

GIM-1 is a member of the class B carbapenemases and has a wide spectrum of activity against carbapenems, penicillins and extended-spectrum cephalosporins, but not aztreonam. In this study, GIM-1 was cloned, overexpressed and crystallized.
Keywords: antibiotic resistance, metallo-β-lactamases, GIM-1, drug targets
Abstract
GIM-1 is a member of the class B carbapenemases (metallo-β-lactamases; MBLs) and has a wide spectrum of activity against carbapenems, penicillins and extended-spectrum cephalosporins, but not aztreonam. GIM-1 presents an enormous challenge to infection control, particularly in the eradication of Gram-negative pathogens including Enterobacteriaceae, Pseudomonas aeruginosa, Acinetobacter baumannii and nonfermenters. There are presently few or no drugs in late-stage development for these pathogens and GIM-1 is a potential target for the development of antimicrobial agents against pathogens producing MBLs. In this study, GIM-1 was cloned, overexpressed and crystallized. The GIM-1 crystals diffracted to 1.4 Å resolution and belonged to the orthorhombic space group P212121, with unit-cell parameters a = 38.5, b = 67.6, c = 72.8 Å. One molecule is present in the asymmetric unit, with a corresponding V M of 1.69 Å3 Da−1 and a solvent content of 27.1%.
1. Introduction
Carbapenems are increasingly being utilized against a variety of infections owing to the emergence of bacteria that produce extended-spectrum β-lactamases (ESBLs) in the Enterobacteriaceae, in particular Escherichia coli, Klebsiella pneumoniae and other enteric bacteria (Lee et al., 2009 ▶, 2012 ▶; Overturf, 2010 ▶). Carbapenems such as imipenem and meropenem are often the drugs of last resort for ESBL-producing pathogens (Overturf, 2010 ▶). Presently, β-lactamases are classified into four distinct classes based on structural similarity (classes A, B, C and D; Bush & Jacoby, 2010 ▶; Frère, 1995 ▶). Class B β-lactamases are zinc-dependent enzymes that are also known as metallo-β-lactamases (MBLs) and are able to inactivate almost all β-lactams, including carbapenems (Crowder et al., 2006 ▶). Increasing numbers (more than 100 variants) of these β-lactamases are being found in the clinic; their genes are primarily located on plasmids (Papp-Wallace et al., 2011 ▶; Queenan & Bush, 2007 ▶). Recently, the continued occurrence of GIM-1, a class B MBL, has been reported and understanding the molecular details of GIM-1 has become more important (Rieber, Frontzek & Pfeifer, 2012 ▶; Rieber, Frontzek, von Baum et al., 2012 ▶). Their potential for wide dispersal together with their broad substrate specificity highlights the importance of investigating the structural details of these enzymes.
There are over 100 known MBL variants and numerous crystal structures have been reported, including those of IMP-1 (active on imipenem; Concha et al., 2000 ▶), VIM-2 (Verona integron-encoded metallo-β-lactamase; Yamaguchi et al., 2007 ▶) and SPM-1 (Sao Paulo metallo-β-lactamase; Murphy et al., 2006 ▶). GIM-1 has 44.3, 27.2 and 22.8% sequence identity to mature IMP-1, VIM-1 and SPM-1, respectively. These three MBLs are clinically related to GIM-1 (German imipenemase; Castanheira et al., 2004 ▶). Unlike the other MBLs, which showed a more significant activity towards imipenem than towards meropenem, GIM-1 demonstrated similar kinetic (k cat/K m) ratios with imipenem and meropenem (Castanheira et al., 2004 ▶). In order to elucidate the differing kinetic properties of GIM-1, we expressed, purified, crystallized GIM-1 and performed a preliminary X-ray crystallographic analysis.
2. Materials and methods
2.1. Cloning
The class B carbapenemase GIM-1 was first isolated from Pseudomonas aeruginosa 73-5671 (Castanheira et al., 2004 ▶). To express the GIM-1 gene (bla GIM-1) in E. coli, a codon-optimized bla GIM-1 gene was chemically synthesized using GenScript technology (GenScript, Piscataway, New Jersey, USA) and amplified by polymerase chain reaction (PCR). The sequences of the forward and reverse oligonucleotide primers designed from the bla GIM-1 gene for PCR were 5′-ATA CAT ATG CAC CAT CAT CAT CAT CAT GAC GAC GAC GAC AAG CAG GGT CAT AAA CCG CTG GA-3′ (NdeI restriction site in bold) and 5′-GAG CTC GAG TTA GTC TGC AGA TGC TTC GGC A-3′ (XhoI restriction site in bold), respectively. The underlined bases represent a hexahistidine-tag site and the bases in italics indicate an enterokinase-recognition site. The amplified gene was double-digested with NdeI and XhoI and then inserted into the pET-30a expression vector (Novagen, Madison, Wisconsin, USA) which was digested with the same DNA restriction enzymes to produce the pET-30a/His6-bla GIM-1 plasmid. After verifying the DNA sequence, the plasmid DNA was transformed into E. coli strain BL21 (DE3) for overexpression of His6-GIM-1.
2.2. Overexpression and purification
The transformed cells were grown to an OD600 of 0.5 at 310 K in Luria–Bertani medium (Difco, Detroit, Michigan, USA) containing 50 µg ml−1 kanamycin. Expression of His6-GIM-1 was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 16 h at 289 K. The cells were harvested by centrifugation at 3000g (Supra 30K A1000S-4 rotor, Hanil, Seoul, Republic of Korea) for 30 min at 277 K. The resulting cell pellet was resuspended in ice-cold lysis buffer (10 mM Tris–HCl pH 7.0) and homogenized by sonication (Sibata, Saitama, Japan). The crude lysate was centrifuged at 19 960g (Hanil) for 30 min at 277 K and the clarified supernatant was loaded onto a His-Bind column (Novagen, Wisconsin, USA) equilibrated with binding buffer (20 mM Tris–HCl, 10 mM imidazole, 500 mM NaCl pH 7.9). His6-GIM-1 was eluted with binding buffer containing 1 M imidazole. Eluted fractions containing His6-GIM-1 were pooled and concentrated to a volume of approximately 1 ml using a Vivaspin 20 concentrator (Sartorius, Göttingen, Germany). The His6 tag was removed using enterokinase according to the manufacturer’s instructions (Roche Molecular Biochemicals, Mannheim, Germany). The reaction mixture was desalted and concentrated using a Fast Desalting column (GE Healthcare Biosciences, Uppsala, Sweden) and then loaded onto a Superdex 200 prep-grade column (GE Healthcare) previously equilibrated with buffer A (10 mM MES pH 6.8) for further purification by gel filtration. The homogeneity of the purified protein was analyzed via SDS–PAGE (Fig. 1 ▶ a). The purified GIM-1 without the His6 tag was dialyzed against buffer A (10 mM MES pH 6.8) and was subsequently concentrated to 7 mg ml−1 for crystallization trials.
Figure 1.

Purification and crystallization of GIM-1. (a) 12% SDS–PAGE of purified GIM-1. The left lane contains molecular-mass markers (labelled in kDa). (b) Crystal of GIM-1 with dimensions of 0.3 × 0.3 × 0.2 mm.
2.3. Crystallization
Crystallization conditions were initially screened by the sitting-drop vapour-diffusion method using a Hydra II eDrop automated pipetting system (Matrix) at 287 K. The initial crystallization conditions tested were from the Index kit (Hampton Research). After one week, crystals were observed in Index kit condition No. 96 [0.15 M potassium bromide, 30%(w/v) PEG MME 2000]. We optimized the initial crystals using the sitting-drop vapour-diffusion method in Cryschem plates (Hampton Research). Drops consisted of 0.5 µl protein solution (7 mg ml−1) and 0.5 µl reservoir solution and were equilibrated against 70 µl reservoir solution at 287 K. Crystallization conditions were optimized by changing the concentrations of the major precipitant PEG MME 2000 and the salt potassium bromide from 27 to 32% and from 0.05 to 0.30 M, respectively. Various pH values from pH 5.5 to 8.0 were also used in the optimization. Only in 0.1 M bis-tris pH 5.5 were single crystals observed. Using a higher protein concentration of 18 mg ml−1, larger crystals were obtained. The final well diffracting crystals were obtained from sitting drops consisting of 1 µl protein solution (18 mg ml−1) and 1 µl reservoir solution [0.1 M bis-tris pH 5.5, 0.25 M potassium bromide, 27%(w/v) PEG MME 2000]. Crystals were cryoprotected in reservoir solution supplemented with 17–21%(v/v) PEG 400.
2.4. X-ray data collection
Crystals were mounted in a loop and transferred into cryoprotectant solution prior to cooling in liquid nitrogen. The cooled crystals were mounted on the goniometer in a stream of cold nitrogen at 100 K. X-ray diffraction data were collected using an ADSC Q315r detector on beamline 5C SB II at the Pohang Light Source (PLS), Republic of Korea. X-ray diffraction data were collected to 1.4 Å resolution for the native protein. All data were collected with 1° oscillation per frame to give a total of 360° of data. All data were integrated and scaled using DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
Initially, intergrown crystals were obtained by mixing 0.5 µl protein solution with 0.5 µl reservoir solution consisting of 0.15 M potassium bromide, 30%(w/v) PEG MME 2000. Although the intergrown crystals could be reproduced using the hanging-drop vapour-diffusion method, they could not be improved to single crystals. We switched the crystallization method from hanging drops to sitting drops, which led to the appearance of single crystals. After optimization, the best crystals were generated in sitting drops with a reservoir solution consisting of 0.1 M bis-tris pH 5.5, 0.25 M potassium bromide, 27%(w/v) PEG MME 2000. The crystals grew to maximum dimensions of approximately 0.3 × 0.3 × 0.2 mm in two weeks (Fig. 1 ▶ b).
X-ray diffraction data were collected at the Pohang Light Source (PLS), Republic of Korea. Data-collection and processing statistics are given in Table 1 ▶. Auto-indexing was performed with DENZO and the results indicated that the crystals belonged to space group P212121, with unit-cell parameters a = 38.5, b = 67.6, c = 72.8 Å. For space group P212121 one monomer is present in the asymmetric unit with a corresponding V M of 1.69 Å3 Da−1 and a solvent content of 27.1%. Molecular replacement (MR) was performed with MOLREP (Vagin & Teplyakov, 2010 ▶) using the IMP-1 metallo-β-lactamase from P. aeruginosa (44.3% sequence identity; PDB entry 1dd6; Concha et al., 2000 ▶) as the search model. The MR solution provided informative 2F o − F c and F o − F c electron-density maps for model improvement. The initial R and R free values from the MR solution were 29.4% and 32.1%, respectively, and the figure of merit was 77.2. The structural details will be described in a separate paper.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Beamline | 5C SB II, PAL |
| Wavelength (Å) | 0.97951 |
| Resolution range (Å) | 50.00–1.40 (1.42–1.40) |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 38.5, b = 67.6, c = 72.8 |
| Total No. of reflections | 439195 |
| No. of unique reflections | 37231 |
| Completeness (%) | 97.5 (82.7) |
| Molecules per asymmetric unit | 1 |
| Solvent content (%) | 27.1 |
| Average I/σ(I) | 77.8 (16.0) |
| R merge † (%) | 7.0 (21.6) |
| Multiplicity | 11.8 (7.8) |
R
merge = 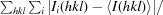
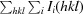 , where I(hkl) is the intensity of reflection hkl,
, where I(hkl) is the intensity of reflection hkl, 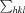 is the sum over all reflections and
is the sum over all reflections and 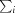 is the sum over i measurements of reflection hkl.
is the sum over i measurements of reflection hkl.
Acknowledgments
We warmly acknowledge the staff at beamline 5C SB II of the Pohang Light Source (PLS), Republic of Korea for their assistance. This study was supported by research grants from the National Research Foundation of Korea (NRF) funded by the Korean government (MEST; No. 2012-0008737), the Marine and Extreme Genome Research Center Program of the Ministry of Land, Transport and Maritime Affairs, Republic of Korea and the Next Generation BioGreen 21 Program (Nos. SA00005735 and PJ008174) of the Rural Development Administration of the Republic of Korea.
References
- Bush, K. & Jacoby, G. A. (2010). Antimicrob. Agents Chemother. 54, 969–976. [DOI] [PMC free article] [PubMed]
- Castanheira, M., Toleman, M. A., Jones, R. N., Schmidt, F. J. & Walsh, T. R. (2004). Antimicrob. Agents Chemother. 48, 4654–4661. [DOI] [PMC free article] [PubMed]
- Concha, N. O., Janson, C. A., Rowling, P., Pearson, S., Cheever, C. A., Clarke, B. P., Lewis, C., Galleni, M., Frère, J.-M., Payne, D. J., Bateson, J. H. & Abdel-Meguid, S. S. (2000). Biochemistry, 39, 4288–4298. [DOI] [PubMed]
- Crowder, M. W., Spencer, J. & Vila, A. J. (2006). Acc. Chem. Res. 39, 721–728. [DOI] [PubMed]
- Frère, J.-M. (1995). Mol. Microbiol. 16, 385–395. [DOI] [PubMed]
- Lee, J. H., Bae, I. K. & Lee, S. H. (2012). Med. Res. Rev. 32, 216–232. [DOI] [PubMed]
- Lee, J. H., Jeong, S. H., Cha, S.-S. & Lee, S. H. (2009). PLoS Pathog. 5, e1000221. [DOI] [PMC free article] [PubMed]
- Murphy, T. A., Catto, L. E., Halford, S. E., Hadfield, A. T., Minor, W., Walsh, T. R. & Spencer, J. (2006). J. Mol. Biol. 357, 890–903. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 277, 307–326. [DOI] [PubMed]
- Overturf, G. D. (2010). Pediatr. Infect. Dis. J. 29, 68–70. [DOI] [PubMed]
- Papp-Wallace, K. M., Endimiani, A., Taracila, M. A. & Bonomo, R. A. (2011). Antimicrob. Agents Chemother. 55, 4943–4960. [DOI] [PMC free article] [PubMed]
- Queenan, A. M. & Bush, K. (2007). Clin. Microbiol. Rev. 20, 440–458. [DOI] [PMC free article] [PubMed]
- Rieber, H., Frontzek, A. & Pfeifer, Y. (2012). Antimicrob. Agents Chemother. 56, 4945–4947 [DOI] [PMC free article] [PubMed]
- Rieber, H., Frontzek, A., von Baum, H. & Pfeifer, Y. (2012). J. Antimicrob. Chemother. 67, 1043–1045. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Yamaguchi, Y., Jin, W., Matsunaga, K., Ikemizu, S., Yamagata, Y., Wachino, J., Shibata, N., Arakawa, Y. & Kurosaki, H. (2007). J. Med. Chem. 50, 6647–6653. [DOI] [PubMed]