

The crystallization and initial diffraction analysis of human Drp1 GTPase-GED fusion protein are reported.
Keywords: dynamin-related protein 1, GTPase domain, GTPase effector domain
Abstract
The mechano-enzyme dynamin-related protein 1 plays an important role in mitochondrial fission and is implicated in cell physiology. Dysregulation of Drp1 is associated with abnormal mitochondrial dynamics and neuronal damage. Drp1 shares structural and functional similarities with dynamin 1 with respect to domain organization, ability to self-assemble into spiral-like oligomers and GTP-cycle-dependent membrane scission. Structural studies of human dynamin-1 have greatly improved the understanding of this prototypical member of the dynamin superfamily. However, high-resolution structural information for full-length human Drp1 covering the GTPase domain, the middle domain and the GTPase effector domain (GED) is still lacking. In order to obtain mechanistic insights into the catalytic activity, a nucleotide-free GTPase-GED fusion protein of human Drp1 was expressed, purified and crystallized. Initial X-ray diffraction experiments yielded data to 2.67 Å resolution. The hexagonal-shaped crystals belonged to space group P21212, with unit-cell parameters a = 53.59, b = 151.65, c = 43.53 Å, one molecule per asymmetric unit and a solvent content of 42%. Expression of selenomethionine-labelled protein is currently in progress. Here, the expression, purification, crystallization and X-ray diffraction analysis of the Drp1 GTPase-GED fusion protein are presented, which form a basis for more detailed structural and biophysical analysis.
1. Introduction
Mitochondria play a critical role in diverse biological processes such as ATP synthesis, calcium homeostasis and apoptosis (Wallace & Fan, 2010 ▶). These highly dynamic organelles undergo frequent fusion and fission to regulate their number, morphology, distribution and function (Bereiter-Hahn & Vöth, 1994 ▶; Youle, 2005 ▶; Chen & Chan, 2009 ▶). The dynamin-related proteins (DRPs) are responsible for maintaining this fusion–fission equilibrium. They represent a subfamily of the dynamin superfamily, a group of highly conserved large GTPases which are known for their ability to self-assemble into high-order ring-like or spiral-like oligomers and to display oligomerization-dependent GTPase activity (Praefcke & McMahon, 2004 ▶; Lackner et al., 2009 ▶). Despite these common biochemical features, DRPs contain only three of the five domains typically found in the classical dynamins, e.g. dynamin-1: an N-terminal GTPase domain, a middle domain (MID) and a C-terminal GTPase effector domain (GED). Whereas the GTPase domain shows a high degree of sequence conservation throughout the superfamily, the MID and GED are more variable. Interestingly, the MID, together with intramolecular and intermolecular interactions formed by the GED, plays important roles during self-assembly (Hoppins et al., 2007 ▶).
Dynamin-related protein 1 (Drp1), a key regulator of mitochondrial fission, has an additional unstructured region between the MID and GED. This so-called insert B or variable domain (VD) has been suggested to play an important role in Drp1 regulation and self-assembly (Heymann & Hinshaw, 2009 ▶; Strack & Cribbs, 2012 ▶; Fig. 1 ▶ a). Drp1 is evolutionarily conserved throughout most kingdoms of life, indicating its fundamental importance to cell biology (Westermann, 2010 ▶; Reddy et al., 2011 ▶). In vivo studies with drp1 knockout mice or mice carrying drp1 mutations confirmed its essential role in cell survival (Wakabayashi et al., 2009 ▶; Waterham et al., 2007 ▶; Ishihara et al., 2009 ▶). In humans, at least six different Drp1 isoforms are known, which are expressed in a tissue-specific and cell-type-specific manner (Reddy et al., 2011 ▶; Ishihara et al., 2009 ▶; Chang & Blackstone, 2010 ▶). In vivo and in vitro studies showed that Drp1 exists in a dimer–tetramer equilibrium in the cytoplasm (Shin et al., 1999 ▶; Zhu et al., 2004 ▶; Chang et al., 2010 ▶; Bossy et al., 2010 ▶). As a response to cellular stimuli, Drp1 is recruited to the mitochondrial outer membrane, where it self-assembles into spiral-like higher oligomeric structures at mitochondrial fission sites. These Drp1 clusters can be visualized as ribbons surrounding mitochondria or foci (Zhu et al., 2004 ▶; Chang et al., 2010 ▶; Frank et al., 2001 ▶). Negative-stain electron microscopy revealed that Drp1 self-assembles into large spiral-shaped oligomers in the presence of GTP or nonhydrolysable GTP in vitro (Smirnova et al., 2001 ▶; Song et al., 2011 ▶; Yoon et al., 2001 ▶). Studies using liposomes, which mimic the mitochondrial outer membrane, revealed Drp1 aggregation at membrane liposomes concurrent with membrane deformation (Yoon et al., 2001 ▶). This finding suggests the involvement of Drp1 in membrane remodelling and scission. However, in vitro studies using Drp1 mutants and GTP analogues showed that GTP binding, but not GTP hydrolysis, is essential for oligomer assembly (Yoon et al., 2001 ▶; Smirnova et al., 2001 ▶). Indeed, it has been reported for the yeast homologue Dnm1 that the GTPase activity increases after assembly on liposomes followed by disassembly of the high-order oligomers (Ingerman et al., 2005 ▶; Mears et al., 2011 ▶; Mears & Hinshaw, 2008 ▶). Thus, GTP hydrolysis might induce a conformational change providing the mechanochemical work necessary for the mitochondrial fission process (Knott & Bossy-Wetzel, 2008 ▶; Otera & Mihara, 2011 ▶; Mears et al., 2011 ▶).
Figure 1.

Construct design and schematic representation of the Drp1 GG fusion protein. (a) Full-length Drp1 consists of an N-terminal GTPase domain (green) and a helical middle domain (blue) followed by an unstructured region (insert B or variable domain) and a C-terminal GED (pink). (b) The GG fusion protein consists of the complete GTPase domain (amino acids 1–327) fused to a C-terminal fragment of the GED (amino acids 711–736) via a (GS)4 linker.
Several factors that are required for membrane binding and regulation of Drp1 during this dynamic mechanism have already been identified (Dikov & Reichert, 2011 ▶; Otera et al., 2010 ▶; Palmer et al., 2011 ▶; Zhao et al., 2011 ▶). Recent evidence also suggests that diverse post-translational modifications of Drp1 regulate mitochondrial fission (Chang & Blackstone, 2010 ▶; Oettinghaus et al., 2012 ▶; Otera & Mihara, 2012 ▶; Elgass et al., 2012 ▶; Chang et al., 2010 ▶). However, their effects on Drp1 activity are incompletely understood and are a matter of current debate (Cho et al., 2009 ▶; Bossy et al., 2010 ▶; Chang & Blackstone, 2010 ▶).
Unravelling the molecular mechanism of human Drp1-mediated mitochondrial fission will allow us to modulate Drp1 activity in the pathogenesis of disease (Song et al., 2011 ▶). For example, recent studies have revealed that excessive mitochondrial fission in degenerative diseases is caused by increased Drp1 activity (Knott et al., 2008 ▶; Elgass et al., 2012 ▶; Manczak & Reddy, 2012 ▶).
High-resolution structural data for the human Drp1 GTPase domain will provide us with important insights into the mechanistic similarities and differences compared with dynamin-1. Construct design for the Drp1 GTPase domain was carried out on the basis of previously described GTPase-GED fusion protein structures of other dynamin superfamily members, such as human dynamin-1 (Chappie et al., 2010 ▶) and a plant Drp1 homologue (Chen et al., 2012 ▶). The Drp1 GG fusion protein shares 48 and 41% sequence identity with the GTPase-GED fusion proteins of human dynamin-1 (PDB entries 2x2e and 3zyc; Chappie et al., 2010 ▶, 2011 ▶) and Arabidopsis thaliana Drp1A (AtDrp1A; PDB entries 3t34 and 3t35; Yan et al., 2011 ▶), respectively. Here, we report the expression, purification, crystallization and X-ray crystallographic characterization of a native fusion protein containing the GTPase domain and the C-terminal part of the GED of human Drp1.
2. Experimental methods
2.1. Cloning
To produce a stable and homogenous but still functional protein for crystallization experiments, the GTPase domain of human Drp1 (amino acids 1–327) was fused to the C-terminal part of the GED (amino acids 711–736) by a (GS)4 linker (Fig. 1 ▶ b). By removing the MID and the unstructured insert B the formation of higher-order oligomers was prevented and protein stability was increased. To generate the Drp1 GTPase-GED (GG) fusion construct the following four primers were designed: 5′-CTGGTCATATGGAGGCGCTAATTCCTGTC-3′ containing an NdeI restriction site, 5′-CTGGTCTCGAGCCAAAGATGAGTCTCCCG-3′ containing an XhoI restriction site, 5′-GGTGAACCCGTGGATGGATCAGGATCAGGATCAGGATCA-3′ and 5′-ATCAGCTGCTTCTTTTGATCCTGATCCTGATCCTGATCC-3′. The K38A mutation was introduced by overlap PCR using the mutagenic primers 5′-GAACGCAGAGCAGCGGAGCAAGCTCAGTGCTAGAAAG-3′ and 5′-CTTTCTAGCACTGAGCTTGCTCCGCTGCTCTGCGTTC-3′. Drp1 fragments were PCR-amplified from template DNA containing full-length Drp1 isoform 2 in pET21 (GenBank Accession No. NM_012063.2) using iProof High-Fidelity DNA Polymerase (Bio-Rad). The PCR product was digested with NdeI and XhoI and cloned into pET21 (Novagen), resulting in the attachment of two non-native amino acids (leucine and glutamic acid) and a His6 tag at the C-terminus of the protein. The sequence was verified by DNA sequencing (Eurofins MWG Operon).
2.2. Expression
For protein expression, the Drp1 GG fusion construct and a construct containing a K38A substitution mutation in the GTPase domain rendering the Drp1 enzymatically inactive were transformed into Escherichia coli BL21 (DE3) Star cells and grown on LB plates containing 50 µg ml−1 ampicillin (Carl Roth) overnight at 310 K. Subsequently, cells were transferred into two Erlenmeyer flasks containing 600 ml LB medium with 50 µg ml−1 ampicillin and grown at 310 K with agitation at 230 rev min−1 until an OD600 of 0.8 was attained. After induction with 0.5 mM isopropyl β-thiogalactopyranoside (Carl Roth), expression was performed overnight at 298 K. The cells were harvested by centrifugation at 4500g for 20 min at 277 K. The cell pellets were dissolved in a 1/20 volume of lysis buffer [50 mM KH2PO4 pH 7.8, 300 mM NaCl, 10%(v/v) glycerol, 5 mM imidazole, 0.1 mg ml−1 lysozyme] and frozen at 253 K overnight before purification.
2.3. Protein purification
After thawing, the cells were lysed on ice by two cycles of sonication (Bandelin Electronic UW2070) at 70% of the maximum intensity amplitude for 3 min. DNA digestion was performed with 10 µg ml−1 DNase I (Fermentas) for 30 min at 293 K. The cell lysates were cleared by centrifugation at 18 000g for 20 min at 277 K. The supernatant was loaded onto Ni–NTA resin (GE Healthcare) pre-equilibrated in wash buffer [20 mM Tris pH 7.8, 300 mM NaCl, 40 mM imidazole, 10%(v/v) glycerol, 10%(w/v) sucrose]. To remove impurities, the nickel beads were washed with 30 column volumes of wash buffer. The protein was eluted with at least two column volumes of elution buffer [20 mM Tris pH 7.8, 300 mM NaCl, 10%(v/v) glycerol, 500 mM imidazole] and further purified on an ÄKTA FPLC system using a Superdex 75 10/300 GL column (GE Healthcare) pre-equilibrated with gel-filtration buffer (30 mM Tris pH 7.8, 100 mM NaCl, 2 mM DTT, 1 mM EDTA). The Drp1 GG fusion protein eluted as a monomer at a retention volume of approximately 11 ml (Fig. 2 ▶ a) and displayed virtually no impurities (Fig. 2 ▶ b). Amicon Ultra centrifugal filter units (10 kDa cutoff, Millipore) were used to concentrate the protein to 10 mg ml−1 and it was stored at 193 K. The protein concentration was determined by UV absorbance at 280 nm. Peptide mass fingerprinting (Toplab, Munich, Germany) unambiguously identified the native protein as the Drp1 GG fusion protein. Furthermore, the intact mass of 40.923 kDa determined by mass spectrometry (Toplab, Munich, Germany) corresponded well to the calculated mass of 40.956 kDa. Drp1 full-length isoform 2 was expressed and purified as described elsewhere (Song et al., 2011 ▶).
Figure 2.

Purification of the Drp1 GG fusion protein. (a) Size-exclusion analysis. After Ni–NTA purification, proteins were loaded onto a Superdex 75 gel-filtration column and fractions containing protein were collected. In the absence of nucleotides, the protein eluted as a monomer at a retention volume of approximately 11 ml, which corresponds to a molecular mass of approximately 40 kDa. The retention volumes of molecular-mass standards (GE Healthcare) are displayed at the top. (b) SDS–PAGE analysis. Protein fractions were loaded onto a 15% SDS–PAGE gel and visualized by Coomassie Brilliant Blue staining. Lane E, eluted protein; lane M, protein molecular-mass markers (labelled in kDa on the left). The retention volumes are indicated at the top.
2.4. Drp1 GG fusion protein shows GTPase activity
The Drp1 GG fusion protein was tested for GTPase activity using the continuous regenerative coupled GTPase assay (Ingerman & Nunnari, 2005 ▶; Fig. 3 ▶). The protein was equilibrated in a buffer consisting of 25 mM HEPES/PIPES pH 7.0, 150 mM NaCl and was diluted to a concentration of 0.1 mg ml−1. The absorbance (340 nm) correlating with NADH depletion at increasing GTP concentrations (0, 10, 20, 30, 50, 100, 250, 500 and 1000 µM) was measured at 303 K for 85 min with 32 s per cycle using an Infinite M200 plate reader (Tecan). Drp1 full-length isoform 2 and the enzymatically inactive K38A GG fusion protein (Smirnova et al., 1998 ▶) were used as positive and negative controls, respectively.
Figure 3.

The Drp1 GG fusion protein displays GTPase activity: steady-state GTPase activities of Drp1 GG fusion protein (native Drp1 GG fusion protein) in comparison to Drp1 full-length isoform 2 and enzymatically inactive K38A mutant (Drp1 GG fusion protein K38A). Data are the mean ± standard deviation from three independent measurements.
2.5. Protein crystallization
Crystallization of the Drp1 GG fusion protein (10 mg ml−1) was performed in gel-filtration buffer using the sitting-drop vapour-diffusion method at 293 K. Commercial sparse-matrix screens such as Index (Hampton Research) and JCSG-plus (Molecular Dimensions) were used to screen for initial crystal hits. Drops were prepared by mixing 0.4 µl reservoir solution with 0.4 µl protein solution using a Hydra II Plus One (Matrix Ltd) liquid-handling system and were equilibrated against 50 µl reservoir solution in 96-well sitting-drop plates (Greiner). Stacks of thin crystal plates (Fig. 4 ▶ a) were observed within 9 d in condition A [0.2 M lithium sulfate, 0.1 M bis-Tris pH 5.5, 25%(v/v) PEG 3350]. Furthermore, initial crystals in the form of clusters of needles were found in condition B (0.1 M sodium citrate pH 5.5, 20% PEG 3000).
Figure 4.

Crystal forms of the Drp1 GG fusion protein. (a) In condition A only star-shaped intergrown crystals were present which could not be reproduced. (b) Reproducible crystals from condition B were only about 50 × 40 × 10 µm in size.
Initial hits were refined manually by varying the buffer pH, the precipitant concentration or the protein concentration in 96-well plates. The crystals found in condition A were not reproducible, whereas a refined condition B (0.1 M sodium citrate pH 5, 27.5% PEG 3000) with a protein concentration of 1.2 mg ml−1, a drop size of 0.8 µl and a 1:1 protein:reservoir solution ratio resulted in thin hexagonal-shaped crystals (Fig. 4 ▶ b).
2.6. Data collection and processing
Refined crystals obtained using condition B were flash-cooled in liquid nitrogen without the addition of cryoprotectant. Diffraction data were collected on BL14.1 operated by the Helmholtz-Zentrum Berlin (HZB) at the BESSY II electron-storage ring (Berlin-Adlershof, Germany; Mueller et al., 2012 ▶) at a wavelength of 0.91841 Å. A total of 180 diffraction images (Fig. 5 ▶) with an oscillation range of 1.0° were recorded using a Rayonics MX225 3 × 3 CCD detector at a distance of 236.4 mm.
Figure 5.

Diffraction image of the Drp1 GG fusion protein: a diffraction image of the crystals produced in condition B collected on BL14.1 at BESSY II, Berlin, Germany.
All data were indexed, integrated, scaled and merged using XDS (Kabsch, 2010 ▶) using the XDSAPP graphical user interface (Krug et al., 2012 ▶). For further information regarding data-collection and processing statistics, see Table 1 ▶.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| Crystallization condition | 0.1 M sodium citrate pH 5.5, 20% PEG 3000 [condition B] |
| Unit-cell parameters (Å, °) | a = 53.59, b = 151.65, c = 43.53, α = β = γ = 90.00 |
| X-ray source | BL14.1, HZB, Berlin |
| Detector | Rayonics MX225 3 × 3 CCD |
| Space group | P21212 |
| Wavelength (Å) | 0.91841 |
| Crystal-to-detector distance (mm) | 236.4 |
| Exposure time per image (s) | 15 |
| Δϕ (°) | 1.0 |
| No. of images | 180 |
| Resolution (Å) | 43.76–2.67 (2.83–2.67) |
| Reflections observed | 76144 (11938) |
| Unique reflections | 10686 (1678) |
| Multiplicity | 7.13 (7.11) |
| Completeness of data (%) | 99.8 (99.6) |
| 〈I/σ(I)〉 | 9.73 (2.16) |
| R merge † (%) | 18.2 (93.6) |
| R r.i.m. ‡ (%) | 19.6 (100.9) |
| R p.i.m. § (%) | 7.3 (37.3) |
| Mosaicity (°) | 0.35 |
| Molecules per asymmetric unit | 1 |
| Solvent content (%) | 42 |
The merging R factor R
merge = 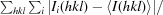
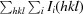 , where 〈I(hkl)〉 is the mean of the i individual measurements Ii(hkl) of the intensity of reflection hkl.
, where 〈I(hkl)〉 is the mean of the i individual measurements Ii(hkl) of the intensity of reflection hkl.
The redundancy-independent merging R factor R
r.i.m. = 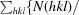
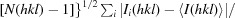 , where 〈I(hkl)〉 is the mean of the N(hkl) individual measurements Ii(hkl) of the intensity of reflection hkl.
, where 〈I(hkl)〉 is the mean of the N(hkl) individual measurements Ii(hkl) of the intensity of reflection hkl.
The precision-indicating merging R factor R
p.i.m. = 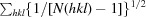
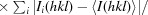 (Weiss, 2001 ▶) calculated with the program RMERGE available upon request from M. S. Weiss.
(Weiss, 2001 ▶) calculated with the program RMERGE available upon request from M. S. Weiss.
3. Results and discussion
Structural information from other DRP-family members indicates that the C-terminus of the GED is necessary in order to interact with both the carboxy-terminus and the amino-terminus of the GTPase domain and to stabilize the GTPase domain (Chappie et al., 2009 ▶). Therefore, a Drp1 GG fusion protein was designed by fusing the GTPase domain to the C-terminal part of the GED. The protein was cloned, expressed in E. coli and purified using a two-step procedure. A continuous coupled GTPase activity assay showed that the human Drp1 GG fusion protein is an active enzyme. However, the GG fusion protein showed reduced GTPase activity in comparison to the full-length protein (Fig. 3 ▶). The observed difference in GTPase activities might be attributed to the inability of the Drp1 GG fusion protein to assemble into higher-order oligomers, preventing it from reaching maximal GTPase activity. According to the recently published GG fusion protein structures of human dynamin and AtDrp1A (Chappie et al., 2010 ▶; Yan et al., 2011 ▶), we expect that the Drp1 GG fusion protein is able to dimerize via the GTPase domains but cannot form tetramers or GTPase-stimulating spiral-like structures.
Initial crystallization experiments were set up and after 9 d crystal plates could be observed in condition A consisting of 0.2 M lithium sulfate, 0.1 M bis-Tris pH 5.5, 25%(v/v) PEG 3350. Clusters of needles could be obtained in condition B consisting of 0.1 M sodium citrate pH 5.5, 20% PEG 3000 and gave three-dimensional hexagonal crystals after manual refinement of the conditions.
Crystals grown in condition B showed diffraction to 2.67 Å resolution and belonged to space group P21212, with unit-cell parameters a = 53.59, b = 151.65, c = 43.53 Å. Calculation of the Matthews coefficient revealed one molecule in the asymmetric unit with a solvent content of 42% (Matthews, 1968 ▶). For additional information on data collection and processing, see Table 1 ▶. As molecular replacement using human dynamin GG fusion protein (PDB entry 2x2e) as the search model failed, expression of selenomethionine-labelled protein has been initiated.
Acknowledgments
Data collection was performed at the Helmholtz-Zentrum Berlin für Materialien und Energie Elektronenspeicherring BESSY II, Berlin, Germany and was supported by European Community’s Seventh Framework Program (FP7/2007-2013) under grant agreement No. 226716. SP was supported by Erwin-Schrödinger Postdoc Fellowships from the Austrian Science Fund (J3173-N17). We thank the Austrian Science Fund (FWF W_01213) for financial support. EBW was supported by NIH grant R01 NS055195. Furthermore we would like to thank Christina Doppler for cloning the K38A mutant.
References
- Bereiter-Hahn, J. & Vöth, M. (1994). Microsc. Res. Tech. 27, 198–219. [DOI] [PubMed]
- Bossy, B., Petrilli, A., Klinglmayr, E., Chen, J., Lutz-Meindl, U., Knott, A. B., Masliah, E., Schwarzenbacher, R. & Bossy-Wetzel, E. (2010). J. Alzheimers Dis. 20, S513–S526. [DOI] [PMC free article] [PubMed]
- Chang, C.-R. & Blackstone, C. (2010). Ann. N. Y. Acad. Sci. 1201, 34–39. [DOI] [PMC free article] [PubMed]
- Chang, C.-R., Manlandro, C. M., Arnoult, D., Stadler, J., Posey, A. E., Hill, R. B. & Blackstone, C. (2010). J. Biol. Chem. 285, 32494–32503. [DOI] [PMC free article] [PubMed]
- Chappie, J. S., Acharya, S., Leonard, M., Schmid, S. L. & Dyda, F. (2010). Nature (London), 465, 435–440. [DOI] [PMC free article] [PubMed]
- Chappie, J. S., Acharya, S., Liu, Y. W., Leonard, M., Pucadyil, T. J. & Schmid, S. L. (2009). Mol. Biol. Cell, 20, 3561–3571. [DOI] [PMC free article] [PubMed]
- Chappie, J. S., Mears, J. A., Fang, S., Leonard, M., Schmid, S. L., Milligan, R. A., Hinshaw, J. E. & Dyda, F. (2011). Cell, 147, 209–222. [DOI] [PMC free article] [PubMed]
- Chen, H. & Chan, D. C. (2009). Hum. Mol. Genet. 18, R169–R176. [DOI] [PMC free article] [PubMed]
- Chen, X., Xu, X., Sun, Y., Zhou, J., Ma, Y., Yan, L. & Lou, Z. (2012). Acta Cryst. F68, 69–72. [DOI] [PMC free article] [PubMed]
- Cho, D. H., Nakamura, T., Fang, J., Cieplak, P., Godzik, A., Gu, Z. & Lipton, S. A. (2009). Science, 324, 102–105. [DOI] [PMC free article] [PubMed]
- Dikov, D. & Reichert, A. S. (2011). EMBO J. 30, 2751–2753. [DOI] [PMC free article] [PubMed]
- Elgass, K., Pakay, J., Ryan, M. T. & Palmer, C. S. (2012). Biochim. Biophys. Acta, doi:10.1016/j.bbamcr.2012.05.002. [DOI] [PubMed]
- Frank, S., Gaume, B., Bergmann-Leitner, E. S., Leitner, W. W., Robert, E. G., Catez, F., Smith, C. L. & Youle, R. J. (2001). Dev. Cell, 1, 515–525. [DOI] [PubMed]
- Heymann, J. A. & Hinshaw, J. E. (2009). J. Cell Sci. 122, 3427–3431. [DOI] [PMC free article] [PubMed]
- Hoppins, S., Lackner, L. & Nunnari, J. (2007). Annu. Rev. Biochem. 76, 751–780. [DOI] [PubMed]
- Ingerman, E. & Nunnari, J. (2005). Methods Enzymol. 404, 611–619. [DOI] [PubMed]
- Ingerman, E., Perkins, E. M., Marino, M., Mears, J. A., McCaffery, J. M., Hinshaw, J. E. & Nunnari, J. (2005). J. Cell Biol. 170, 1021–1027. [DOI] [PMC free article] [PubMed]
- Ishihara, N. et al. (2009). Nature Cell Biol. 11, 958–966. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Knott, A. B. & Bossy-Wetzel, E. (2008). Ann. N. Y. Acad. Sci. 1147, 283–292. [DOI] [PMC free article] [PubMed]
- Knott, A. B., Perkins, G., Schwarzenbacher, R. & Bossy-Wetzel, E. (2008). Nature Rev. Neurosci. 9, 505–518. [DOI] [PMC free article] [PubMed]
- Krug, M., Weiss, M. S., Heinemann, U. & Mueller, U. (2012). J. Appl. Cryst. 45, 568–572.
- Lackner, L. L., Horner, J. S. & Nunnari, J. (2009). Science, 325, 874–877. [DOI] [PMC free article] [PubMed]
- Manczak, M. & Reddy, P. H. (2012). Hum. Mol. Genet. 21, 2538–2547. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Mears, J. A. & Hinshaw, J. E. (2008). Methods Cell Biol. 88, 237–256. [DOI] [PMC free article] [PubMed]
- Mears, J. A., Lackner, L. L., Fang, S., Ingerman, E., Nunnari, J. & Hinshaw, J. E. (2011). Nature Struct. Mol. Biol. 18, 20–26. [DOI] [PMC free article] [PubMed]
- Mueller, U., Darowski, N., Fuchs, M. R., Förster, R., Hellmig, M., Paithankar, K. S., Pühringer, S., Steffien, M., Zocher, G. & Weiss, M. S. (2012). J. Synchrotron Rad. 19, 442–449. [DOI] [PMC free article] [PubMed]
- Oettinghaus, B., Licci, M., Scorrano, L. & Frank, S. (2012). Acta Neuropathol. 123, 189–203. [DOI] [PubMed]
- Otera, H. & Mihara, K. (2011). Small GTPases, 2, 167–172. [DOI] [PMC free article] [PubMed]
- Otera, H. & Mihara, K. (2012). Int. J. Cell Biol. 2012, 821676. [DOI] [PMC free article] [PubMed]
- Otera, H., Wang, C., Cleland, M. M., Setoguchi, K., Yokota, S., Youle, R. J. & Mihara, K. (2010). J. Cell Biol. 191, 1141–1158. [DOI] [PMC free article] [PubMed]
- Palmer, C. S., Osellame, L. D., Laine, D., Koutsopoulos, O. S., Frazier, A. E. & Ryan, M. T. (2011). EMBO Rep. 12, 565–573. [DOI] [PMC free article] [PubMed]
- Praefcke, G. J. & McMahon, H. T. (2004). Nature Rev. Mol. Cell Biol. 5, 133–147. [DOI] [PubMed]
- Reddy, P. H., Reddy, T. P., Manczak, M., Calkins, M. J., Shirendeb, U. & Mao, P. (2011). Brain Res. Rev. 67, 103–118. [DOI] [PMC free article] [PubMed]
- Shin, H.-W., Takatsu, H., Mukai, H., Munekata, E., Murakami, K. & Nakayama, K. (1999). J. Biol. Chem. 274, 2780–2785. [DOI] [PubMed]
- Smirnova, E., Griparic, L., Shurland, D. L. & van der Bliek, A. M. (2001). Mol. Biol. Cell, 12, 2245–2256. [DOI] [PMC free article] [PubMed]
- Smirnova, E., Shurland, D. L., Ryazantsev, S. N. & van der Bliek, A. M. (1998). J. Cell Biol. 143, 351–358. [DOI] [PMC free article] [PubMed]
- Song, W. et al. (2011). Nature Med. 17, 377–382. [DOI] [PMC free article] [PubMed]
- Strack, S. & Cribbs, J. T. (2012). J. Biol. Chem. 287, 10990–11001. [DOI] [PMC free article] [PubMed]
- Wakabayashi, J., Zhang, Z., Wakabayashi, N., Tamura, Y., Fukaya, M., Kensler, T. W., Iijima, M. & Sesaki, H. (2009). J. Cell Biol. 186, 805–816. [DOI] [PMC free article] [PubMed]
- Wallace, D. C. & Fan, W. (2010). Mitochondrion, 10, 12–31. [DOI] [PMC free article] [PubMed]
- Waterham, H. R., Koster, J., van Roermund, C. W., Mooyer, P. A., Wanders, R. J. & Leonard, J. V. (2007). N. Engl. J. Med. 356, 1736–1741. [DOI] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst. 34, 130–135.
- Westermann, B. (2010). Nature Rev. Mol. Cell Biol. 11, 872–884. [DOI] [PubMed]
- Yan, L., Ma, Y., Sun, Y., Gao, J., Chen, X., Liu, J., Wang, C., Rao, Z. & Lou, Z. (2011). J. Mol. Cell. Biol. 3, 378–381. [DOI] [PubMed]
- Yoon, Y., Pitts, K. R. & McNiven, M. A. (2001). Mol. Biol. Cell, 12, 2894–2905. [DOI] [PMC free article] [PubMed]
- Youle, R. J. (2005). Dev. Cell, 8, 298–299. [DOI] [PubMed]
- Zhao, J., Liu, T., Jin, S., Wang, X., Qu, M., Uhlén, P., Tomilin, N., Shupliakov, O., Lendahl, U. & Nistér, M. (2011). EMBO J. 30, 2762–2778. [DOI] [PMC free article] [PubMed]
- Zhu, P.-P., Patterson, A., Stadler, J., Seeburg, D. P., Sheng, M. & Blackstone, C. (2004). J. Biol. Chem. 279, 35967–35974. [DOI] [PubMed]