

A putative human d-xylulokinase enzyme has been expressed, purified and crystallized using a repetitive seeding protocol. The trigonal crystals belonged to space group P31 or P32, with unit-cell parameters a = b = 101.87, c = 158.85 Å, and diffracted X-rays to at least 2.7 Å resolution.
Keywords: d-xylulokinases, d-xylulose, d-xylulose 5-phosphate, lipogenesis, glucuronate–xylulose pathway, pentose phosphate pathway
Abstract
In mammals, the enzyme d-xylulokinase (XK; EC 2.7.1.17) catalyses the last step of the glucuronate–xylulose pathway, in which the ketopentose sugar d-xylulose is phosphorylated to yield d-xylulose 5-phosphate (Xu5P). Xu5P is also a metabolite of the pentose phosphate pathway and acts as a signalling molecule that regulates lipogenesis and glycolysis in the liver. To date, no eukaryotic XK has been structurally characterized. A putative human XK was expressed in Escherichia coli aided by molecular chaperones, purified and crystallized. A seeding procedure involving repeated rounds of seeding was developed and proved to be essential for obtaining diffraction-quality crystals. Preliminary X-ray diffraction analysis was performed using synchrotron radiation. This resulted in the collection of a complete diffraction data set to 2.7 Å resolution from a crystal belonging to the trigonal space group P31 or P32 with unit-cell parameters a = b = 101.87, c = 158.85 Å.
1. Introduction
d-Xylulokinase (XK; EC 2.7.1.17) catalyses the ATP-dependent phosphorylation of d-xylulose, a ketopentose sugar, to produce xylulose 5-phosphate (Xu5P). XK is the final enzyme in the glucuronate–xylulose (GX) pathway (Touster, 1959 ▶), which in mammals is primarily active in the kidneys and the liver, and which is the only known catabolic pathway for key sugars, notably myo-inositol. For this reason, the enzymes of the GX pathway are thought to have potential as drug targets for the control of diabetic complications (Brown et al., 2006 ▶). In addition, Xu5P, the product of XK activity, is also produced by the pentose phosphate pathway and has been shown to modulate the expression of several glycolytic- and lipogenic-related genes, resulting in lipogenesis in mammals (Kabashima et al., 2003 ▶). d-Xylulokinase may therefore be an important regulator of Xu5P levels and hence of sugar and fat levels in mammals, emphasizing its potential importance for controlling metabolic disease.
To date, the identity and the functional properties of the human d-xylulokinase (hXK) enzyme have not been defined experimentally and the enzyme is uncharacterized. Functionally, XK enzymes belong to a broad superfamily of kinases known as the sugar kinase/Hsp70/actin superfamily (Hurley, 1996 ▶). This comprises several subfamilies, of which XK belongs to the FGGY carbohydrate kinase family, as defined in the InterPro database (Apweiler et al., 2001 ▶); well known members of this family include glycerol kinase and glucokinase. XK enzymes have been functionally characterized from several sources, but structural information is limited to the XKs from two bacterial species: Escherichia coli (PDB entry 2itm; di Luccio et al., 2007 ▶) and Chromobacterium violaceum (PDB entry 3hz6; New York SGX Research Center for Structural Genomics, unpublished work). These have little sequence identity with putative mammalian XKs, typically less than 20%.
To clarify the identity and function of hXK, and with the aim of determining the first structure of a mammalian XK, a putative hXK gene product that had been annotated on the basis of its 22% sequence identity to XK from Haemophilus influenzae (Tamari et al., 1998 ▶) was selected for investigation. The predicted polypeptide comprises 536 amino acids with an expected mass of 58.4 kDa. Here, we report the molecular cloning, expression, purification, crystallization and preliminary X-ray crystallographic analysis of this putative d-xylulokinase from Homo sapiens (hXK).
2. Materials and methods
2.1. Cloning and expression
A cDNA clone containing the hXK coding sequence was obtained from Invitrogen (clone IOH46573; NCBI Reference Sequence NM_005108.2). The hXK-coding sequence was amplified by PCR using PrimeSTAR HS DNA polymerase (Takara) with specific primers (5′-CGGTAGGCCCATGGCGGAGCACGCCCC-3′ and 5′-GCCTACCGGGTACCCTACTCCGGAGGCCCC-3′; Invitrogen). The PCR product was inserted into the bacterial expression plasmid pProEX HTb (Invitrogen) using restriction-ligation cloning with the restriction endonucleases NcoI and KpnI (Roche). The resulting construct expresses hXK with an N-terminal hexahistidine fusion tag that facilitates immobilized metal-affinity purification (IMAC). The fusion tag can be removed by proteolysis with recombinant tobacco etch virus (rTEV) protease, leaving a Gly-Ala dipeptide immediately N-terminal to the hXK amino-acid sequence. The hXK/pProEX plasmid construct was verified by DNA sequencing.
Expression of the hXK/pProEX plasmid construct was carried out using E. coli BL21 (DE3) cells. However, soluble protein was only obtained when this construct was co-expressed with high levels of the plasmid-encoded E. coli molecular chaperones DnaK–DnaJ–GrpE, ClpB and GroEL–GroES provided under the inducible control of the lac operon by plasmids pBB540 and pBB542 (Tomoyasu et al., 2001 ▶). E. coli BL21 (DE3) cells containing pBB540 and pBB542 were therefore co-transformed with hXK/pProEX by heat shock and selected on lysogen broth–Lennox (LB–Lennox) formulation agar with the following antibiotics: 100 µg ml−1 ampicillin, 50 µg ml−1 spectinomycin and 10 µg ml−1 chloramphenicol.
Beginning from a single bacterial colony, a starter culture was grown overnight at 310 K in non-inducing MDG medium (Studier, 2005 ▶) with antibiotics. For expression, LB–Lennox medium (Invitrogen) supplemented with antibiotics was inoculated 1:100 with starter culture and grown at 310 K until the OD600 reached approximately 0.6. After transfer to 293 K, protein expression was induced by the addition of 1 mM isopropyl β-d-1-thiogalactopyranoside. Cells were harvested after 18 h by centrifugation (3500g, 15 min, 273 K), washed by resuspension in 4 ml 50 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.50 mM tris(2-carboxyethyl)phosphine (TCEP) (buffer A) per gram of wet cell mass, pelleted by centrifugation (2400g, 20 min, 277 K) and stored at 253 K.
2.2. Protein purification
Purification steps were carried out at 277 K unless otherwise stated. Cell pellets, typically 10 g wet weight for each preparation, were resuspended in 4 ml lysis buffer [buffer A including 25 mM imidazole pH 8.0, 2 mg ml−1 lysozyme, 2 µg ml−1 DNase I and half a Mini EDTA-free protease-inhibitor tablet (Roche)] per gram and lysed by cell disruption at 18 kPa and 293 K.
The lysate was clarified by centrifugation (30 000g, 30 min). The supernatant was then filtered (0.45 µm) and loaded at 2 ml min−1 onto a HisTrap FF 5 ml IMAC column (GE Healthcare) charged with Ni2+ ions (100 mM nickel chloride) and equilibrated with buffer A including 25 mM imidazole pH 8.0. The column was washed with 20 ml buffer A including 25 mM imidazole pH 8.0, after which the protein was eluted with a 50 ml linear gradient of 25–500 mM imidazole pH 8.0 in buffer A (2 ml min−1). To remove the hexahistidine fusion tag, the protein solution was mixed 1:15 with rTEV protease solution [1 mg ml−1 rTEV protease in 20 mM Tris–HCl pH 8.0, 150 mM NaCl, 50%(v/v) glycerol, 2 mM TCEP] and dialyzed against 1 l buffer A with 1 mM TCEP overnight. Uncleaved protein was removed by passing the protein solution through a 1 ml HiTrap Chelating HP IMAC column (2 ml min−1, 293 K; GE Healthcare) charged with Ni2+ ions (100 mM nickel chloride) and equilibrated with buffer A. The column was washed with 5 ml buffer A including 25 mM imidazole pH 8.0 and pooled with the column-flowthrough eluate. The protein solution was then concentrated approximately sixfold to 5 ml by ultrafiltration in Vivaspin 20 ml 10 kDa molecular-weight cutoff (MWCO) concentrators at 1600g (GE Healthcare). The concentrated protein solution was applied onto a Superdex 200 16/60 gel-filtration column (GE Healthcare) equilibrated with buffer A containing 1 mM TCEP and operated at 1.2 ml min−1. The column eluate was collected in 2 ml fractions. Fractions containing hXK with greater than 98% purity were identified by SDS–PAGE. The two most monodisperse fractions, as determined by dynamic light scattering, were concentrated by ultrafiltration [2.5 ml 10 kDa MWCO Vivaspin concentrators (GE Healthcare), 1600g] to approximately 16 mg ml−1 (estimated spectrophotometrically assuming an extinction constant ∊280 of 64 580 M cm−1, A 280 = 17.2).
2.3. Crystallization
Crystallization experiments were performed at 293 K. Initial crystallization trials were performed by sitting-drop vapour diffusion in 96-well Intelli-Plates (Art Robbins) using a Cartesian nanolitre dispensing robot (Genomic Solutions) with a 480-component screen built up from a combination of locally developed and commercially available screens (Moreland et al., 2005 ▶). Equal volumes of protein solution (16 mg ml−1 protein in 50 mM Tris–HCl, 150 mM NaCl, 1 mM TCEP pH 8.0) and well solution were mixed (100 nl + 100 nl) and equilibrated against 100 µl well solution. No crystals were obtained, but one condition that gave promising phase separation was discovered after 24 h incubation. This observation was followed up by hanging-drop vapour-diffusion experiments in 24-well NeXtal plates, in which protein was screened against the same condition with different protein:well solution ratios. Two types of crystals grew spontaneously within 24 h: small prismatic crystals when 2 µl protein solution was mixed with 1 µl well solution and clusters of needle-like crystals when 1 µl protein solution was mixed with 2 or 3 µl well solution. In both cases the well solution comprised 200 mM MES–KOH pH 6.0, 16%(w/v) PEG 6000, 0.3 mM NaN3. The needle-like crystals (Fig. 1 ▶ a) were subsequently optimized by seeding.
Figure 1.

(a) Initial crystals of hXK. These crystals were obtained in a hanging-drop vapour-diffusion experiment by spontaneous nucleation in well solution comprising 200 mM MES–KOH pH 6.0, 16% PEG 6000 when 1 µl protein solution (16 mg ml−1 protein in 50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM TCEP) was mixed with 2 µl well solution. The image was recorded one week following preparation, when the crystals had reached their maximum dimensions. (b) An optimized XK crystal obtained in the fifth generation of repeat seeding. One crystal was present in this drop, as was common in the later rounds of seeding.
2.4. Seeding protocols
In an effort to improve their X-ray diffraction quality, the initial needle-like crystals were propagated by streak-seeding into sitting drops. Crystal seed stocks were prepared by transferring 1–3 crystals into a 2.5 µl drop of a seed-stock solution made up to replicate the crystal-growth condition as far as possible: 200 mM MES–KOH pH 5.9, 16%(w/v) PEG 6000, 0.22 mM NaN3 supplemented with 25 mM Tris–HCl and 75 mM NaCl. The crystals were broken up using a fine needle and the drop containing crystal fragments was transferred into a tube containing 47.5 µl seed-stock solution, which was then vortex-mixed in 3 × 5 s bursts. Although crystal seed stocks can be successfully stored frozen (Shaw Stewart et al., 2011 ▶), in the present case the seed stocks were freshly prepared for each round of crystallization, with the seeds for the next round being prepared from the crystals obtained in the previous round.
All subsequent crystallization experiments were prepared in sitting drops (CrystalClear D plates, Douglas Instruments) using protein solutions mixed with well solution at 75% of the protein concentration that generally yielded crystals spontaneously. After the drops had been incubated for 16–24 h, they were seeded by passing the tip of a cat’s whisker that had previously been dipped into a fresh seed-stock solution through the drop. A similar seeding protocol that is suitable for automation has been described by Khurshid et al. (2010 ▶). After seeding, the sitting drops were resealed and kept undisturbed to allow crystal growth. These experiments were carried out as optimization screens, in which protein concentration, protein:well solution ratio, pH and PEG concentration were varied systematically from those in the initial crystallization condition.
2.5. Data collection and processing
A useful cryoprotectant solution comprising 20%(v/v) ethylene glycol, 25 mM Tris–HCl pH 8.0, 75 mM NaCl, 200 mM MES–KOH pH 5.9, 16%(w/v) PEG 6000 was determined by screening glycerol, PEG 400 and ethylene glycol concentrations in combination with differing well solutions and buffer conditions. A single crystal was harvested, submerged in cryoprotectant solution for approximately 30 s and flash-cooled in liquid nitrogen. A complete preliminary X-ray diffraction data set was collected on beamline PX2 of the Australian Synchrotron using an ADSC Quantum 315r detector. Diffraction data were indexed and integrated with XDS (Kabsch, 2010 ▶) and scaled by SCALA (Evans, 2011 ▶), with supplementary procedures performed using programs from the CCP4 suite (Winn et al., 2011 ▶).
3. Results and discussion
The putative human d-xylulokinase (hXK) was readily expressed as soluble protein from an E. coli expression vector in the presence of added chaperones. The protein was then purified and subjected to crystallization trials after removal of its N-terminal polyhistidine tag. Crystals were obtained from only one condition, which was identified from the observation of phase separation in the initial screening experiments. Most of the subsequent crystallization trials only yielded prismatic crystals that never gave any visible diffraction. On just one occasion, clusters of long thin needle-like crystals were obtained using 200 mM MES–KOH pH 6.0, 16%(w/v) PEG 6000, 0.3 mM NaN3 as the precipitant; although these initial crystals did not give visible diffraction of X-rays beyond 20 Å resolution, they provided the basis for subsequent successful crystal optimization by repetitive rounds of seeding.
Crystals of adequate quality for X-ray diffraction analysis were obtained after four or five rounds of repeated streak-seeding from drops prepared with 1 µl protein solution (12 mg ml−1 protein in 50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM TCEP pH 8.0) mixed with 2 µl well solution [200 mM MES–KOH pH 5.9, 14%(w/v) PEG 6000, 0.22 mM NaN3] and equilibrated against a 100 µl reservoir of well solution. Needle-like crystals were visible after 3 d and grew to their maximum dimensions, typically 15 × 15 × 150 µm, after 5 d. Further rounds of seeding beyond five produced no further obvious improvement in crystal size or diffraction.
The crystals proved to be trigonal, with point-group symmetry 3 and unit-cell parameters a = b = 101.87, c = 158.85 Å. Inspection of the reflections in the 00l zone showed that the systematic absence conditions (l = 3n) for a threefold screw axis were met, indicating that the crystals belonged to space group P31 or P32. The most probable Matthews coefficients (V M) of 2.72 or 2.04 Å3 Da−1 for the crystal suggest three molecules with 55% solvent content or four molecules with 40% solvent content in the asymmetric unit, respectively. The crystals diffracted well (Fig. 2 ▶), but the initial data set (see Table 1 ▶ for details) was limited to 2.7 Å resolution because of the small size of the crystal used. We anticipate that higher resolution data will be obtainable with further refinement of the seeding protocol.
Figure 2.

A representative X-ray diffraction image collected from the hXK crystal used to generate the native data set. The image was collected with a 0.5° oscillation in 1 s on beamline PX2 of the Australian Synchrotron using an ADSC Quantum 315r detector with radiation of wavelength 0.9537 Å attenuated by 85%. A ring shows the 3.0 Å resolution limit.
Table 1. Crystallographic data-collection statistics for hXK.
Values in parentheses are for the highest resolution shell.
| Wavelength (Å) | 0.9537 |
| Space group | P31 or P32 |
| Unit-cell parameters (Å) | a = b = 101.87, c = 158.85 |
| Resolution (Å) | 45–2.71 (2.86–2.71) |
| R merge † (%) | 9.7 (48.6) |
| R meas (%) | 12.0 (60.1) |
| R p.i.m. (%) | 5.3 (25.0) |
| Overall B factor from Wilson plot‡ (A2) | 53.7 |
| Completeness (%) | 100 (100) |
| No. of observations | 287860 (42307) |
| No. of unique reflections | 49860 (7298) |
| Multiplicity | 5.8 (5.8) |
| Mean I/σ(I) | 14.6 (3.6) |
R
merge = 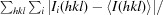
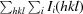 , where Ii(hkl) is the ith measurement and 〈I(hkl)〉 is the weighted mean of all measurements of I(hkl).
, where Ii(hkl) is the ith measurement and 〈I(hkl)〉 is the weighted mean of all measurements of I(hkl).
As calculated by TRUNCATE (French & Wilson, 1978 ▶).
Efforts to determine the hXK structure by molecular replacement have so far been unsuccessful, which is not surprising given that no homologous structure is available with a sequence identity of greater than 20%. Growth of selenomethionine-substituted hXK crystals is currently being explored in an effort to determine the crystal structure using anomalous dispersion methods. Use of the expression and purification protocol that has been established here will also enable future biochemical characterization of this prospective hXK protein.
Acknowledgments
We thank Heather Baker for help in developing the seeding procedures. This research was funded by the Tertiary Education Commission through funding of the Maurice Wilkins Centre for Molecular Biodiscovery. RDB gratefully acknowledges support from a Doctorate Scholarship awarded by the University of Auckland. Access to the Australian Synchrotron was supported by the New Zealand Synchrotron Program.
References
- Apweiler, R. et al. (2001). Nucleic Acids Res. 29, 37–40. [DOI] [PMC free article] [PubMed]
- Brown, P. M., Caradoc-Davies, T. T., Dickson, J. M., Cooper, G. J., Loomes, K. M. & Baker, E. N. (2006). Proc. Natl Acad. Sci. USA, 103, 15032–15037. [DOI] [PMC free article] [PubMed]
- Di Luccio, E., Petschacher, B., Voegtli, J., Chou, H.-T., Stahlberg, H., Nidetzky, B. & Wilson, D. K. (2007). J. Mol. Biol. 365, 783–798. [DOI] [PMC free article] [PubMed]
- Evans, P. R. (2011). Acta Cryst. D67, 282–292. [DOI] [PMC free article] [PubMed]
- French, S. & Wilson, K. (1978). Acta Cryst. A34, 517–525.
- Hurley, J. H. (1996). Annu. Rev. Biophys. Biomol. Struct. 25, 137–162. [DOI] [PubMed]
- Kabashima, T., Kawaguchi, T., Wadzinski, B. E. & Uyeda, K. (2003). Proc. Natl Acad. Sci. USA, 100, 5107–5112. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Khurshid, S., Haire, L. F. & Chayen, N. E. (2010). J. Appl. Cryst. 43, 752–756.
- Moreland, N., Ashton, R., Baker, H. M., Ivanovic, I., Patterson, S., Arcus, V. L., Baker, E. N. & Lott, J. S. (2005). Acta Cryst. D61, 1378–1385. [DOI] [PubMed]
- Shaw Stewart, P. D., Kolek, S. A., Briggs, R. A., Chayen, N. E. & Baldock, P. F. M. (2011). Cryst. Growth Des. 11, 3432–3441.
- Studier, F. W. (2005). Protein Expr. Purif. 41, 207–234. [DOI] [PubMed]
- Tamari, M., Daigo, Y., Ishikawa, S. & Nakamura, Y. (1998). Cytogenet. Cell Genet. 82, 101–104. [DOI] [PubMed]
- Tomoyasu, T., Mogk, A., Langen, H., Goloubinoff, P. & Bukau, B. (2001). Mol. Microbiol. 40, 397–413. [DOI] [PubMed]
- Touster, O. (1959). Am. J. Med. 26, 724–739. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.