

Abstract
We describe a method for modifying proteins site-specifically using a chemoenzymatic bioconjugation approach. Formylglycine generating enzyme (FGE) recognizes a pentapeptide consensus sequence, CxPxR, and it specifically oxidizes the cysteine in this sequence to an unusual aldehyde-bearing formylglyine. The FGE recognition sequence, or aldehyde tag, can be inserted into heterologous recombinant proteins produced in either prokaryotic or eukaryotic expression systems. The conversion of cysteine to formylglycine is accomplished by co-overexpression of FGE, either transiently or as a stable cell line, and the resulting aldehyde can be selectively reacted with α-nucleophiles to generate a site-selectively modified bioconjugate. This protocol outlines both the generation and the analysis of proteins aldehyde-tagged at their termini and the methods for chemical conjugation to the formylglycine. The process of generating aldehyde-tagged protein followed by chemical conjugation and purification takes 20 d.
INTRODUCTION
A number of synthetic transformations have been developed for protein chemical modification1, enabling the attachment of small synthetic molecules such as drugs and imaging agents or bulky polymers such as polyethylene glycol (PEG)2. Traditionally, chemical modification of proteins is accomplished by attaching synthetic cargo to nucleophilic amino acid side chains on protein surfaces, such as those associated with lysine or cysteine residues. These methods inherently lack single-site specificity and produce product mixtures with heterogeneity in both the regiochemistry and stoichiometry of the modification. Moreover, the precise location of chemical conjugation can markedly alter protein function and illustrates a need for methods to modify proteins with site specificity3.
A number of methodologies developed over the last decade address the issue of site-specific protein modification4. These techniques primarily focus on introducing orthogonal functionality to amino acid side chains or utilizing enzymes to catalyze selective bond formation. In the former instance, an orthogonal chemical functionality, or `chemical handle', can be further elaborated through highly selective covalent reactions. Aldehydes and ketones are examples of such chemical handles, and their addition to proteins has achieved using both genetic5,6 and chemical7 methods.
We reported the use of the hexapeptide sequence LCTPSR, derived from the active sites of human type I sulfatases, as a genetically encoded tag for site-specific chemical modification of proteins6,8–10. This short amino acid sequence (defined by the broader consensus sequence CxPxR) is recognized by the FGE, which catalyzes the co-translational oxidation of the cysteine residue to an aldehyde-bearing Cα-formylglycine (FGly) residue11–13, a modification that is required for sulfatase activity. Type 1 sulfatases are found in most organisms, and likewise the amino acid modification and FGEs that generate it occur in all domains of life14. Notably, FGEs can recognize and modify this short consensus sequence within the context of heterologous proteins15. Thus, the sequence can be introduced into recombinant proteins as a means to introduce FGly residues at chosen sites. We exploited this phenomenon for site-specific protein chemical modification as shown in Figure 1.
Figure 1.
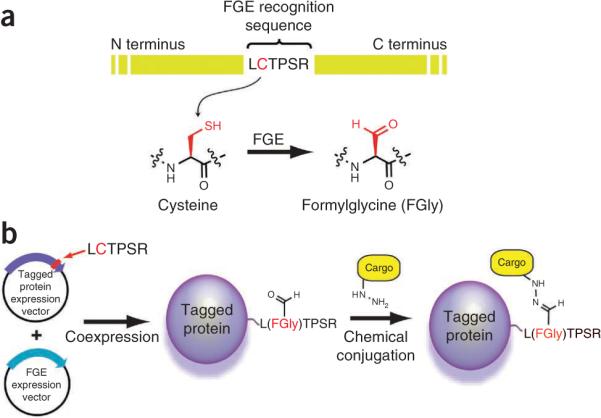
Schemes illustrating our approach to chemical modification of proteins. (a) Conversion of cysteine to formylglycine within the FGE recognition peptide sequence. (b) Coexpressing an aldehyde-tagged protein with FGE results in a site-specifically modified protein with an aldehyde that can be subsequently modified with aldehyde-specific chemical cargo.
The protocol presented here describes two methods for incorporating the aldehyde tag–coding nucleotides into a plasmid-resident gene. We then provide examples of expressing aldehyde-tagged proteins in prokaryotic (E. coli) and eukaryotic (CHO) cell lines and their subsequent site-specific labeling. A method for mass spectrometry (MS)-based analysis of the extent of Cys-to-FGly conversion is also described.
Aldehyde-tagging proteins in an expression vector
We use two methods for introducing aldehyde tag–coding nucleotides into a gene of interest. If there is a convenient restriction site, ligating annealed nucleotides is the preferred method. If no such restriction site exists, we use a site-directed mutagenesis approach.
Prokaryotic expression
Genome mining using human FGE as a query has identified putative FGE orthologs in Mycobacterium tuberculosis and other organisms. Early expression experiments in E. coli, which does not code for an apparent FGE, nonetheless indicated the presence of an FGE-like activity12. We found incomplete FGly formation on an aldehyde-tagged model protein, maltose binding protein (MBP). However, if an arabinose-inducible expression plasmid containing M. tuberculosis FGE is co-transformed with the expression vector coding for the aldehyde-tagged protein target (under alternate induction), the increased amount of FGE present in the cells maximizes FGly formation efficiency7. A MS-based assay revealed that C-terminal aldehyde-tagged MBP, when co-expressed with FGE, had a FGly formation efficiency of >85%. This coexpression protocol is now routinely used for the production of aldehyde-tagged proteins in prokaryotes7,9.
Eukaryotic expression
The aldehyde tag technology can be adapted to mammalian expression systems. In eukaryotic cells, endogenous FGE is located in the endoplasmic reticulum lumen, where it normally acts on sulfatases destined for lysosomes or for secretion16. This placement allows facile modification of heterologously expressed, secreted, aldehyde-tagged proteins. The protocol presented here uses a secreted human IgG Fc domain as an example in mammalian cells. This secreted protein is a disulfide-bound homodimer that possesses a conserved N-linked glycan at the N-terminal hinge region. Notably, the Fc domain expresses robustly in many mammalian host cells, facilitating biochemical characterization. By using the pFuse-Fc vector (Invivogen), expression plasmids can be generated containing the gene encoding the Fc protein fused at the N or C terminus to the six-residue FGE recognition motif. These plasmids also encode the interleukin-2 (IL2) signal sequence to promote robust secretion into the medium. Although Chinese Hamster Ovary (CHO) cells possess endogenous FGE, we have found that additional FGE expression is required for optimum FGly formation on a highly expressed protein such as IgG Fc6. The additional FGE expression may be transient or in a cell line stably expressing human FGE (Box 1).
Conjugation and analysis of aldehyde-tagged proteins
The aldehyde functionality in a protein can be selectively modified with a diverse array of aminooxy- or hydrazide-functionalized molecules. This includes, but is not limited to, fluorescent imaging probes such as Alexa Fluor 647 C5-aminooxyacetamide, affinity probes such as biotin-hydrazide, aminooxy-functionalized PEG polymers or aminooxy-bearing peptides (Table 1 and refs. 6–9,10). As a control for labeling experiments, we always generate proteins in parallel with a nonfunctional aldehyde tag, where the crucial cysteine is mutated to alanine (LATPSR). These proteins demonstrate significant labeling with aldehyde-specific reagents.
Table 1.
Examples of aldehyde-tag conjugation conditions.
| aldehyde reactive compound | chemical structure | protein | reagent concentration | conditions |
|---|---|---|---|---|
| Biotin hydrazide |
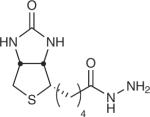
|
MBP6, GFP6, PDGFR-TM6 | 30–300 μM | 100 mM MES, pH 5.5, 37 °C, 2 h (in vitro) 50 μM, 10% FBS in PBS, pH 6.4, 1 h (cell culture) |
| AO-Flag |
|
MBP7, HGH7, IgG Fc6 | 300 μM | 100 mM MES, pH 5.5, 37 °C, 2 h |
| AO-Alexa Fluor 647 |
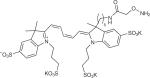
|
Stf07, MBP7, hGH7 | 300 μM | 100 mM MES, pH 5.5, 37 °C, 2 h |
| AO-PEG |
|
Stf07, MBP7 | 10 mM | 1:1 MeCN/H2O, 0.1% (wt/vol) TFA, 1 h followed by lyophilization |
| AO-Alexa Fluor 488 |
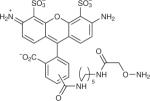
|
IgG6, mouse-CD4 (ref. 6) | 300 μM | 100 mM MES, pH 5.5, 37 °C, 2 h (in vitro) 50 μM, 10% (vol/vol) FBS in PBS, pH 6.4, 1 h (cell culture) |
| 1 AO-glycan |
|
hGH9 | 1 mM | 5% (vol/vol) MeCN, 0.02% formic acid or, 5% MeCN, 50 mM sodium citrate |
| AO-azide |
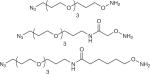
|
hIgG10, MBP10, hGH10 | 0.2–1 mM 10–20 equiv. | 100 mM KOAc, pH 4.6, 16 h, 35 °C |
| AO-DIBAC |
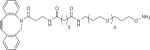
|
hIgG10, MBP10 | 0.2–1 mM 10–20 equiv. | 100 mM KOAc, pH 4.6, 16 h, 35 °C |
Conjugation to FGly using aminooxy- or hydrazide-bearing reagents can be performed in a number of aqueous buffers in slightly acidic pH. The kinetics of the condensation reaction is profoundly affected by the pH and at a pH greater than 6.0, the formation of oximes and hydrazines is considerably slower17. Concentration of the reagents is also important. When the protein concentration is below 10 μM, the small-molecule concentration should be above 500 μM in order for the reaction to proceed to completion. At these concentrations, the reactions take 12 h at 37 °C and pH 5.5. Table 1 provides examples of small-molecule payloads and conjugation conditions used to chemically elaborate aldehyde-tagged proteins.
To assess the efficiency of FGly formation, MS can be used. Tryptic digestion of aldehyde-tagged proteins is followed by high-performance liquid chromatography (HPLC)-coupled MS and allows for the direct comparison of FGly- or Cys- containing fragments with the quantification of the extent of conversion6. It is important to note that the formylglycine is hydrated in water to give the geminol diol (Fig. 2). Both the aldehyde and the diol will be observed as two distinct molecular weights and both chemical species should be factored into the MS analysis.
Figure 2.

Anticipated results of LC-MS analysis of tryptic peptides from an aldehyde-tagged protein. Protein was reduced with DTT, alkylated with IAA and digested with trypsin. The sample was analyzed by LC-MS on a Waters QToF mass spectrometer. (a) Expected tag peptides. To determine conversion rates, we monitored the presence of ions corresponding to the peptides containing the formylglycine modification as the aldehyde (i) or the diol (ii) or the unconverted peptide with the Cys protected with iodoacetamide (iii). (b) Illustration of extracted ion currents (XIC) corresponding to the peptides indicated: (i), M + 2H at m/z 693.9; (ii), M + 2H at m/z 702.9; and (iii), M + 2H at m/z 731.4. Note that the carbamidomethylated Cys-containing peptide elutes slightly later than its aldehyde-containing counterpart. (c) Shows mass spectra of the same peptides. The area under the curve (AUC) of the relevant XICs was used to determine the conversion rates as follows: i + ii/i + ii + iii.
A schematic illustrating both the ease of modification and the method of chemical modification is shown in Figure 1b. Because the aldehyde tag can be used on target proteins produced in mammalian cells, and thereby it can also make use of endogenous mammalian protein folding and post-translational modification machinery, the ability of native FGE to recognize and modify the aldehyde tag sequence in multiple contexts is noteworthy. Although it is not highlighted in this protocol, incorporation of the aldehyde tag is not limited to the termini of proteins. The aldehyde tag sequence can be incorporated into internal regions of a protein and, if the sequence is solvent accessible, can be conjugated with an aldehyde reactive compound. Furthermore, multiple tags can be inserted into a protein of interest, increasing the amount of chemical payload on an aldehyde-tagged protein of interest. We have, for example, simultaneously inserted three aldehyde tag sequences in human serum albumin and IgG Fc domains (D.R. and G.W.D., unpublished data). Using available structural data, such as X-ray crystallography or NMR spectroscopy, we inserted the FGE recognition sequence in unstructured loop regions located on the protein surface. We did not insert the recognition sequence into structured regions of a protein, such as α-helices and β-sheets, where altering the peptide sequence could impact overall protein structure and function. However, if the site of modification is appropriately selected, we have found that internal aldehyde tag placement and the addition of multiple tags into a protein backbone did not adversely affect protein folding or expression titers.
Given the versatility of tag number and position of placement, the utility of the aldehyde tag extends far beyond the applications described herein. For example, the technique could have widespread utility for imaging cellular proteins and chemical modification of therapeutic proteins3,18–20.
Protocol overview
In this article, protocols are provided for plasmid construction and insertion of the sulfatase motif into two model heterologous proteins. The expression and purification of an aldehyde-tagged protein in E. coli is described, as well as the expression and purification of an aldehyde-tagged protein in mammalian expression systems, specifically in CHO cells. Analysis of the modified proteins by fluorescence is discussed, as is protein analysis and quantification of the conversion of Cys to FGly by MS. For research projects that will require larger amounts of converted protein over longer periods of time, a protocol for generation and propagation of a CHO-K1 cell line stably expressing FGE is provided.
MATERIALS
REAGENTS
Acetic acid (AcOH; Fisher Scientific, cat. no. A38-212)
Acetic acid in deionized water (DI H2O; 7.5%, vol/vol)
Acetonitrile (ACN; Sigma-Aldrich, cat. no. 271004)
Agar (Fisher Scientific, cat. no. BP2641)
Alexa Fluor 647 C5-aminooxyacetamide (Molecular Probes, cat. no. A30632)
Alexa Fluor 647 C5-aminooxyacetamide (10 mM in DMSO)
Alkylation buffer
Annealing buffer
l-Arabinose (Sigma-Aldrich Chemical, cat. no. A3256)
l-arabinose (20%, wt/vol)
BamHI (NEB, cat. no. R0136)
BglII (NEB, cat. no. R0104)
Biomax 5K filter (Millipore, cat. no. UFV5BCC00)
Bromophenol blue (Sigma-Aldrich Chemical, cat. no. B8026)
Calf intestinal alkaline phosphatase (CIP; NEB, cat. no. M0290)
Carbenicillin (Sigma-Aldrich Chemical, cat. no. C9231)
CHO K1 cells (ATCC, cat. no. CCL-61)
Complete EDTA-free protease inhibitor cocktail (Roche, cat. no. 11 873 580 001)
Criterion XT MES running buffer (20×; Bio-Rad, cat. no. 161-0789)
1× Criterion XT MES running buffer
Deionized water (DI H2O)
Denaturing buffer
Dimethyl sulfoxide (DMSO; Sigma-Aldrich Chemical, cat. no. D8418)
Dithiothreitol (DTT; Sigma-Aldrich Chemical, cat. no. 43815)
DnaseI (NEB, cat. no. M0303)
DpnI (NEB, cat. no. R0176)
dNTPs (2.5 mM; Invitrogen, cat. no. R72501)
EcoRV (NEB, cat. no. R0104)
Elution buffer
Ethylenediamine tetraacidic disodium salt (EDTA; Fisher Scientific, cat. no. BP120-1)
Ex-Cell 325 PF CHO serum-free medium (Sigma-Aldrich, cat. no. 14340C)
Fetal bovine serum (Mediatech, cat. no. 35-010-CV)
l-Glutamine (Mediatech, cat. no. 25-005-CI)
Glycerol (Sigma-Aldrich Chemical, cat. no. G5516)
Glycerol (50%, vol/vol)
Guanidine hydrochloride (Sigma-Aldrich, cat. no. G4505)
HindIII (NEB, cat. no. R0104)
Hydrochloric acid (HCl; EMD, cat. no. 3120-500)
IgG elution buffer (Thermo Fisher Scientific, cat. no. 21004)
Imidazole (Sigma-Aldrich Chemical, cat. no. I2399)
Iodoacetamide (IAA; Sigma-Aldrich, cat. no. 068k53022)
Isopropyl β-d-1-thiogalactopyranoside (IPTG; Sigma-Aldrich Chemical, cat. no. I5502)
IPTG (isopropyl-1-thio-β-d-galactopyranoside, 1 M)
Kanamycin (Sigma-Aldrich Chemical, cat. no. K1876)
Labeling buffer (2×, 500 mM NaOAc, pH = 4.6)
Lipofectamine 2000 (Invitrogen, cat. no. 11668019)
Liquid nitrogen
Lysis buffer
Lysozyme (Sigma-Aldrich Chemical, cat. no. L6876)
Methoxyamine hydrochloride (Sigma-Aldrich, cat. no. M1139)
Ni-NTA agarose (Qiagen, cat. no. 30230)
Oligonucleotides (see Table 2)
One Shot BL21(DE3) chemically competent E. coli (Invitrogen, cat. no. C6000-03)
One Shot TOP10 chemically competent E. coli (Invitrogen, cat. no. C4040-03)
Opti-MEM I medium (Invitrogen, cat. no. 51985034)
pBAD-Mtb-FGE plasmid5. Can be obtained from a nonprofit plasmid repository (AddGene; http://www.addgene.org/)
pcDNA3.1-hFGE plasmid (Available from AddGene)
Penicillin/streptomycin solution (Mediatech, cat. no. 30-002-CI)
pFuse-mFc2 (ILss) vector (InvivoGen, cat. no. pfuse-mfc2)
Phosphate-buffered saline (PBS), sterile, pH 7.4, without calcium or magnesium (Mediatech, cat. no. 21-040)
Phusion hot start polymerase (Finnzymes, cat. no. F-540)
pMAL-c4E vector (NEB, cat. no. N8105S)
pMALc-H vector20
Protein A–agarose 50% slurry (Thermo Fisher Scientific, cat. no. 20333)
Protein A–IgG binding buffer (Thermo Fisher Scientific, cat. no. 21001)
Reducing buffer
QIAprep miniprep kit (Qiagen, cat. no. 27104)
QIAquick PCR purification kit (Qiagen, cat. no. 28104)
SDS loading buffer, 4×
Sodium acetate (NaOAc; Fisher Scientific, cat. no. S209)
Sodium chloride (NaCl; Fisher Scientific, cat. no. S271)
Sodium dodecyl sulfate (SDS; Sigma-Aldrich Chemical, cat. no. L6026)
Sodium hydroxide (NaOH; Fisher Scientific, cat. no. S318)
Sodium phosphate monobasic (NaH2PO4; Fisher Scientific, cat. no. S369)
Sterile deionized water
Storage buffer
Sypro Orange (Molecular Probes, cat. no. S6650)
Sypro Orange working solution
T4 DNA ligase (Invitrogen, cat. no. 15224-041)
TE buffer
Trifluoroacetic acid (TFA; Thermo Scientific, prod. no. 28904)
Tris (Trizma or Tris(hydroxymethyl)aminomethane) base (Sigma-Aldrich Chemical, cat. no. T1503)
Trypsin (Promega, cat. no. V5113)
Trypsin-EDTA solution (1×, 0.25% (wt/vol); Mediatech, cat. no. 25-053-CI)
Tryptone (Fisher Scientific, cat. no. BP1421)
Wash buffer
Yeast extract (Fisher Scientific, cat. no. BP1422)
TABLE 2.
OLigonudeotides used.
| Oligo | Sequence 5′→3′ |
|---|---|
| 1 | [Phos]GATCCCTGTGCACACCATCGCGGTGAGCGGCCGCAA |
| 2 | [Phos]AGCTtTGCGGCCGCTCACCGCGATGGTGTGCACAGG |
| 3 | GGATCCCTGGCCACACCATCGCGGTGAGCGGC |
| 4 | GTTCCGCGTGGATCCCTGGCCACACCATC |
| 5 | GCGCAGACTCTGTGCACACCATCGCGGTGAAATTCGAGCTCGAACAAC |
| 6 | CGAATTTCACCGCGATGGTGTGCACAGAGTCTGCGCGTCTTTCAGGGCTTCATCGACAGTCTG |
| 7 | [Phos]ATCGCTGTGCACCCCCAGCCGGGCCGCCCTGCTGACCGGCCGGA |
| 8 | [Phos]GATCTCCGGCCGGTCAGCAGGGCGGCCCGGCTGGGGGTGCACAGCGAT |
| 9 | ATCGCTGGCCACCCCCAGCCGGGCCGCCCTGCTGACCGGCCGGA |
| 10 | GATCTCCGGCCGGTCAGCAGGGCGGCCCGGCTGGGGGTGGCCAGCGAT |
Bold lettering indicates regions coding for the aldehyde tag.
EQUIPMENT
Amicon Ultra centrifugal filters (10 kDa molecular weight cutoff, Millipore, cat. no. UFC5010BK)
Benchtop centrifuge (Sorvall Legend Mach 1.6 refrigerated, cat. no. 75004337)
Centrifuge (Sorvall RC5C plus, cat. no. 74505)
Criterion cell (Bio-Rad, cat. no. 165-6001)
Criterion XT 4–12% (wt/vol) Bis-Tris gel (Bio-Rad, cat. no. 345-0123)
Falcon tubes (15 ml; Fisher Scientific, cat. no. 05-527-90)
Falcon tubes (50 ml, Fisher Scientific, cat. no. 14-432-22)
Fast protein liquid chromatography (FPLC; GE Healthcare, ÄKTA purifier)
HPLC (Waters)
Microcentrifuge (Eppendorf 5417c, cat. no. 5417 000.315)
Microcentrifuge tubes (2 ml; Fisher Scientific, cat. no. 02-681-291)
NanoVue spectrophotometer (GE)
PCR machine (Eppendorf, Mastercycler gradient, cat. no. 950000015)
PCR tubes (Fisher Scientific, cat. no. 14-230-225)
PD SpinTrap G-25 (GE Healthcare Life Sciences, cat. no. 28-9180-04)
Reverse-phase HPLC column (Phenomenex, part: 00G-4396-B0)
Shaking incubator (set at 220 r.p.m., refrigerated 4230; New Brunswick Scientific, cat. no. M1335-0012)
Sterile 16-ml culture tubes (Fisher Scientific, cat. no. 05-540-6)
Sterile 100 mm × 25 mm Petri dishes (Fisher Scientific, cat. no. 08-757-13)
Sterile 0.22-μm polyethersulfone filters (Millipore, cat. no. SLGP033RS)
Spin concentrator (Amicon Ultra 30kDa; Millipore, cat. no. UFC903008)
Sonicator (Misonix sonicator 3000; Misonix)
Tabletop rocking platform (Fisher Scientific, cat. no. 1404123Q)
Typhoon variable mode imager (Amersham Bioscience, cat. no. 63-0055-80)
Vortex mixer (Vortex Genie-2, Mo Bio Laboratories, cat. no. 13111)
Waters QToF premier mass spectrometer (Waters) and Masslynx software
REAGENT SETUP
Acetic acid, 7.5% (vol/vol) Acetic acid (7.5%, vol/vol) is prepared using DI H2O. Store for up to 1 year.
Alexa Fluor 647 C5-aminooxyacetamide Add DMSO to a final concentration of 10 mM directly in the manufacturer-provided vial. Store for 6 months at − 20 °C in the absence of light.
Alkylation buffer Mix 0.5 M IAA in DI H2O. ▲ CRITICAL Prepare immediately prior to use.
Annealing buffer Combine 10 mM Tris, 50 mM NaCl (adjusted to pH 7.5). Store for up to 1 year at room temperature (25 °C).
l-Arabinose, 20% (wt/vol) l-Arabinose (20%, wt/vol) is prepared by dissolving in DI H2O. Filter-sterilize and store at − 20 °C for up to 1 year.
Carbenicillin To prepare a 1,000× stock solution, dissolve at 50 mg ml− 1 and filter sterilize. Store at − 20 °C for up to 1 year.
Criterion XT MES running buffer Running buffer is prepared by diluting the 20× solution provided by the manufacturer. Store for up to 1 year.
Denaturing buffer Combine 6 M guanidine HCl and 0.5 M Tris-HCl (adjusted to pH 8). Store at room temperature for up to 1 year.
Elution buffer Combine 50 mM NaH2PO4, 300 mM NaCl, 10% (vol/vol) glycerol and 250 mM imidazole (adjusted to pH 8.0). Store at 4 °C for up to 1 year.
Glycerol, 50% Glycerol (50%, vol/vol) is prepared using DI H2O. Filter-sterilize the solution and store it at room temperature for up to 1 year.
HPLC mobile phase A Add TFA to 0.1% (wt/vol) in DI H2O. Freshly prepare.
HPLC mobile phase B Add TFA to 0.1% (wt/vol) in ACN. Freshly prepare.
IPTG, 1 M IPTG (1 M) is prepared by dissolving in DI H2O. Filter-sterilize and store at − 20 °C.
Kanamycin To prepare a 1,000× stock solution, dissolve at 50 mg ml− 1 and filter sterilize. Store at −20 °C for up to 1 year.
Labeling buffer, 2× Labeling buffer is 500 mM NaOAc, adjusted to pH 4.6 with AcOH.
LB agar plates Add 15 g of agar to 1,000 ml of LB medium and then autoclave the suspension. Cool the autoclaved solution to 50 °C, add appropriate antibiotic(s), mix and then add to sterile Petri dishes.
LB medium Add 10 g of tryptone, 5 g of yeast extract and 10 g of NaCl to 1,000 ml of DI H2O. Autoclave to sterilize. Add antibiotics to cooled sterile medium as needed. Final concentrations are 50 μg ml− 1 kanamycin and/or 50 μg ml− 1 carbenicillin.
Lysis buffer Combine 50 mM NaH2PO4, 300 mM NaCl, 10% glycerol and 10 mM imidazole (adjusted to pH 8.0). Add protease inhibitor cocktail immediately prior to use. Store at 4 °C for up to 1 year.
Reducing buffer Reducing buffer is 0.5 M DTT in DI H2O.
▲ CRITICAL Prepare immediately prior to use.
SDS loading buffer, 4× Mix 0.18 M Tris, 40% (vol/vol) glycerol, 4% (wt/vol) SDS, 0.04% (wt/vol) bromophenol blue, 0.4 M DTT (adjusted to pH 6.8). Store at −20 °C for up to 1 year.
Storage buffer Combine 50 mM NaH2PO4, 300 mM NaCl and 10% (vol/vol) glycerol (adjusted to pH 8.0). Store at 4 °C for up to 1 year.
Sypro Orange working solution Make a 1:5,000 dilution of the 5,000× concentrate (supplied by Molecular Probes), in 7.5% (vol/vol) acetic acid.
TE buffer Combine 10 mM Tris and 1 mM EDTA (adjusted to pH 8.0). Store at room temperature for up to 1 year.
Wash buffer Combine 50 mM NaH2PO4, 300 mM NaCl, 10% (vol/vol) glycerol, 20 mM imidazole (adjusted to pH 8.0). Store at 4 °C for up to 1 year.
PROCEDURE
Creating aldehyde-tagged genes
1| To create an aldehyde-tagged gene by ligation of annealed nucleotides, begin at Step 2. To create an aldehyde-tagged gene by site-directed mutagenesis, proceed to Step 25.
Constructing an aldehyde-tagged gene by ligation of annealed nucleotides ● TIMING 5 d
2| For proteins to be expressed in E. coli digest an aliquot of pMALc-H vector with BamHI and HindIII. For proteins to be expressed in CHO-K1 cells (or other eukaryote cell lines), digest an aliquot of pFuse-mFc2 (IL2ss) vector with EcoRV and BglII.
| Components for prokaryote method | Amount (μl) | Final |
|
| ||
| pMALc-H (100 μg ml−1) | 5 | 0.5 μg |
| BSA (1 mg mL−1) | 3 | 100 μg ml−1 |
| NEB buffer 2 (10×) | 3 | 1× |
| BamHI (20,000 U ml−1) | 1 | 10 U |
| HindIII (20,000 U ml−1) | 1 | 10 U |
| H20 | 17 | |
| Components for eukaryote method | Amount (μl) | Final |
|
| ||
| pFuse-mFc2 (100 μg ml−1) | 5 | 0.5 μg |
| BSA (1 mg mL−1) | 3 | 100 μg ml−1 |
| NEB buffer 3 (10×) | 3 | 1× |
| BgLII (20,000 U ml−1) | 1 | 10 U |
| EcoRV (20,000 U ml−1) | 1 | 10 U |
| H2O | 17 | |
3| Incubate the digest reaction at 37 °C for 3 h in a microcentrifuge tube.
▲ CRITICAL STEP Sticky-ended DNA fragments are required. Do not use blunt cutting restriction endonucleases or nucleases that leave overhangs of less than four bases.
4| Run digested DNA on a 1% (wt/vol) agarose gel. Cut out the desired band and purify it using a gel extraction kit as instructed by the manufacturer. Elute the purified DNA in 50 μl of elution buffer. This will be used directly in the ligation below.
5| Separately dissolve the appropriate 5'-phosphorylated oligonucleotides in annealing buffer (refer to Table 1).
| Oligonucleotides for prokaryote method | 1 and 2, at 200 μM |
| Oligonucleotides for eukaryote method | 7 and 8, at 200 μM |
▲ CRITICAL STEP The oligonucleotides must be phosphorylated at the 5' ends for the ligation to proceed. The oligonucleotides can be purchased in this form or phosphorylated enzymatically by T4 polynucleotide kinase (NEB, cat. no. M0201).
6| Mix 5 μl of each oligonucleotide solution from either the prokaryote method or the eukaryote method in a PCR tube. Add 1 μl of 5 M NaCl, 1 μl of 100× TE buffer and 89 μl of H2O to the oligonucleotide solution.
7| By using a PCR machine, heat the oligonucleotide mixture to 95 °C for 1 min, cool to 85 °C for 1 min, and then cool to 4 °C.
■ PAUSE POINT The purified digested DNA and the annealed oligonucleotides can be stored at −20 °C for several weeks.
8| According to the manufacturer's instructions for T4 DNA ligase, set up 10-μl ligation reactions according to the table below. This is a general method that can be used for both prokaryotes and eukaryotes.
| Component | Amount (μl) Ligation | Amount (μl) Mock ligation | Final |
|
| |||
| Annealed oLigonucLeotides (50 μM) | 5.5 | 27.5 μM | |
| Digested and CIP-treated pLasmid | 1.5 | 1.5 | |
| NucLease-free H20 | 5.5 | ||
| T4 DNA Ligase buffer (5×) | 2 | 2 | 1× |
| T4 DNA Ligase (5 U μL−1) | 1 | 1 | 0.5 U |
9| Incubate the ligation reaction at 16 °C for 12–16 h.
10| Transform 3 μl of each ligation reaction into chemically competent TOP10 E. coli according to the manufacturer's instructions.
11| Plate the transformed cells on LB agar plates supplemented with 50 μg ml−1 kanamycin for pMALc-H or 50 μg ml−1 Zeocin for pFuse-mFc2. Incubate the plates at 37 °C overnight.
12| Pick several colonies from the real ligation and inoculate 5 ml of LB medium supplemented with 50 μg ml−1 kanamycin for pMALc-H or 50 μg ml−1 Zeocin for pFuse-mFc2. Grow the cultures overnight.
? TROUBLESHOOTING
13| Mix 750 μl of the overnight culture with 750 μl of sterile 50% (vol/vol) glycerol. Store the bacteria glycerol stocks at −80 °C for future use.
14| Pellet the remainder of the culture by centrifugation at 4,000g at 4 °C for 3 min and purify the recombinant plasmid DNA using a plasmid miniprep kit.
15| Verify the presence of the insert by DNA sequencing.
■ PAUSE POINT The DNA can be stored at −20 °C in TE buffer for several years. The frozen glycerol stocks should be stored at −80 °C.
? TROUBLESHOOTING
Constructing a Cys-to-Ala mutant by site-directed mutagenesis ● TIMING 4 d
16| In a PCR tube, set up the following reaction using the materials provided with Phusion hot start polymerase. The components for prokaryotes and eukaryotes are tabulated separately below.
| Components for prokaryote method | Amount (μl) | Final |
|
| ||
| 5× Buffer | 10.0 | 1× |
| PLasmid from Step 15 (100 μg ml−1) | 0.25 | 25 ng |
| Primer 3 (5 mM) | 3.0 | 300 μM |
| Primer 4 (5 mM) | 3.0 | 300 μM |
| dNTP (2.5 mM) | 5.0 | 250 μM |
| NucLease-free H20 | 25.25 | |
| DMSO | 2.5 | 5.0% (voL/voL) |
| Phusion hot start (2 U μl−1) | 1.0 | 2 U |
| Components for eukaryote method | Amount (μl) | Final |
|
| ||
| 5× Buffer | 10.0 | 1× |
| PLasmid from Step 15 (100 μg ml−1) | 0.25 | 25 μg |
| Primer 9 (5 mM) | 3.0 | 300 μM |
| Primer 10 (5 mM) | 3.0 | 300 μM |
| dNTP (2.5mM) | 5.0 | 250 μM |
| NucLease-free H2O | 25.25 | |
| DMSO | 2.5 | 5.0% (vol/vol) |
| Phusion hot start (2 U μl−1) | 1.0 | 2 U |
17| Use the following temperature cycling program for the mutagenesis reactions. The program is the same for prokaryotes and eukaryotes.
| Cycle | Denature | Anneal | Extend | Hold |
|
| ||||
| 1 | 98 °C, 2 min | |||
| 2–19 | 98 °C, 30 s | 50 °C, 30 s | 72 °C, 3 min 45 s | |
| 20 | 72 °C, 10 min | |||
| 21 | 4 °C | |||
18| When the PCR is complete, add 1 μl of DpnI (20,000 U ml−1) and incubate for 1 h at 37 °C.
19| Transform 3 μl of the DpnI-treated PCR into chemically competent TOP10 E. coli according to the manufacturer's instructions.
20| Plate the transformed cells on LB agar plates supplemented with 50 μg ml−1 kanamycin. Incubate the plates at 37 °C overnight.
21| Pick several colonies and inoculate 5 ml of LB medium supplemented with 50 μg ml−1 kanamycin. Grow the cultures overnight.
22| Mix 750 μl of overnight culture with 750 μl of sterile 50% (vol/vol) glycerol. Store the bacteria glycerol stocks at −80 °C for future use.
23| Pellet the remainder of the culture by centrifugation at 4,000g and purify the recombinant plasmid DNA using a plasmid miniprep kit.
24| Verify the presence of the insert by DNA sequencing.
■ PAUSE POINT The DNA can be stored at −20 °C in TE buffer for several years. The frozen glycerol stocks should be stored at −80 °C.
? TROUBLESHOOTING
Constructing an aldehyde-tagged gene by site-directed mutagenesis ● TIMING 4 d
25| This is done in the same manner as described above (Steps 16–24), with modifications to the PCR components and temperature cycling program as listed below. In addition, the antibiotic used in the LB agar and LB medium is 50 μg ml−1 carbenicillin.
The components for PCR and the temperature cycling program are as follows:
| Component | Amount (μl) | Final |
|
| ||
| 5× Buffer | 10.0 | 1× |
| pMAL-c4E (100 μg ml−1) | 1.0 | 100 ng |
| Oligonucleotide 5 (5 mM) | 5.0 | 500 μM |
| Oligonucleotide 6 (5 mM) | 5.0 | 500 μM |
| dNTP (2.5 mM/base) | 5.0 | 250 μM/base |
| Nuclease-free H20 | 20.5 | |
| DMSO | 2.5 | 5.0% (vol/vol) |
| Phusion hot start (2 U μl−1) | 1.0 | 2 U |
| Cycle | Denature | Anneal | Extend | Hold |
|
| ||||
| 1 | 98 °C, 2 min | |||
| 2–19 | 98 °C, 30 s | 50 °C, 30 s | 72 °C, 3 min 45 s | |
| 20 | 72 °C, 10 min | |||
| 21 | 4 °C | |||
Expression of aldehyde-tagged genes
26| Express the aldehyde-tagged genes in E. coli or CHO-K1 cells by following option A or option B, respectively.
(A) Expression of aldehyde-tagged genes in E. coli ● TIMING 4 d
-
(i)
Co-transform chemically competent BL21(DE3) E. coli, according to the manufacturer's instructions, with 1–3 μl (5 pg to 0.5 μg) of pBAD-Mtb-FGE plasmid and 1–3 μl (5 pg to 0.5 μg) of recombinant plasmid from Step 25 or 26.
-
(ii)
Plate the transformed cells on LB agar supplemented with 50 μg ml−1 carbenicillin and 50 μg ml−1 kanamycin, and then incubate the plate at 37 °C overnight.
-
(iii)
Select a colony from each plate and inoculate 10 ml of LB medium supplemented with 50 μg ml−1 carbenicillin and 50 μg ml−1 kanamycin. Grow the culture overnight at 37 °C with shaking.
-
(iv)
Make frozen glycerol stocks. Use the remainder of the cultures to inoculate 1,000 ml of LB medium supplemented with 50 μg ml−1 carbenicillin and 50 μg ml−1 kanamycin.
▲ CRITICAL STEP The aldehyde-tagged protein and its Cys-to-Ala mutant are taken through Step 26 in parallel. The Cys-to-Ala negative control can be conducted on a smaller scale for convenience.
-
(v)
Grow the cultures with shaking at 37 °C until optical density at 600 nm (OD600) is 0.5. Induce FGE expression by adding l-arabinose to a final concentration of 0.02% (wt/vol) and grow the cultures with shaking at 37 °C for an additional 30 min before cooling the culture to 18 °C.
-
(vi)
Induce expression of the aldehyde-tagged protein (and its Cys-to-Ala mutant) by addition of IPTG to a final concentration of 100 μM and grow the culture with shaking at 18 °C for 12 – 16 h.
▲ CRITICAL STEP The optimal concentrations of l-arabinose and IPTG may vary depending on the protein expressed.
-
(vii)
Collect the cells by centrifugation at 4,000g for 20 min at 4 °C.
■ PAUSE POINT After decanting the supernatant, the cell pellets can be stored at −80 °C for months.
-
(viii)
Resuspend the cells in 25 ml of lysis buffer, then add lysozyme to 10 μg ml−1 and DnaseI to 10 μg ml−1.
-
(ix)
While maintaining the samples on ice, lyse the cells by sonication and then clarify the lysates by centrifugation at 30,000g for 40 min at 4 °C.
-
(x)
Transfer the clarified lysates to separate 50-ml conical tubes containing 10 ml of Ni-NTA agarose equilibrated in lysis buffer. Save the insoluble fraction for later analysis.
-
(xi)
Incubate the conical tubes at 4 °C for 2 h with gentle agitation.
-
(xii)
Transfer the slurry to a column and drain the resin; retain the eluates.
-
(xiii)
Add 40 ml of wash buffer to each column to drain by gravity; retain the eluates. Repeat this step once more.
-
(xiv)
Add 5 ml of elution buffer to each column and collect the column eluates as they drain by gravity. Repeat this step three times.
-
(xv)
Analyze the saved eluates on an SDS-PAGE gel to confirm that the aldehyde-tagged protein is present primarily in the elution fractions.
? TROUBLESHOOTING
-
(xvi)
By using a spin concentrator or dialysis membrane, exchange the lysis buffer for storage buffer and reduce the volume to a desired concentration. For later use, snap-freeze the concentrated protein aliquots in liquid nitrogen and store at −80 °C.
■ PAUSE POINT The protein aliquots can be stored at −80 °C for months.
(B) Expression of aldehyde-tagged genes in CHO-K1 cells ● TIMING 10 d
-
(i)
Thaw vial of CHO-K1 cells quickly in a water bath at 37 °C.
-
(ii)
Dilute cells in 10 ml of growth medium (Ex-Cell 325 PF CHO serum-free medium supplemented with 10% (vol/vol) fetal bovine serum, 1 mM l-glutamine, 100 i.u. penicillin and 100 μg ml−1 streptomycin); gently pipette to completely resuspend the cells.
-
(iii)
Gently pellet cells at 800g for 5 min at 4 °C and aspirate the supernatant.
-
(iv)
Transfer cells to a 100-mm tissue culture dish and add 10 ml of growth medium.
-
(v)
Incubate cells at 37 °C to a confluence of 90–95%. This step can take 3–4 d.
-
(vi)
Aspirate medium and wash with 5 ml of PBS warmed to 37 °C.
-
(vii)
Aspirate PBS and add 12 ml of Opti-MEM I medium warmed to 37 °C.
-
(viii)
Add 12 μg each of pcDNA3.1-hFGE and pFuse-mFc2 (ILss) to 1.5 ml of Opti-MEM I in a 10-ml conical vial. At the same time, in a 1.5-ml microcentrifuge tube add 60 μl of Lipofectamine 2000 to 1.5 ml of Opti-MEM I.
-
(ix)
Incubate both vials for 5 min at room temperature.
-
(x)
Add the diluted Lipofectamine 2000 to the tube of diluted DNA, mix by inverting the tube four times, and then incubate at room temperature for 20 min.
-
(xi)
Add the mixture containing DNA/Lipofectamine 2000 complexes to the tissue culture dish containing the CHO-K1 cells in Opti-MEM I (the total volume in the dish should be 15 ml).
-
(xii)
Incubate the cells at 37 °C for 4 h.
-
(xiii)
Aspirate DNA/Lipofectamine complex-containing medium, and add a fresh 10 ml of Ex-Cell 325 PF CHO serum-free medium without serum or antibiotics, prewarmed to 37 °C. Incubate the cells at 37 °C for 72 h.
-
(xiv)
Collect conditioned medium from the cells and spin for 5 min at 4 °C in a 10-ml conical vial at 800g to remove cellular debris.
-
(xv)
Prepare protein A agarose beads: briefly spin down 100 μl of a 50% slurry of protein A-agarose beads at 800g in a microcentrifuge to remove the ethanol solution. Add 500 μl of Protein A-IgG binding buffer and spin again. Aspirate the supernatant. Repeat twice, and then resuspend the beads in 100 μl of PBS.
-
(xvi)
Combine the clarified conditioned medium (Step 26B(xiv)), 10 ml of Protein A-IgG binding buffer and 100 μl of protein A-agarose beads (Step 26B(xv)) in a 50-ml conical vial.
-
(xvii)
Incubate the beads with the conditioned medium at room temperature for 1 h with gentle rocking using a tabletop rocking platform, and then add in portions to a 10-ml chromatography column.
-
(xviii)
Wash the protein A-agarose beads with 5 ml of binding buffer at 4 °C.
-
(xix)
Add ten times the bead volume (500 μl) of elution buffer to the beads and collect the flow-through in a 1.5-ml vial.
-
(xx)
Immediately neutralize the pH of Fc-containing eluate by adding 1 M Tris-HCl (pH = 8.0) to 10% of the original volume of eluate (50 μl).
-
(xxi)
Concentrate the eluate using Amicon Ultra centrifugal filters (10,000 Da molecular weight cutoff) to a volume of 50 μl.
-
(xxii)
Equilibrate a mini desalting column (PD SpinTrap G-25) by spinning five times with 300 μl of PBS at 800g at room temperature for 1 min. Discard the waste after each spin.
-
(xxiii)
To exchange the buffer of Fc solution to PBS: add 70 μl of PBS to the equilibrated desalting column, then add the Fc solution and spin at 800g at room temperature in a microcentrifuge. The eluate contains the purified aldehyde-tagged Fc domain.
-
(xxiv)
Flash-freeze protein samples in liquid nitrogen and store them at −80 °C.
■ PAUSE POINT The protein can be stored at −80 °C for several months before chemical labeling.
Conjugation of aldehyde-tagged proteins ● TIMING 6 h
27| In a microcentrifuge tube, set up the conjugation reactions detailed in the table below, and then incubate the reactions for 6 h at 37 °C. The amounts of reagent and protein can be scaled up accordingly if one desires higher amounts of labeled protein for MS analysis (Step 32) or if one desires larger amounts of purified bioconjugate (Step 35).
| Component | Amount (LCTPSR) | Amount (LATPSR) |
|
| ||
| Aldehyde-tagged protein | 10 μg | 10 μg |
| Labeling buffer (2×) | 10 μl | 10 μl |
| Alexa Fluor 647 hydroxylamine (10 mM) | 1 μl | 1 μl |
| DI H2O | To 20 μl | To 20 μl |
Detection of conjugated proteins ● TIMING 3 h
28| Add 6.67 μl of 4× SDS loading buffer to the microcentrifuge tubes from Step 27 and load the samples on a Criterion XT 4 – 12% (wt/vol) Bis-Tris gel.
▲ CRITICAL STEP Boiling samples in reducing SDS loading buffer can destroy the oxime linkage. If this is required, an unheated sample should be run in parallel as a control.
29| Resolve the samples for 90 min at 150 V.
▲ CRITICAL STEP The gel must be run for a longer than average time in order to elute excess dye, which can cause high background when imaging the gel.
30| Image gel fluorescence on a Typhoon 9410 scanner using the manufacturer's recommended laser and filter settings for the Alexa Fluor 647 fluorophore.
? TROUBLESHOOTING
31| Assess total protein loading by staining the gel with Sypro Orange according the manufacturer's instructions and then image gel fluorescence again, using the manufacturer's recommended laser and filter settings for Sypro Orange.
▲ CRITICAL STEP If alternate fluorophores are used, ensure that the total protein stain (e.g., Sypro Orange) does not have overlapping emission spectra with the aminooxy-functionalized fluorophores used.
32| For MS analysis (box 2): In a microcentrifuge tube, set up the reactions detailed in the table below, then incubate the reactions for 6 h at 37 °C.
| Component | Amount (LCTPSR) |
|
| |
| Aldehyde-tagged protein | 200 μg |
| Labeling buffer (2×) | 25 μl |
| Methoxyamine (10 M) | 5 μl |
| DI H2O | To 50 μl |
33| The solution is concentrated using Amicon Ultra centrifugal filters to a volume of 20 μl and is exchanged in PBS using a desalting column (PD SpinTrap G-25).
34| The sample is frozen and stored at −80 °C.
■ PAUSE POINT The protein sample can be stored for over 6 months before analysis (described in box 2).
Purification of conjugated proteins ● TIMING 3 h
35| Add PBS (pH 7.0) to the reaction mixture from Step 27 up to a final volume of 400 μl.
36| The aldehyde-tagged protein solution is purified by size-exclusion fast protein liquid chromatography using the following conditions:
| Instrument | GE Healthcare ÄKTA purifier |
| Flow rate | 0.8 ml min−1 |
| Column | GE Healthcare Superdex 200 10/300 GL |
| Column temperature | 4 °C |
| Particle size | 13 μm |
| Binding/elution buffer | PBS, pH 7.0 |
| Injection volume | 0.5 ml |
| Elution fraction size | 0.5 ml |
| Elution volume | 1.5 column volumes |
37| Elution fractions containing the conjugated protein are combined and concentrated using an Amicon Ultra centrifugal filter (10 kDa cutoff) by spinning at 3,500g for 25 min at 4 °C to a 100-μl final volume.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 3.
TABLE 3.
Troubleshooting table.
| Step | Problem | Potential solution (s) |
|---|---|---|
| 12 | No colonies | Try an alternate source of DNA ligase |
| Use more DNA ligase | ||
| Include a crowding agent such as polyethylene glycol 400 (PEG400) | ||
| Conduct the ligation reaction at a lower temperature to aid sticky-end annealing | ||
| Equal number of colonies on real and mock Ligation | Extend the CIP treatment | |
| 15, 24 | Plasmid does not contain insert | Anneal the nucleotides in a slower manner. Turn off the PCR machine after heating to 95 °C and allow it to slowly cool to room temperature |
| Extend the CIP treatment | ||
| Increase the ratio of annealed oligonucleotides to digested plasmid | ||
| None of the colonies or plasmid contain insert | Vary the concentration of DMSO | |
| Use an alternate polymerase | ||
| Add or extend 5' overhangs on the oligonucleotides to increase their affinity toward the template relative to each other | ||
| Use a gradient of annealing temperatures | ||
| 26A(xv) | Aldehyde-tagged protein primarily in the insoluble fraction | Use a lower concentration of IPTG during induction |
| If the nontagged protein is soluble under these expression conditions, try an alternative aldehyde tag | ||
| Aldehyde-tagged protein primarily in the wash fractions | Lower the concentration of imidazole in the lysis and wash buffers | |
| 30 | Intense signaL at the bottom of the gel complicates detection | Repeat and run the gel longer to eLute excess fluorophore |
| Use a desalting spin column to reduce the amount of fluorophore present before SDS-PAGE |
● TIMING
Steps 2–15, constructing an aldehyde-tagged gene by ligation: 5 d
Steps 16–24, construction of a Cys-to-Ala mutant: 4 d
Step 25, construction of an aldehyde-tagged gene: 4 d
Step 26, expression of aldehyde-tagged genes: 4–10 d
Step 27, conjugation of aldehyde-tagged proteins: 6 h
Steps 28–34, detection of conjugated proteins: 3 h
Steps 35–37, purification of conjugated proteins: 3 h
Box 1, generation of stably expressing FGE CHO cell line: 21 d
Box 2, MS analysis of aldehyde-tagged proteins: 30 h
ANTICIPATED RESULTS
Figure 3 depicts expected results obtained after Step 27. While staining of the gel with Sypro Orange shows that both LCTPSR- and LATPSR-tagged proteins are present, gel fluorescence measurements show protein conjugation of an aldehyde-specific fluorophore to the LCTPSR-tagged protein, whereas the LATPSR-tagged protein was not converted to a formylglycine and hence not conjugated to the fluorophore.
Figure 3.

Detection of conjugation: results expected when an aldehyde-tagged protein (LCTPSR) and the Cys-to-Ala control (LATPSR) from Step 27 are run on an SDS-PAGE gel. (a) The gel is stained with Sypro Orange to determine protein loading. (b) The fluorescence of the gel is measured to determine aldehyde-specific protein conjugation of an aldehyde-specific fluorophore to the LCTPSR-tagged protein; LATPSR-tagged protein is not converted to a formylglycine and will not be conjugated to the fluorophore.
Chemical labeling efficiency is dependent on the pH of the reaction buffer and we have observed substantially decreased conjugation efficiency when the reaction buffer has a pH >6. As a first step in troubleshooting, if no labeling is observed we confirm that the pH of the reaction mixture is as expected. It should be noted that this labeling technique is not effective for proteins that are sensitive to slightly acidic pH.
Labeling efficiency is additionally affected by reagent concentration. If poor labeling is observed, subsequent reactions can be carried out in smaller volumes or with increased concentration of both the aldehyde-tagged protein and the aldehyde-specific small molecule. We have also observed that larger molecules, for example, peptides that are greater than 5 kDa in size, did not label efficiently and required additional optimization. The chemical optimization is somewhat dependent on both the aldehyde-tagged protein and the aldehyde-specific molecule. We have found that the addition of organic solvent such as ACN (5% (vol/vol) solution) or dimethylformamide (5% (vol/vol) solution) can increase conjugation efficiency. In addition, we often will screen different conjugation buffers, for example, PBS, MES, KOAc or sodium citrate, and have observed that conjugation efficiency can be improved by varying buffer composition (Table 1). For example, conjugations to IgG Fc domains are best carried out in MES buffer, whereas conjugations with hGH are more efficient in KOAc buffers. Finally, conjugation efficiency can be enhanced by increasing reaction time and reaction temperature.
Poor observed labeling efficiency could also be the result of poor conversion of cysteine to formylglycine. Poor conversion efficiency can quickly be assessed using MS (box 2). Often poor conversion is the result of poor FGE induction during protein expression. Poor enzyme production can be quickly assessed by lysis of the cell pellet (Step 26A(xv)) and analysis of the crude lysate by SDS-PAGE. Ideally, one should observe the appearance of a pronounced band in the gel corresponding to FGE (35 kDa).
Box 1 | Generation of stably expressing FGE CHO cell line ● TIMING 21 d.
REAGENTS
CHO-K1 cells (ATCC, cat. no. CCL-61)
Ex-Cell 325 PF CHO serum-free medium (Sigma-Aldrich, cat. no. 14340C)
FBS (Mediatech, cat. no. 35-010-CV)
G418 (Geneticin; Invitrogen, cat. no. 10131035)
l-Glutamine (Mediatech, cat. no. 25-005-CI)
Opti-MEM I medium (Invitrogen, cat. no. 51985034)
pcDNA3.1-hFGE plasmid
Penicillin/streptomycin solution (Mediatech, cat. no. 30-002-CI)
PROCEDURE
Thaw a vial of CHO-K1 cells (in 1 ml of growth medium containing 10% (vol/vol) DMSO) quickly in a water bath at 37 °C.
Gently pellet cells at 800g for 1 min at 4 °C and aspirate the supernatant.
Dilute cells in 10 ml of growth medium (Ex-Cell 325 PF CHO serum-free medium supplemented with 10% (vol/vol) fetal bovine serum, 1 mM l-glutamine, 100 i.u. penicillin and 100 μg ml − 1 streptomycin), gently pipetting to completely resuspend cells.
Transfer cells to a 100-mm tissue culture dish and add 10 ml of growth medium.
Incubate cells at 37 °C to a confluence of 90–95%. This step can take 3–4 d.
Aspirate the medium and wash with 10 ml of PBS warmed to 37 °C.
Decant the PBS and add 12 ml of Opti-MEM I medium warmed to 37 °C.
Add 12 μg each of pcDNA3.1-hFGE to 1.5 ml of Opti-MEM I in a 10-ml conical vial. At the same time, in a microcentrifuge tube add 60 μl of Lipofectamine 2000 to 1.5 ml of Opti-MEM I. Incubate each for 5 min at room temperature.
Add the diluted Lipofectamine 2000 to the tube of diluted DNA, mix by inverting four times, and then incubate for 20 min at room temperature.
Add the mixture of DNA/Lipofectamine 2000 complexes dropwise to the tissue culture dish containing the CHO-K1 cells (the total volume in the dish should be 15 ml).
Incubate the cells at 37 °C for 4 h.
Aspirate the medium and add 10 ml of growth medium containing 500 μg ml − 1 G418 for 72 h.
Repeat Step 12 every 48 h until clonal G418-resistant cell populations are observed (this is generally 10–14 d after transfection).
When the cells are ~75% confluent, aspirate the medium and add 2 ml of trypsin-EDTA solution and incubate at 37 °C in the CO2 incubator for 3 min or until nearly all the cells have lifted off the plate as observed under a microscope.
Gently pipette the cells into a conical vial and spin down the cells at 800g for 5 min at 4 °C.
Aspirate the medium and resuspend cells in 10 ml of growth medium containing 500 μg ml − 1 G418.
Add the resuspended cells to a dish and incubate at 37 °C for 72 h.
Repeat Steps 14 through 18 as required.
To bank cells, repeat Steps 15 through 17.
Aspirate the medium and resuspend the cells from each dish in 1 ml of growth medium containing 10% (vol/vol) DMSO.
Transfer to a 1.5-ml vial and freeze slowly at − 80 °C. If possible, for maximal cell viability upon thawing, we recommend freezing the cells at a rate of − 1 °C min − 1 in an isopropanol/CO2(s) bath. Alternatively, although initial viability may be reduced after thawing, cell vials can be frozen in styrofoam overnight.
-
Store frozen vials in liquid nitrogen.
∎ PAUSE POINT The vials can be stored for many years in liquid nitrogen.
Box 2 | MS analysis of aldehyde-tagged proteins ● TIMING 30 h.
REAGENTS
ACN (Sigma-Aldrich, cat. no. 271004)
Aldehyde-tagged protein of interest (15 mg ml − 1)
DTT (Sigma-Aldrich, cat. no. D0632)
Guanidine hydrochloride (Sigma-Aldrich, cat. no. G4505)
IAA (Sigma-Aldrich, cat. no. 068k53022)
Tris (hydroxymethyl) amionmethane (Sigma-Aldrich, cat. no. 252859)
1M Tris-HCl solution, pH = 8 (Sigma, cat. no. T2694)
Trifluoroacetic acid (TFA) (Thermo Scientific, prod. no. 28904)
Trypsin (Promega, cat. no. V5113)
EQUIPMENT
Biomax 5K filter (Millipore, cat. no. UFV5BCC00)
Centrifuge (Beckman)
Reverse-phase HPLC column (Phenomenex, part: 00G-4396-B0)
Heating block (VWR)
HPLC (Waters)
Micropipette (Fisher)
Vial (1.5 ml; Fisher, cat. no. 02-682-550)
Vortex (VWR)
Waters QToF premier mass spectrometer (Waters)
Procedure for reduction, alkylation and digestion of aldehyde-tagged protein
Pipette 11 μl of protein solution (from PROCEDURE Step 27; ~200 μg) to a 1.5-ml vial and add 100 μl of denaturing buffer.
Add 1 μl of 0.5 M DTT, mix well, and incubate at 37 °C for 40 min.
Add 3 μl of 0.5 M of IAA solution, and incubate in the dark for 45 min.
Add 2 μl of 0.5M DTT to quench the alkylation.
Remove the cap from a Biomax 5K filter and cut the tip.
Put the filter in a receiving vial.
Pipette all the solution to the filter and centrifuge at 10,000g for 10 min at 4 °C.
Remove the filter and transfer the solution in the receiving vial to a waste container.
Add 500 μl of 50 mM Tris-HCl (pH 8) solution to the filter and centrifuge at 10,000g for 10 min at 4 °C.
Transfer the solution left in the filter to a 1.5-ml vial; the volume should be ~50 μl.
Add 10 μl of 1 M Tris-HCl, pH 8, solution to the vial containing the desalted sample.
Add 20 μl of H2O.
Add 20 μl of trypsin and digest at 37 °C for 18 h using a heating block.
Procedure for Hplc-Ms of the peptide fragments
- Use the conditions and gradient below to resolve and analyze the peptide fragments from Step 13 and above.
Mobile phase A: 0.1% (wt/vol) TFA in H2O B: 0.1% (wt/vol) TFA in ACN Flow rate 0.2 ml min−1 Columns Phenomenex Jupiter Column Proteo 90A (4 μm, 90 Å, 250 × 2.0 mm) Column temperature 37 °C Autosampler temperature 5 ° ± 3 °C Injection volume 100 μl Detector wavelength 214 nm Run time 90 min Time (min) %A %B 0 98 2 5 98 2 10 89 11 54 78 22 80 70 30 97 55 45 107 40 60 117 10 90 119 10 90 120 98 2 130 98 2 - Use the Waters premier QToF mass spectrometer operating conditions as follows:
Ionization Electrospray positive, V Mode Scan type MS scan Mass range 100–4,000 Capillary voltage 3.0 kV Sampling cone voltage 25.0 V Source temperature 110 °C Desolvation temperature 300 °C Cone gas flow 40.0 liters h−1 Desolvation gas flow 800 liters h−1 LM resolution 4.7 V HM resolution 15.0 V Collision cell entrance 2.0 V Collision exit −10.0 V Collision gas flow 0.35 liters per h Reference scan frequency 10 s Analyze the peptide fragments including formylglycine containing fragments (Fig. 2), capped cysteine fragments, and methoxyamine-conjugated fragments to assess percent conversion and conjugation using Masslynx software.
ACKNOWLEDGMENTS
D.R. and C.R.B. would like to thank the US National Institutes of Health for financial support (1RC1EB010344-01 and R01GM059907). We also thank S. Zheng and P. Drake for helpful discussions and assistance with the MS protocols.
Footnotes
AUTHOR CONTRIBUTIONS All authors discussed all steps of the protocol and applications. D.R. wrote the manuscript and J.S.R., G.W.d., P.W. and C.R.B. revised it.
COMPETING FINANCIAL INTERESTS The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Sletten EM, Bertozzi CR. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem.-Int. Edit. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs. 2008;22:315–329. doi: 10.2165/00063030-200822050-00004. [DOI] [PubMed] [Google Scholar]
- 3.Cho H, et al. Optimized clinical performance of growth hormone with an expanded genetic code. Proc. Natl. Acad. Sci. USA. 2011;108:9060–9065. doi: 10.1073/pnas.1100387108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stephanopoulos N, Francis MB. Choosing an effective protein bioconjugation strategy. Nat. Chem. Biol. 2011;7:876–884. doi: 10.1038/nchembio.720. [DOI] [PubMed] [Google Scholar]
- 5.Rabuka D. Chemoenzymatic methods for site-specific protein modification. Curr. Opin. Chem. Biol. 2010;14:790–796. doi: 10.1016/j.cbpa.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu P, et al. Site-specific chemical modification of recombinant proteins produced in mammalian cells by using the genetically encoded aldehyde tag. Proc. Natl. Acad. Sci. USA. 2009;106:3000–3005. doi: 10.1073/pnas.0807820106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrico IS. Chemoselective modification of proteins: hitting the target. Chem. Soc. Rev. 2008;37:1423–1431. doi: 10.1039/b703364h. [DOI] [PubMed] [Google Scholar]
- 8.Carlson BL, et al. Function and structure of a prokaryotic formylglycine-generating enzyme. J. Biol. Chem. 2008;283:20117–20125. doi: 10.1074/jbc.M800217200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hudak JE, Yu HH, Bertozzi CR. Protein glycoengineering enabled by the versatile synthesis of aminooxy glycans and the genetically encoded aldehyde tag. J. Am. Chem. Soc. 2011;133:16127–16135. doi: 10.1021/ja206023e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hudak JE, et al. Synthesis of Heterobifunctional protein fusions using copper-free click chemistry and the aldehyde Tag. Angew. Chem. Int. Ed. Engl. 2012;124:4237–4241. doi: 10.1002/anie.201108130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dierks T, et al. Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell. 2005;121:541–552. doi: 10.1016/j.cell.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Dierks T, et al. Posttranslational formation of formylglycine in prokaryotic sulfatases by modification of either cysteine or serine. J. Biol. Chem. 1998;273:25560–25564. doi: 10.1074/jbc.273.40.25560. [DOI] [PubMed] [Google Scholar]
- 13.Frese MA, Dierks T. Formylglycine aldehyde Tag-protein engineering through a novel post-translational modification. Chembiochem. 2009;10:425–427. doi: 10.1002/cbic.200800801. [DOI] [PubMed] [Google Scholar]
- 14.Sardiello M, Annunziata I, Roma G, Ballabio A. Sulfatases and sulfatase modifying factors: an exclusive and promiscuous relationship. Hum. Mol. Genet. 2005;14:3203–3217. doi: 10.1093/hmg/ddi351. [DOI] [PubMed] [Google Scholar]
- 15.Rush JS, Bertozzi CR. New aldehyde tag sequences identified by screening formylglycine generating enzymes in vitro and in vivo. J. Am. Chem. Soc. 2008;130:12240–12241. doi: 10.1021/ja804530w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.von Figura K, Schmidt B, Selmer T, Dierks T. A novel protein modification generating an aldehyde group in sulfatases: its role in catalysis and disease. BioEssays. 1998;20:505–510. doi: 10.1002/(SICI)1521-1878(199806)20:6<505::AID-BIES9>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 17.Kalia J, Raines RT. Hydrolytic stability of hydrazones and oximes. Angew. Chem. Int. Ed. Engl. 2008;47:7523–7526. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010;14:529–537. doi: 10.1016/j.cbpa.2010.06.170. [DOI] [PubMed] [Google Scholar]
- 19.Junutula JR, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008;26:925–932. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 20.Pryor KD, Leiting B. High-level expression of soluble protein in Escherichia coli using a His6-tag and maltose-binding-protein double-affinity fusion system. Protein Expr. purify. 1997;10:309–319. doi: 10.1006/prep.1997.0759. [DOI] [PubMed] [Google Scholar]