases the unfolding rate in the A7C/N59C background, similar to the effect observed on the low-force shearing regime (Fig. 6 and Table 1). It is not clear if the rollover at high [GdmCl] is related to the biphasic behavior observed in the single-molecule experiments. It is also worth noting that the S47A mutation shows slightly different effects in the two cysteine backgrounds.
Probing the detailed structural features of each pathway will require evaluating the effect of many mutations, defining the mechanical transition state along the two pulling geometries by using an analysis analogous to the ϕ-value methodology (5, 54) shown in previous AFM studies (15, 23, 24). Such an approach is particularly complex, because mutations can affect the force range at which the protein unfolds; therefore, a detailed comparison between variants requires the methods used here, which measure the unfolding over a broad range of forces. In addition to providing structural details about the differences between the unfolding mechanism in each geometry, such “mechanical ϕ-values” will also enable a comparison between the mechanical and ensemble transition states. The results from these experiments will need to be incorporated into the development of a quantitative description of a protein’s energy landscape (55).
Conclusion
Our results clearly demonstrate that even under this low-force regime, using a small, cooperatively folded single domain protein such as src SH3, pulling geometry plays a crucial role in mechanical unfolding. The protein responds very differently when pulled along either a shearing or unzipping axis with respect to the terminal β strand. As anticipated from theoretical studies, the shearing axis appears more mechanically stable. Furthermore, when pulled in the shearing direction, the protein shows biphasic behavior, suggesting two different trajectories. The simple notion that certain folds have evolved to be mechanically resistant must take into account the position of the applied force during specific biological processes. Even within a specific geometry, the barriers traversed in mechanically induced conformational changes will depend on the force and may be unrelated to the barriers traversed in the absence of force. Thus, the regions of a protein tuned to respond or resist force in the cell will depend greatly on both the direction and magnitude of the applied force.
Materials and Methods
Protein Expression and Purification.
Site-directed cysteine mutations in the chicken src SH3 domain sequence were introduced using QuikChange mutagenesis. The variant proteins were expressed and purified as described previously (32).
Optical Tweezers.
DNA handles were attached to the protein as described previously (37). The data were recorded using the optical tweezers instrument described in previous studies (6, 30, 36). The optical trap is made of two coaxial, counter-propagating lasers holding a 3.2-μm, anti-digoxigenin–coated bead at the focus. This bead is tethered via the DNA–protein–DNA chimera to a 2.1-μm streptavidin-coated bead, which is held on a micropipette via suction. The micropipette is stationary, and the trapped bead is manipulated by steering the optical trap, which samples data at 1 kHz and has a spring constant of approximately 0.08 pN/nm.
Ensemble Equilibrium and Kinetic Studies.
Chemical denaturant melts were performed as described previously using a Horiba FloroMax-3 fluorimeter (31). Kinetic data for ensemble chevron plots were collected on a BioLogic SFM-400/MOS 200 stopped-flow fluorescence system as described previously (31).
Supplementary Material
ACKNOWLEDGMENTS.
We thank L. Rosen and P. Onuchic for assistance with stopped-flow kinetics and protein purification, respectively. We thank D. King for assistance with mass spectrometry of proteins. We also thank C. Kaiser, J. Chodera, D. Goldman, and the entire Marqusee group for helpful discussions. The research was supported by grants from the National Institutes of Health and the National Science Foundation (S.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1201800109/-/DCSupplemental.
References
- 1.Bustamante C, Chemla YR, Forde NR, Izhaky D. Mechanical processes in biochemistry. Annu Rev Biochem. 2004;73:705–748. doi: 10.1146/annurev.biochem.72.121801.161542. [DOI] [PubMed] [Google Scholar]
- 2.Oberhauser AF, Carrion-Vazquez M. Mechanical biochemistry of proteins one molecule at a time. J Biol Chem. 2008;283:6617–6621. doi: 10.1074/jbc.R700050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maillard RA, et al. ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell. 2011;145:459–469. doi: 10.1016/j.cell.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aubin-Tam ME, Olivares AO, Sauer RT, Baker TA, Lang MJ. Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell. 2011;145:257–267. doi: 10.1016/j.cell.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Best RB, Fowler SB, Toca-Herrera JL, Clarke J. A simple method for probing the mechanical unfolding pathway of proteins in detail. Proc Natl Acad Sci USA. 2002;99:12143–12148. doi: 10.1073/pnas.192351899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cecconi C, Shank EA, Bustamante C, Marqusee S. Direct observation of the three-state folding of a single protein molecule. Science. 2005;309:2057–2060. doi: 10.1126/science.1116702. [DOI] [PubMed] [Google Scholar]
- 7.Stigler J, Ziegler F, Gieseke A, Gebhardt JC, Rief M. The complex folding network of single calmodulin molecules. Science. 2011;334:512–516. doi: 10.1126/science.1207598. [DOI] [PubMed] [Google Scholar]
- 8.Tinoco I, Jr, Li PT, Bustamante C. Determination of thermodynamics and kinetics of RNA reactions by force. Q Rev Biophys. 2006;39:325–360. doi: 10.1017/S0033583506004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Best RB, Paci E, Hummer G, Dudko OK. Pulling direction as a reaction coordinate for the mechanical unfolding of single molecules. J Phys Chem B. 2008;112:5968–5976. doi: 10.1021/jp075955j. [DOI] [PubMed] [Google Scholar]
- 10.Dudko OK, Hummer G, Szabo A. Intrinsic rates and activation free energies from single-molecule pulling experiments. Phys Rev Lett. 2006;96:108101. doi: 10.1103/PhysRevLett.96.108101. [DOI] [PubMed] [Google Scholar]
- 11.Gebhardt JCM, Bornschlögl T, Rief M. Full distance-resolved folding energy landscape of one single protein molecule. Proc Natl Acad Sci USA. 2010;107:2013–2018. doi: 10.1073/pnas.0909854107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crampton N, Brockwell DJ. Unravelling the design principles for single protein mechanical strength. Curr Opin Struct Biol. 2010;20:508–517. doi: 10.1016/j.sbi.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 13.Dietz H, Berkemeier F, Bertz M, Rief M. Anisotropic deformation response of single protein molecules. Proc Natl Acad Sci USA. 2006;103:12724–12728. doi: 10.1073/pnas.0602995103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Carrion-Vazquez M, Oberhauser AF, Marszalek PE, Fernandez JM. Point mutations alter the mechanical stability of immunoglobulin modules. Nat Struct Biol. 2000;7:1117–1120. doi: 10.1038/81964. [DOI] [PubMed] [Google Scholar]
- 15.Best RB, et al. Mechanical unfolding of a titin Ig domain: Structure of transition state revealed by combining atomic force microscopy, protein engineering and molecular dynamics simulations. J Mol Biol. 2003;330:867–877. doi: 10.1016/s0022-2836(03)00618-1. [DOI] [PubMed] [Google Scholar]
- 16.Brockwell DJ, et al. Mechanically unfolding the small, topologically simple protein L. Biophys J. 2005;89:506–519. doi: 10.1529/biophysj.105.061465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paci E, Karplus M. Forced unfolding of fibronectin type 3 modules: An analysis by biased molecular dynamics simulations. J Mol Biol. 1999;288:441–459. doi: 10.1006/jmbi.1999.2670. [DOI] [PubMed] [Google Scholar]
- 18.Forman JR, et al. Non-native interactions are critical for mechanical strength in PKD domains. Structure. 2009;17:1582–1590. doi: 10.1016/j.str.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams PM, et al. Hidden complexity in the mechanical properties of titin. Nature. 2003;422:446–449. doi: 10.1038/nature01517. [DOI] [PubMed] [Google Scholar]
- 20.Fowler SB, et al. Mechanical unfolding of a titin Ig domain: Structure of unfolding intermediate revealed by combining AFM, molecular dynamics simulations, NMR and protein engineering. J Mol Biol. 2002;322:841–849. doi: 10.1016/s0022-2836(02)00805-7. [DOI] [PubMed] [Google Scholar]
- 21.Lu H, Isralewitz B, Krammer A, Vogel V, Schulten K. Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. Biophys J. 1998;75:662–671. doi: 10.1016/S0006-3495(98)77556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marszalek PE, et al. Mechanical unfolding intermediates in titin modules. Nature. 1999;402:100–103. doi: 10.1038/47083. [DOI] [PubMed] [Google Scholar]
- 23.Ng SP, et al. Mechanical unfolding of TNfn3: The unfolding pathway of a fnIII domain probed by protein engineering, AFM and MD simulation. J Mol Biol. 2005;350:776–789. doi: 10.1016/j.jmb.2005.04.070. [DOI] [PubMed] [Google Scholar]
- 24.Brockwell DJ, et al. The effect of core destabilization on the mechanical resistance of I27. Biophys J. 2002;83:458–472. doi: 10.1016/S0006-3495(02)75182-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Best RB, Li B, Steward A, Daggett V, Clarke J. Can non-mechanical proteins withstand force? Stretching barnase by atomic force microscopy and molecular dynamics simulation. Biophys J. 2001;81:2344–2356. doi: 10.1016/S0006-3495(01)75881-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graham TG, Best RB. Force-induced change in protein unfolding mechanism: Discrete or continuous switch? J Phys Chem B. 2011;115:1546–1561. doi: 10.1021/jp110738m. [DOI] [PubMed] [Google Scholar]
- 27.Brockwell DJ, et al. Pulling geometry defines the mechanical resistance of a beta-sheet protein. Nat Struct Biol. 2003;10:731–737. doi: 10.1038/nsb968. [DOI] [PubMed] [Google Scholar]
- 28.Carrion-Vazquez M, et al. The mechanical stability of ubiquitin is linkage dependent. Nat Struct Biol. 2003;10:738–743. doi: 10.1038/nsb965. [DOI] [PubMed] [Google Scholar]
- 29.Junker JP, Rief M. Evidence for a broad transition-state ensemble in calmodulin folding from single-molecule force spectroscopy. Angew Chem Int Ed Engl. 2010;49:3306–3309. doi: 10.1002/anie.200905747. [DOI] [PubMed] [Google Scholar]
- 30.Shank EA, Cecconi C, Dill JW, Marqusee S, Bustamante C. The folding cooperativity of a protein is controlled by its chain topology. Nature. 2010;465:637–640. doi: 10.1038/nature09021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grantcharova VP, Baker D. Folding dynamics of the src SH3 domain. Biochemistry. 1997;36:15685–15692. doi: 10.1021/bi971786p. [DOI] [PubMed] [Google Scholar]
- 32.Grantcharova VP, Riddle DS, Santiago JV, Baker D. Important role of hydrogen bonds in the structurally polarized transition state for folding of the src SH3 domain. Nat Struct Biol. 1998;5:714–720. doi: 10.1038/1412. [DOI] [PubMed] [Google Scholar]
- 33.Rohs R, Etchebest C, Lavery R. Unraveling proteins: A molecular mechanics study. Biophys J. 1999;76:2760–2768. doi: 10.1016/S0006-3495(99)77429-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Street TO, Courtemanche N, Barrick D. Protein folding and stability using denaturants. Methods Cell Biol. 2008;84:295–325. doi: 10.1016/S0091-679X(07)84011-8. [DOI] [PubMed] [Google Scholar]
- 35.Pace CN, Shaw KL. Linear extrapolation method of analyzing solvent denaturation curves. Proteins. 2000;41(Suppl 4):1–7. doi: 10.1002/1097-0134(2000)41:4+<1::aid-prot10>3.3.co;2-u. [DOI] [PubMed] [Google Scholar]
- 36.Smith SB, Cui Y, Bustamante C. Optical-trap force transducer that operates by direct measurement of light momentum. Methods Enzymol. 2003;361:134–162. doi: 10.1016/s0076-6879(03)61009-8. [DOI] [PubMed] [Google Scholar]
- 37.Cecconi C, Shank EA, Dahlquist FW, Marqusee S, Bustamante C. Protein-DNA chimeras for single molecule mechanical folding studies with the optical tweezers. Eur Biophys J. 2008;37:729–738. doi: 10.1007/s00249-007-0247-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crooks GE. Entropy production fluctuation theorem and the nonequilibrium work relation for free energy differences. Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Topics. 1999;60:2721–2726. doi: 10.1103/physreve.60.2721. [DOI] [PubMed] [Google Scholar]
- 39.Collin D, et al. Verification of the Crooks fluctuation theorem and recovery of RNA folding free energies. Nature. 2005;437:231–234. doi: 10.1038/nature04061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bell GI. Models for the specific adhesion of cells to cells. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 41.Hyeon C, Thirumalai D. Measuring the energy landscape roughness and the transition state location of biomolecules using single molecule mechanical unfolding experiments. J Phys Condens Matter. 2007;19:1–27. [Google Scholar]
- 42.Matthews JM, Fersht AR. Exploring the energy surface of protein folding by structure-reactivity relationships and engineered proteins: Observation of Hammond behavior for the gross structure of the transition state and anti-Hammond behavior for structural elements for unfolding/folding of barnase. Biochemistry. 1995;34:6805–6814. doi: 10.1021/bi00020a027. [DOI] [PubMed] [Google Scholar]
- 43.Dudko OK, Hummer G, Szabo A. Theory, analysis, and interpretation of single-molecule force spectroscopy experiments. Proc Natl Acad Sci USA. 2008;105:15755–15760. doi: 10.1073/pnas.0806085105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li PT, Collin D, Smith SB, Bustamante C, Tinoco I., Jr Probing the mechanical folding kinetics of TAR RNA by hopping, force-jump, and force-ramp methods. Biophys J. 2006;90:250–260. doi: 10.1529/biophysj.105.068049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evans E, Ritchie K. Dynamic strength of molecular adhesion bonds. Biophys J. 1997;72:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Merkel R, Nassoy P, Leung A, Ritchie K, Evans E. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature. 1999;397:50–53. doi: 10.1038/16219. [DOI] [PubMed] [Google Scholar]
- 47.Matouschek A, Otzen DE, Itzhaki LS, Jackson SE, Fersht AR. Movement of the position of the transition state in protein folding. Biochemistry. 1995;34:13656–13662. doi: 10.1021/bi00041a047. [DOI] [PubMed] [Google Scholar]
- 48.Suzuki Y, Dudko OK. Single-molecule rupture dynamics on multidimensional landscapes. Phys Rev Lett. 2010;104:048101. doi: 10.1103/PhysRevLett.104.048101. [DOI] [PubMed] [Google Scholar]
- 49.Wright CF, Lindorff-Larsen K, Randles LG, Clarke J. Parallel protein-unfolding pathways revealed and mapped. Nat Struct Biol. 2003;10:658–662. doi: 10.1038/nsb947. [DOI] [PubMed] [Google Scholar]
- 50.Fersht AR, Itzhaki LS, elMasry NF, Matthews JM, Otzen DE. Single versus parallel pathways of protein folding and fractional formation of structure in the transition state. Proc Natl Acad Sci USA. 1994;91:10426–10429. doi: 10.1073/pnas.91.22.10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schlierf M, Yew ZT, Rief M, Paci E. Complex unfolding kinetics of single-domain proteins in the presence of force. Biophys J. 2010;99:1620–1627. doi: 10.1016/j.bpj.2010.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elms P, Chodera J, Bustamante C, Marqusee S. The limitations of constant-force-feedback experiments. Biophys J. 2012 doi: 10.1016/j.bpj.2012.06.051. 10.1016/j.bpj.2012.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Best RB, Hummer G. Protein folding kinetics under force from molecular simulation. J Am Chem Soc. 2008;130:3706–3707. doi: 10.1021/ja0762691. [DOI] [PubMed] [Google Scholar]
- 54.Fersht AR, Matouschek A, Serrano L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J Mol Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 55.Liu Z, Reddy G, O'Brien EP, Thirumalai D. Collapse kinetics and chevron plots from simulations of denaturant-dependent folding of globular proteins. Proc Natl Acad Sci USA. 2011;108:7787–7792. doi: 10.1073/pnas.1019500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.