

Crystals of S1-domain-truncated X. campestris PNPase were grown in the presence of c-di-GMP and diffracted to a resolution of 2.00 Å.
Keywords: PNPases, c-di-GMP, Xanthomonas campestris, pathogens
Abstract
Bacterial polynucleotide phosphorylase (PNPase) is a 3′–5′ processive exoribonuclease that participates in mRNA turnover and quality control of rRNA precursors in many bacterial species. It also associates with the RNase E scaffold and other components to form a multi-enzyme RNA degradasome machinery that performs a wider regulatory role in degradation, quality control and maturation of mRNA and noncoding RNA. Several crystal structures of bacterial PNPases, as well as some biological activity studies, have been published. However, how the enzymatic activity of PNPase is regulated is less well understood. Recently, Escherichia coli PNPase was found to be a direct c-di-GMP binding target, raising the possibility that c-di-GMP may participate in the regulation of RNA processing. Here, the successful cloning, purification and crystallization of S1-domain-truncated Xanthomonas campestris PNPase (XcPNPaseΔS1) in the presence of c-di-GMP are reported. The crystals belonged to the monoclinic space group C2, with unit-cell parameters a = 132.76, b = 128.38, c = 133.01 Å, γ = 93.3°, and diffracted to a resolution of 2.00 Å.
1. Introduction
RNA turnover is an important mechanism in gene regulation and quality control in almost all kingdoms of life (Carpousis, 2007 ▶; Gorna et al., 2011 ▶; Kaberdin et al., 2011 ▶). It enables bacteria to adjust their gene products in response to environmental change more rapidly and economically than translational regulation (Grunberg-Manago, 1999 ▶). While eukaryotic mRNAs are mainly degraded by the protein complex exosome (Symmons et al., 2002 ▶; Raijmakers et al., 2004 ▶; Houseley et al., 2006 ▶), bacterial mRNAs are degraded by PNPase, which adopts a similar domain organization to the eukaryotic exosome core complex (Marcaida et al., 2006 ▶; Lin-Chao et al., 2007 ▶). Studies have also accumulated showing that some PNPases are correlated with small RNA function (De Lay & Gottesman, 2011 ▶), RNA transport (Wang et al., 2010 ▶) or bacterial virulence-gene expression (Ygberg et al., 2006 ▶). Intriguingly, a small fraction of bacterial PNPases are associated with the RNase E scaffold and other components such as RhlB helicase, enolase etc. to form a large multifunctional degradasome complex that performs a wider regulatory role in degradation, quality control and maturation of mRNA and noncoding RNA (Raijmakers et al., 2004 ▶; Marcaida et al., 2006 ▶; Carpousis, 2007 ▶; Gorna et al., 2011 ▶).
Bacterial PNPase is a 3′–5′ exoribonuclease. However, it uses phosphate instead of water to catalyze RNA phosphorolysis, generating nucleotide diphosphates (NDPs). Under conditions of abundant NDPs and low phosphate, it also catalyzes the reverse reaction to extend RNA from the 3′-end (Mohanty & Kushner, 2006 ▶). Each PNPase protein contains a tandem of two N-terminal RNase PH domains and an α-helical domain followed by C-terminal S1 and KH domains that are involved in RNA binding. Several bacterial PNPase crystal structures with or without the S1/KH domains have been solved to date (Symmons et al., 2000 ▶; Shi et al., 2008 ▶; Nurmohamed et al., 2009 ▶; Hardwick et al., 2012 ▶). They all adopt a homotrimer that reveals a doughnut-shaped structure formed by the six RNase PH domains. The overall organization of the hexameric ring structure comprising the S1/KH domain at the top and the α-helical domain at the bottom is similar to the archaeal and human exosome core structures. This aspect indicates an evolutionary link in structure and function between bacterial PNPases and the human exosome (Symmons et al., 2002 ▶; Lin-Chao et al., 2007 ▶).
However, regulation of PNPase activity has been less investigated to date, although some studies have been published and have started to shed light on this interesting subject. For example, PNPase activity was found to be modulated by cell metabolites and metal-chelated citrate was found to bind in the active site of Escherichia coli PNPase (EcPNPase) to inhibit its enzymatic activity. In contrast, metal-free citrate appears to bind at a vestigial active site and seems to stimulate EcPNPase activity (Nurmohamed et al., 2011 ▶). These studies suggest that RNA degradation is correlated with the central metabolism of the E. coli cell. Furthermore, EcPNPase activity was also found to be allosterically inhibited by ATP (Favero et al., 2008 ▶), and homologous PNPases from Nonomuraea sp. and Streptomyces were inhibited by the signalling molecules (p)ppGpp (Gatewood & Jones, 2010 ▶; Siculella et al., 2010 ▶), further establishing a link between the RNA-degradation machines and the metabolism or energy state in the E. coli cell.
c-di-GMP is a unique bacterial secondary-messenger molecule that was discovered more than 20 years ago. It was found to play a regulatory role in cellulose synthesis in Acetobacter xylinum (Ross et al., 1990 ▶). Recently, however, it has been found to play crucial roles in determining bacterial lifestyle changes, including transformation between biofilm formation and virulence state (Römling et al., 2005 ▶; Jenal & Malone, 2006 ▶; Hengge, 2009 ▶; Schirmer & Jenal, 2009 ▶). Although extensive investigations have been carried out, many intriguing questions regarding c-di-GMP still remain. For example, it is still unclear how many distinct c-di-GMP receptors are available, how these receptors perform their functions and how such diverse functions are controlled in the cell. Some representative receptor examples include transcriptional factors such as Clp (Leduc & Roberts, 2009 ▶; Chin et al., 2010 ▶; Tao et al., 2010 ▶), degenerate GGDEF or EAL domains (Navarro et al., 2009 ▶, 2011 ▶), PilZ domains (Amikam & Galperin, 2006 ▶; Benach et al., 2007 ▶; Li et al., 2009 ▶, 2011 ▶; Habazettl et al., 2011 ▶), RNA riboswitches (Smith et al., 2009 ▶) etc. Indeed, the search for novel c-di-GMP receptors is still ongoing (Römling, 2011 ▶; Sondermann et al., 2011 ▶; Ryan et al., 2012 ▶).
Interestingly, it has very recently been demonstrated that c-di-GMP can bind reasonably well to EcPNPase (K d = 2.9 µM) to mediate signal-dependent RNA processing (Tuckerman et al., 2011 ▶). This novel type of mRNA-degradation control by c-di-GMP may represent a broad and interesting type of RNA regulation by c-di-GMP and is worthy of further investigation. Xanthomonas is a large genus of Gram-negative bacteria that cause serious disease in hundreds of plant hosts, including many economically important crops (Ryan et al., 2011 ▶). It is also a model bacterium for studying bacteria–plant interactions because it contains a significant number of enzymes that are correlated with c-di-GMP metabolism. Hence, it is interesting to investigate how c-di-GMP interacts with XcPNPase to control mRNA turnover. In this work, we report the successful purification of S1-domain-truncated PNPase from X. campestris pv. campestris strain 17 (XcPNPaseΔS1). The crystallization and preliminary X-ray diffraction characterization of the XcPNPaseΔS1 have also been studied in the presence of c-di-GMP.
2. Materials and methods
2.1. Reagents
C-di-GMP was produced by an enzymatic method using an altered thermophilic DGC enzyme as described previously (Rao et al., 2009 ▶).
2.2. Cloning, expression and purification
The PNPaseΔS1 gene fragment was PCR-amplified directly from a local X. campestris genome (Xc17) with a forward 5′-TACTTCCAATCCAATGCTACACCTAAGGAAAAAACCTCCGTGG primer and a reverse 5′-TTATCCACTTCCAATGTTACGAGGTGATCTGCTCGATGCG primer (the linker sequences are italicized). A ligation-independent cloning (LIC) approach (Aslanidis & de Jong, 1990 ▶) was carried out to obtain the desired construct according to a previously published protocol (Wu et al., 2005 ▶). The final construct codes for an N-terminal His6 tag, a 17-amino-acid linker and the XcPNPaseΔS1 target protein (625 amino acids in total) under the control of the T7 promoter. The vector was transformed into E. coli BL21 (DE3) host cells, which were grown in LB medium at 310 K until an OD600 of 0.6 was attained. Overexpression of the His6-tagged target protein was induced by the addition of 0.5 mM IPTG at 293 K for 20 h. The cells were harvested, resuspended in equilibration buffer (50 mM Tris–HCl pH 8.0, 500 mM NaCl, 5% glycerol) and lysed using a microfluidizer (Microfluidics). Most of the tagged target protein was present in the soluble fraction (Fig. 1 ▶ b). After centrifugation, the target protein was purified by immobilized metal-affinity chromatography (IMAC) on a nickel column (Sigma) and eluted with a 50–300 mM imidazole gradient in 50 mM Tris–HCl pH 8.0, 500 mM NaCl, 5% glycerol. Fractions containing XcPNPaseΔS1 were monitored by SDS–PAGE, recombined and dialyzed repeatedly against 20 mM Tris–HCl pH 8.0, 400 mM NaCl, 5% glycerol. After buffer exchange, the His6 tag and linker were cleaved from XcPNPaseΔS1 by TEV (tobacco etch virus) protease at 295 K for 16 h and were removed by immobilized metal-affinity chromatography (IMAC) on a nickel column (Sigma). The final protein had greater than 95% purity and contained a non-native tripeptide (Ser-Asn-Ala) followed by the target protein sequence (Fig. 1 ▶).
Figure 1.

(a) The domain architecture and constructs of XcPNPase used in these studies. The starting and ending residues are numbered. (b) SDS–PAGE (12%) monitoring of the overexpression and purification of XcPNPaseΔS1. Lane M, protein markers (labelled in kDa); lane 1, whole cell lysate before IPTG induction; lane 2, whole cell lysate after IPTG induction; lane 3, nickel-column-purified XcPNPaseΔS1 after IPTG induction; lane 4, nickel column-purified XcPNPaseΔS1 after TEV cleavage.
2.3. Crystallization
For crystallization, the protein was concentrated to 10 mg ml−1 in 20 mM Tris–HCl pH 8.0, 200 mM NaCl, 1% MgCl2 using an Amicon Ultra-10 (Millipore). Screening for crystallization conditions of the XcPNPase–c-di-GMP complex was performed using the sitting-drop vapour-diffusion method in 96-well plates (Hampton Research) at 298 K. 0.8 µl drops comprising equal volumes of protein solution and reservoir solution in the presence of 1 mM c-di-GMP were equilibrated against 50 µl reservoir solution. The initial screening was performed manually using the sparse-matrix screens Crystal Screen and Crystal Screen 2, PEG/Ion (Hampton Research) and a systematic PEG–pH screen. Triangular prism-like crystals appeared in 3 d from a reservoir solution comprising 0.1 M HEPES pH 7.5, 0.2 M ammonium acetate, 25%(w/v) PEG 3350. The crystals reached maximum dimensions of 0.1 × 0.18 × 0.22 mm after 9 d (Fig. 2 ▶).
Figure 2.

Crystal of the XcPNPaseΔS1–c-di-GMP complex grown in 0.1 M HEPES pH 7.5, 0.2 M ammonium acetate, 25%(w/v) PEG 3350 using the sitting-drop vapour-diffusion method at 298 K. These crystals reached average dimensions of 0.1 × 0.18 × 0.22 mm after 9 d.
2.4. Data collection
The XcPNPaseΔS1 crystals were soaked in a cryoprotectant solution comprising reservoir solution plus 15%(v/v) glycerol. X-ray diffraction data were collected on beamline 13B1 at the National Synchrotron Radiation Research Center (NSRRC), Taiwan and also on the SP44XU micro-beamline at SPring-8, Japan. A native data set was collected to a resolution of 2.0 Å and was indexed and integrated using the HKL-2000 processing software (Otwinowski & Minor, 1997 ▶) to yield a data set that was 90% complete with an R merge of 4.6%. The XcPNPaseΔS1 crystals belonged to space group C2. The data-collection statistics are summarized in Table 1 ▶ and an X-ray diffraction image is shown in Fig. 3 ▶.
Table 1. Summary of data collection for native XcPNPaseΔS1–c-di-GMP.
Values in parentheses are for the outermost resolution shell.
| Beamline | 13B1, NSRRC |
| Wavelength (Å) | 0.98922 |
| Space group | C2 |
| Unit-cell parameters (Å, °) | a = 132.76, b = 128.38, c = 133.01, α = γ = 90, β = 93.25 |
| Resolution range (Å) | 30–2.0 (2.07–2.00) |
| Total reflections | 558672 (27656) |
| Unique reflections | 129388 (8134) |
| Multiplicity | 4.3 (3.4) |
| Completeness (%) | 90 (86) |
| R merge † (%) | 4.6 (41) |
| 〈I/σ(I)〉 | 24.8 (2.1) |
| Matthews coefficient (Å3 Da−1) | 2.83 |
| Solvent content (%) | 56.6 |
R
merge = 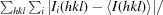
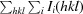 , where I
i(hkl) is the ith intensity measurement of reflection hkl, including symmetry-related reflections, and 〈I(hkl)〉 is its average.
, where I
i(hkl) is the ith intensity measurement of reflection hkl, including symmetry-related reflections, and 〈I(hkl)〉 is its average.
Figure 3.

Diffraction pattern of the XcPNPaseΔS1–c-di-GMP complex collected using a MAR CCD detector on beamline 13B1 at the National Synchrotron Radiation Research Center (NSRRC), Taiwan. The exposure time was 15 s, the oscillation range was 1° per frame and the crystal-to-detector distance was 280 mm. The edge of the detector corresponds to a resolution of 2.0 Å.
3. Results and discussion
In these studies, we successfully cloned, expressed and purified full-length XcPNPase (residues 25–735), XcPNPaseΔS1 (residues 25–649) and XcPNPaseΔKH/S1 (residues 25–579) (Fig. 1 ▶ a). All of these were soluble and formed homotrimers in solution (data not shown). However, only XcPNPaseΔS1 yielded well packed crystals that diffracted to a good resolution of 2.0 Å. Analyses of the diffraction intensities indicated that the space group was C2 and the unit-cell parameters were a = 132.76, b = 128.38, c = 133.01 Å. The Matthews coefficient V M (Matthews, 1968 ▶) suggested the presence of three XcPNPaseΔS1 protein molecules per asymmetric unit and a solvent content of 56.6%. Since XcPNPase has a very high sequence identity to EcPNPase (64%), we surmised that we could use the molecular-replacement approach to solve its phase. We successfully obtained an initial electron-density map using the program PHENIX (Adams et al., 2010 ▶) with the coordinates of EcPNPase (PDB entry 3cdj; Shi et al., 2008 ▶) as the search model (data not shown). Further structural refinement is now in progress.
Acknowledgments
This work was supported in part by the Ministry of Education, Taiwan, ROC under the ATU plan and by the National Science Council, Taiwan, ROC (grant 97-2113-M005-005-MY3 to S-HC). We appreciate the Structural Genomics Database service provided by the GMBD Bioinformatics Core (http://www.tbi.org.tw), NRPGM, Taiwan, ROC. We would also like to thank the Core Facilities for Protein X-ray Crystallography in the Academia Sinica, Taiwan, ROC for help in crystal screening and the National Synchrotron Radiation Research Center (NSRRC), Taiwan and the SPring-8 synchrotron facility, Japan for assistance in the X-ray data collections. Data for the complex were also collected on the SP44XU micro-beamline at SPring-8, Japan. The National Synchrotron Radiation Research Center is a user facility supported by the National Science Council, Taiwan, ROC and the Protein Crystallography Facility is supported by the National Research Program for Genomic Medicine, Taiwan, ROC.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Amikam, D. & Galperin, M. Y. (2006). Bioinformatics, 22, 3–6. [DOI] [PubMed]
- Aslanidis, C. & de Jong, P. J. (1990). Nucleic Acids Res. 18, 6069–6074. [DOI] [PMC free article] [PubMed]
- Benach, J., Swaminathan, S. S., Tamayo, R., Handelman, S. K., Folta-Stogniew, E., Ramos, J. E., Forouhar, F., Neely, H., Seetharaman, J., Camilli, A. & Hunt, J. F. (2007). EMBO J. 26, 5153–5166. [DOI] [PMC free article] [PubMed]
- Carpousis, A. J. (2007). Annu. Rev. Microbiol. 61, 71–87. [DOI] [PubMed]
- Chin, K.-H., Lee, Y.-C., Tu, Z.-L., Chen, C.-H., Tseng, Y.-H., Yang, J.-M., Ryan, R. P., McCarthy, Y., Dow, J. M., Wang, A. H.-J. & Chou, S.-H. (2010). J. Mol. Biol. 396, 646–662. [DOI] [PubMed]
- De Lay, N. & Gottesman, S. (2011). RNA, 17, 1172–1189. [DOI] [PMC free article] [PubMed]
- Del Favero, M., Mazzantini, E., Briani, F., Zangrossi, S., Tortora, P. & Dehò, G. (2008). J. Biol. Chem. 283, 27355–27359. [DOI] [PubMed]
- Gatewood, M. L. & Jones, G. H. (2010). J. Bacteriol. 192, 4275–4280. [DOI] [PMC free article] [PubMed]
- Gorna, M. W., Carpousis, A. J. & Luisi, B. F. (2011). Q. Rev. Biophys. 45, 105–145. [DOI] [PubMed]
- Grunberg-Manago, M. (1999). Annu. Rev. Genet. 33, 193–227. [DOI] [PubMed]
- Habazettl, J., Allan, M. G., Jenal, U. & Grzesiek, S. (2011). J. Biol. Chem. 286, 14304–14314. [DOI] [PMC free article] [PubMed]
- Hardwick, S. W., Gubbey, T., Hug, I., Jenal, U. & Luisi, B. F. (2012). Open Biol. 2, 120028. [DOI] [PMC free article] [PubMed]
- Hengge, R. (2009). Nature Rev. Microbiol. 7, 263–273. [DOI] [PubMed]
- Houseley, J., LaCava, J. & Tollervey, D. (2006). Nature Rev. Mol. Cell Biol. 7, 529–539. [DOI] [PubMed]
- Jenal, U. & Malone, J. (2006). Annu. Rev. Genet. 40, 385–407. [DOI] [PubMed]
- Kaberdin, V. R., Singh, D. & Lin-Chao, S. (2011). J. Biomed. Sci. 18, 23. [DOI] [PMC free article] [PubMed]
- Leduc, J. L. & Roberts, G. P. (2009). J. Bacteriol. 191, 7121–7122. [DOI] [PMC free article] [PubMed]
- Li, T.-N., Chin, K.-H., Fung, K.-M., Yang, M.-T., Wang, A. H.-J. & Chou, S.-H. (2011). PLoS One, 6, e22036. [DOI] [PMC free article] [PubMed]
- Li, T.-N., Chin, K.-H., Liu, J.-H., Wang, A. H.-J. & Chou, S.-H. (2009). Proteins, 75, 282–288. [DOI] [PubMed]
- Lin-Chao, S., Chiou, N. T. & Schuster, G. (2007). J. Biomed. Sci. 14, 523–532. [DOI] [PubMed]
- Marcaida, M. J., DePristo, M. A., Chandran, V., Carpousis, A. J. & Luisi, B. F. (2006). Trends Biochem. Sci. 31, 359–365. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Mohanty, B. K. & Kushner, S. R. (2006). Proc. Natl Acad. Sci. USA, 97, 11966–11971. [DOI] [PMC free article] [PubMed]
- Navarro, M. V., De, N., Bae, N., Wang, Q. & Sondermann, H. (2009). Structure, 17, 1104–1116. [DOI] [PMC free article] [PubMed]
- Navarro, M. V., Newell, P. D., Krasteva, P. V., Chatterjee, D., Madden, D. R., O’Toole, G. A. & Sondermann, H. (2011). PLoS Biol. 9, e1000588. [DOI] [PMC free article] [PubMed]
- Nurmohamed, S., Vincent, H. A., Titman, C. M., Chandran, V., Pears, M. R., Du, D., Griffin, J. L., Callaghan, A. J. & Luisi, B. F. (2011). J. Biol. Chem. 286, 14315–14323. [DOI] [PMC free article] [PubMed]
- Nurmohamed, S., Vaidialingam, B., Callaghan, A. J. & Luisi, B. F. (2009). J. Mol. Biol. 389, 17–33. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Raijmakers, R., Schilders, G. & Pruijn, G. J. (2004). Eur. J. Cell Biol. 83, 175–183. [DOI] [PubMed]
- Rao, F., Pasunooti, S., Ng, Y., Zhuo, W., Lim, L., Liu, A. W. & Liang, Z.-X. (2009). Anal. Biochem. 389, 138–142. [DOI] [PubMed]
- Römling, U. (2011). Environ. Microbiol. 14, 1817–1829. [DOI] [PubMed]
- Römling, U., Gomelsky, M. & Galperin, M. Y. (2005). Mol. Microbiol. 57, 629–639. [DOI] [PubMed]
- Ross, P., Mayer, R., Weinhouse, H., Amikam, D., Huggirat, Y., Benziman, M., de Vroom, E., Fidder, A., de Paus, P., Sliedregt, L. A. J. M., van der Marel, G. A. & van Boom, J. H. (1990). J. Biol. Chem. 265, 18933–18943. [PubMed]
- Ryan, R. P., Tolker-Nielsen, T. & Dow, J. M. (2012). Trends Microbiol. 20, 235–242. [DOI] [PubMed]
- Ryan, R. P., Vorhölter, F. J., Potnis, N., Jones, J. B., Van Sluys, M. A., Bogdanove, A. J. & Dow, J. M. (2011). Nature Rev. Microbiol. 9, 344–355. [DOI] [PubMed]
- Schirmer, T. & Jenal, U. (2009). Nature Rev. Microbiol. 7, 724–735. [DOI] [PubMed]
- Shi, Z., Yang, W.-Z., Lin-Chao, S., Chak, K.-F. & Yuan, H. S. (2008). RNA, 14, 2361–2371. [DOI] [PMC free article] [PubMed]
- Siculella, L., Damiano, F., di Summa, R., Tredici, S. M., Alduina, R., Gnoni, G. V. & Alifano, P. (2010). Mol. Microbiol. 77, 716–729. [DOI] [PubMed]
- Smith, K. D., Lipchock, S. V., Ames, T. D., Wang, J., Breaker, R. R. & Strobel, S. A. (2009). Nature Struct. Mol. Biol. 16, 1218–1223. [DOI] [PMC free article] [PubMed]
- Sondermann, H., Shikuma, N. J. & Yildiz, F. H. (2011). Curr. Opin. Microbiol. 15, 140–146. [DOI] [PMC free article] [PubMed]
- Symmons, M. F., Jones, G. H. & Luisi, B. F. (2000). Structure, 8, 1215–1226. [DOI] [PubMed]
- Symmons, M. F., Williams, M. G., Luisi, B. F., Jones, G. H. & Carpousis, A. J. (2002). Trends Biochem. Sci. 27, 11–18. [DOI] [PubMed]
- Tao, F., He, Y.-W., Wu, D.-H., Swarup, S. & Zhang, L.-H. (2010). J. Bacteriol. 192, 1020–1029. [DOI] [PMC free article] [PubMed]
- Tuckerman, J. R., Gonzalez, G. & Gilles-Gonzalez, M.-A. (2011). J. Mol. Biol. 407, 633–639. [DOI] [PubMed]
- Wang, G., Chen, H.-W., Oktay, Y., Zhang, J., Allen, E. L., Smith, G. M., Fan, K. C., Hong, J. S., French, S. W., McCaffery, J. M., Lightowlers, R. N., Morse, H. C. III, Koehler, C. M. & Teitell, M. A. (2010). Cell, 142, 456–467. [DOI] [PMC free article] [PubMed]
- Wu, Y.-Y., Chin, K.-H., Chou, C.-C., Lee, C.-C., Shr, H.-L., Gao, F. P., Lyu, P.-C., Wang, A. H.-J. & Chou, S.-H. (2005). Acta Cryst. F61, 902–905. [DOI] [PMC free article] [PubMed]
- Ygberg, S. E., Clements, M. O., Rytkönen, A., Thompson, A., Holden, D. W., Hinton, J. C. & Rhen, M. (2006). Infect. Immun. 74, 1243–1254. [DOI] [PMC free article] [PubMed]