

Abstract
Recent studies have highlighted the importance of the human microbiome in host health and disease. However, for the most part the mechanisms by which the microbiome mediates disease, or protection from it, remain poorly understood. The “keystone pathogen” hypothesis holds that certain low-abundance microbial pathogens can orchestrate inflammatory disease by remodelling a normally benign microbiota into a dysbiotic one. In this Opinion, we critically assess the available literature in support of this hypothesis, which may provide a novel conceptual basis for the development of targeted diagnostic and treatment modalities for complex dysbiotic diseases.
In architecture, the keystone is the central supporting stone at the apex of an arch. The term “keystone” has been introduced in the ecological literature to characterize species whose effects on their communities are disproportionately large relative to their abundance and which are thought to form the “keystone” of the community’s structure1–3. Although originally applied to a top predator in the rocky intertidal zone (the starfish Pisaster ochraceus)2,4, the keystone concept has been extended to species across different trophic levels and has been categorized to reflect specific functions. For instance, beavers exert keystone effects on their ecosystem by engineering the environment (“keystone modifiers”), whereas certain plants impact the ecosystem by supporting pollinators and seed dispersers (“keystone hosts”)2. The influence of keystone species contrasts with that of dominant species, which are major energy transformers in an ecosystem and thus influence it by virtue of their large biomass.
If keystone species can be identified in microbial ecology, this may have important implications in terms of enhanced insights into the structure of microbial communities and their interplay with their hosts or their environment. In humans, given the central importance of the microbiome in health and disease, there is currently tremendous interest in elucidating both the mechanisms that maintain host-microbial homeostasis at mucosal surfaces and the mechanisms that disturb this homeostatic balance, leading to dysbiosis and the initiation of inflammatory disease5–10. Keystone microorganisms that support and stabilize a microbiota associated with disease states are referred to here as “keystone pathogens”. Importantly, the capacity of keystone pathogens to instigate inflammation even when present as quantitatively minor components of the microbiota is in stark contrast to inflammation induced by dominant pathogens, which become established as the dominant component of the microbiota whilst simultaneously causing suppression of the commensals11 (Figure 1).
Figure 1. Keystone vs. dominant pathogens.

(a) Keystone pathogen-induced dysbiotic disease. Despite its low-level colonization of the periodontium, P. gingivalis causes inflammatory periodontitis through dysbiosis, i.e., an unbalancing of the relative abundance of individual components of the microbiota compared with their abundancies in health. This activity requires the bacterium’s gingipain, a C5 convertase-like enzyme which cleaves C5 generating high levels of C5a locally. C5a-induced activation of C5aR triggers inflammation but is also critically involved in a subversive crosstalk (with TLR2) that impairs leukocyte killing. The ability of P. gingivalis to orchestrate inflammatory disease via community-wide effects, while being a minor constituent of this community, qualifies it as a keystone pathogen. This process is reversible since C5aR blockade promotes the clearance of P. gingivalis and negates its dysbiotic effects. (b) Dominant pathogen-induced inflammation and effects on the microbiota. Salmonella enterica Serovar Typhimurium (S. Tm) induces and exploits inflammation to alter the composition of and outgrow the indigenous gut microbiota leading to colitis. Therefore, S. Tm incites inflammatory disease while becoming the dominant species, in stark contrast to P. gingivalis which acts as a “keystone” that supports the oral microbiota. Adapted from ref. 78.
The identification of keystone pathogens would have significant clinical benefits as it could facilitate the development of novel treatments for polymicrobial or complex dysbiotic diseases by focusing therapeutic strategies on only on a limited number of bacterial targets that stabilize the dysbiotic microbial community. Moreover, novel, targeted diagnostic tools could be developed if a complex polymicrobial disease is shown to be driven by a keystone pathogen or by a limited number of microorganisms acting in this manner.
This Opinion aims to critically assess and discuss the available evidence regarding the possible presence of keystone or keystone-like microorganisms in the human microbiotas and their role in disease. The evidence is derived mainly from experimental animal models of disease (periodontitis, inflammatory bowel disease, colon cancer and obesity) and is consistent with data derived from studies in humans. The literature is supportive of the “keystone hypothesis” and warrants further research in the quest to identify microorganisms that exert an inordinately large and adverse impact on host-microbe homeostasis.
Periodontitis and keystone pathogen-induced dysbiosis
Periodontitis is a biofilm-induced chronic inflammatory disease which affects the tooth-supporting tissues (periodontium)12 and exerts an impact on systemic health, as it increases a patient’s risk for atherosclerosis, diabetes, and possibly rheumatoid arthritis13–15. Although the tooth-associated biofilm plays a crucial role in the initiation and progression of periodontitis, it is primarily the host inflammatory response that inflicts the irreversible damage to the periodontal tissues leading, in some cases, to tooth loss16,17.
Early bacteriological studies revealed dramatic differences in the composition of the periodontal microbiota in health and disease18,19. This shift in bacterial community composition could be interpreted in two ways. First, it could be taken as a sign that specific bacteria are involved in the etiology of periodontitis, in that the disease-associated microbiota contained novel species – or periodontal pathogens – that were either not present or barely detectable in the healthy state. Second, it could imply that the disease is caused by dysbiosis of the periodontal microbiota, that is, a change in the relative abundance of individual components of the microbiota compared with their abundance in health, leading to alterations in the host-microbial crosstalk sufficient to initiate inflammatory disease.
The quest to identify specific periodontal pathogens has led to significant progress, including the identification of a number of candidates, mostly gram-negative anaerobic bacteria that colonize subgingival tooth sites. Foremost amongst this group are three species which comprise the so-called “red complex”, are frequently isolated together, and are strongly associated with diseased sites in the mouth: Porphyromonas gingivalis (formerly known as Bacteroides gingivalis)20, Treponema denticola and Tannerella forsythia21,22. Much research work has been directed towards understanding the pathogenic mechanisms and virulence determinants of these three bacterial species in the context of a conventional host-pathogen interaction, as exemplified by diseases with a single infective etiology21.
Support for the alternative hypothesis, in which periodontal pathogens transform the normally symbiotic microbiota into a dysbiotic state that leads to a breakdown in the normal homeostatic relationship with the host, came from evidence that P. gingivalis has evolved sophisticated strategies to evade or subvert components of the host immune system (e.g., Toll-like receptors and complement) rather than act directly as a proinflammatory bacterium (reviewed in refs. 9,23,24). Accordingly, it was hypothesized that P. gingivalis impairs innate immunity in ways that alter the growth and development of the entire biofilm, triggering a destructive change in the normally homeostatic host-microbial interplay in the periodontium. In other words, P. gingivalis could be a keystone pathogen of the disease-provoking periodontal microbiota.
The keystone hypothesis was supported by a recent study in the mouse model. This study showed that, at very low colonization levels (<0.01% of the total bacterial count), P. gingivalis induces periodontitis accompanied by significant alterations in the number and community organization of the oral commensal bacteria25 (Figure 1a). These alterations occur soon after P. gingivalis colonization and precede the onset of inflammatory bone loss, suggesting that the dysbiosis is probably the cause of the disease. The obligatory participation of the commensal microbiota in disease pathogenesis was shown by the failure of P. gingivalis alone to cause periodontitis in germ-free mice, despite its ability to colonize this host25.
P. gingivalis failed to cause dysbiosis and periodontitis in conventional mice if they lacked the cellular receptors necessary to subvert leukocyte defenses, or if the bacterium lacked a crucial enzymatic activity involved in leukocyte subversion25,26. In this regard, the Arg-specific cysteine proteinases (gingipains) of P. gingivalis exhibit complement C5 convertase-like activity, which generates high levels of C5a locally to activate the C5a receptor (C5aR) on leukocytes (Figure 1a). C5aR signalling is involved in crosstalk with Toll-like receptor 2 (TLR2), which is activated in parallel by P. gingivalis surface ligands, and the crosstalk leads to enhanced inflammation but impaired leukocyte killing capacity26,27. P. gingivalis may additionally prevent the activation of TLR4-dependent antimicrobial pathways in leukocytes by expressing an atypical lipopolysaccharide with a 4-acyl monophosphorylated lipid A moiety that potently antagonizes TLR4 (ref. 9). P. gingivalis can also inhibit the synthesis of IL-8 by epithelial cells to delay the recruitment of neutrophils and facilitate its initial colonization of the periodontium28,29. This inhibitory effect on IL-8 is mediated by a secreted serine phosphatase (SerB)30. The synthesis of IL-8 by the junctional gingival epithelium, adjacent to the tooth surface, is thought to be an important feature of the healthy periodontium because it generates a gradient for recruitment of neutrophils into the gingival crevice.
The subversion of recruited leukocytes by wild-type P. gingivalis may allow uncontrolled growth of other species in the same biofilm, consistent with the observed elevation of the total microbiota count following P. gingivalis colonization of the murine periodontium25. Moreover, uncontrolled bacterial growth leads to enhanced complement-dependent destructive inflammation, which generates abundant tissue breakdown products (e.g., degraded proteins and hemin) that serve the nutritional needs of the bacteria (Figure 2). This may fuel further changes to the biofilm and stabilize the transition to a disease-provoking microbiota. The inflammatory environmental changes can be better exploited by proteolytic and asaccharolytic bacteria, i.e., those organisms associated with periodontal disease rather than health9. Those species that cannot benefit from the inflammatory exudate-derived nutrients, or for which host inflammation is detrimental, may have a fitness disadvantage and hence be outcompeted or eliminated.
Figure 2. P. gingivalis-induced dysbiosis and periodontal disease.

P. gingivalis subverts complement and impairs host defense leading to overgrowth of oral commensal bacteria, which cause complement-dependent inflammation. Inflammatory tissue destruction is favorable to further bacterial growth as it provides a nutrient-rich gingival inflammatory exudate (degraded host proteins and hemin, a source of essential iron). These environmental changes are better exploited by and thus favor proteolytic and asaccharolytic bacteria, leading to compositional changes in the bacterial community. Inflammatory bone resorption, moreover, provides the dysbiotic microbiota with new niches for colonization. These alterations collectively lead to and sustain periodontal disease. The numbers indicate a possible sequence of events, which set off a self-feeding “vicious” cycle.
Although P. gingivalis exerts a keystone effect via host modulation (Figure 2), one cannot rule out the possibility that this pathogen may additionally modulate the commensal oral microbiota through host-independent, direct effects. Metatranscriptomic analysis of oral microbial community gene expression has shown that the introduction of P. gingivalis into a healthy multispecies biofilm alters the pattern of community gene expression (e.g., upregulation of proteins related to growth and division, chaperones, ABC-transport systems, putative transposases, as well as numerous transcription factors)31.
Although established in the mouse model, the keystone pathogen concept is consistent with observations in other animal models of periodontitis: In rabbits, P. gingivalis causes a shift to a more anaerobic microbiota in the dental biofilm and an overall increase in bacterial load32. In non-human primates, the reduction in the numbers of indigenous P. gingivalis, as a result of specific immunization with a gingipain-based vaccine, is accompanied by a reduction in the total subgingival bacterial load and protection against bone loss33. The keystone concept is furthermore consistent with P. gingivalis being a quantitatively minor constituent of human periodontitis-associated biofilms18,34,35, despite its high prevalence and association with progressive bone loss in periodontitis patients36,37. Importantly, specific removal of P. gingivalis from the periodontal biofilm (by means of a C5aR antagonist) reverses the dysbiotic changes25 (Figure 1a), suggesting that dysbiotic diseases could be treated by specific targeting of keystone pathogens.
Intestinal inflammatory diseases
The commensal microbiota is thought to play a role in the development of inflammatory bowel diseases (IBD)6,8,38,39. A commensal microbiota-dependent ulcerative colitis model was recently developed in T-bet−/− x Rag2−/− mice (TRUC model)40. In these mice, the absence of adaptive immunity (Rag2−/−) combined with lack of the transcription factor T-bet (T-bet−/−) results in spontaneous colitis, which is transmissible to co-housed wild-type mice, and is characterized by compromised colonic barrier function, elevated TNF, and dysfunctional dendritic cells40. It is thought that T-bet plays a role in maintaining a homeostatic relationship between the host and the microbiota; loss of T-bet expression in colonic dendritic cells causes aberrant TNF responses that drive tissue injury and compromise the colonic epithelial barrier function, which precedes the initiation of colitis40. Colitis in TRUC mice correlates strongly with the presence of Klebsiella pneumoniae and Proteus mirabilis, the combination of which induces the disease state even in specific-pathogen-free wild-type mice, hence their ability to instigate disease does not require a host with altered immune function41. Strikingly, the combination of K. pneumoniae and P. mirabilis could not by itself induce colitis when administered to germ-free TRUC mice, suggesting that their colitogenic effect is strictly dependent on the indigenous microbial community. Although K. pneumoniae and P. mirabilis can co-colonize germ-free mice at very high levels (each at ≥1011 cfu/g feces), they constitute less than 1% of the total fecal microbiota (107–109 cfu/g feces) of TRUC mice or of infected wild-type mice which, as mentioned above, develop colitis after inoculation with these two Enterobacteriaceae species41.
This study in the TRUC model41 supports the concept that IBD may not be caused by individual pathogens, but rather by an entire microbial community under the influence of specific organisms that can tip the balance from homeostasis to destructive inflammation. The requirement of the endogenous gut microbiota for the colitogenicity of two low-abundance species, K. pneumoniae and P. mirabilis, and the fact that colitis in TRUC mice is not only characterized by the presence of K. pneumoniae and P. mirabilis but also by a dysbiotic gut microbiota that is quantitatively and qualitatively different from that of healthy controls, suggests that these bacteria could act as keystone pathogens in a mode similar to that of P. gingivalis in periodontitis. However, whether K. pneumoniae and P. mirabilis affect the numbers and/or composition of the endogenous microbiota in order to elicit the colitogenic effect has not yet been addressed and one cannot exclude the possibility that the role of the commensal microbiota in this IBD model is the induction (or priming) of immunological processes that facilitate destructive inflammatory responses to K. pneumoniae and P. mirabilis.
Infection of mice with Citrobacter rodentium is used as a model for enterohemorrhagic and enteropathogenic Escherichia coli infection in humans. In this model, C. rodentium induces gut inflammation and alters the composition of the intestinal microbiota42,43. In terms of quantitative changes, one study has observed overgrowth of the Enterobacteriaceae and a significant reduction in the total number of intestinal bacteria, owing to preferential elimination of a subset of the indigenous microbiota (Cytophaga-Flavobacter-Bacteroides; CFB)42. An independent study using a similar C. rodentium infection model has observed increased abundance of Deferribacteres, Tenericutes, and some Proteobacteria, whereas the abundance of the Lactobacillaceae, which are thought to ameliorate intestinal inflammation, was reduced43. In the same study, C. rodentium comprised 2.8% of the total count in the cecum tissue and 0.23% of its luminal content43. Despite some differences in the observed microbiota compositional changes, the results of both studies suggest that C. rodentium causes global changes in microbial community structure42,43, apparently dependent upon the ability of this pathogen to cause inflammation42.
C. rodentium-induced colitis in mice is transient and the altered microbiota returns to its normal state following resolution of the infection four weeks post-infection42,43. In fact, the C. rodentium load in infected wild-type mice starts to decline two weeks post-infection43,44, although in immunocompromised mice, which develop severe disease and succumb to the infection, it continues to rise by ≈100-fold, reaching ≈1010 CFU/g colon44. Under immunocompromised conditions, therefore, this attaching-and-effacing bacterium appears to act more like a dominant pathogen. In infected normal mice, on the other hand, the relatively low levels (2.8% of the total count) of C. rodentium43 suggest that it might act as a keystone pathogen. However, it remains to be proven whether, and to what extent, the C. rodentium-induced changes to the gut microbiota contribute to inflammatory pathology. It is possible that the C. rodentium-induced changes to the intestinal microbiota occur as a result of, rather than being the cause of, intestinal inflammation42. In this regard, chemical induction of gut inflammation by administration of dextran sodium sulfate leads to a dysbiotic microbiota42, suggesting an intimate relationship between the inflammatory status of the intestine and the gut microbiota.
Until recently, the predominant view of the relationship between the inflammatory status of the intestine and the gut microbiota was that dysbiosis was a consequence and not a cause of increased inflammation. In addition to the findings in TRUC mice40,41, other studies by independent groups pose further challenge to this traditional notion: Reduced production of interleukin 18 by colonic epithelial cells in NLRP6 inflammasome-deficient mice leads to a dysbiotic microbiota (over-representation of members of Prevotellaceae and TM7; under-representation of members of genus Lactobacillus in the Firmicutes phylum), which in turn upregulates CCL5 chemokine expression and inflammatory cell recruitment leading to spontaneous inflammation45. The colitogenic activity of this altered microbiota could be transferred to cross-fostered neonatal or cohoused adult wild-type mice45. Moreover, TLR5-deficient mice exhibit features of metabolic syndrome (e.g., hyperlipidemia, hypertension, insulin resistance, and obesity) that correlate with an altered gut microbiota, which is necessary and sufficient to transfer the metabolic syndrome phenotype to germ-free mice, perhaps through chronic low-grade inflammatory signalling46. The above described studies which have demonstrated that transfer of a dysbiotic gut microbiota from a genetically compromised animal into a healthy normal recipient animal can reproduce the disease phenotype, are leading to a reevaluation of the relationship between dysbiosis and inflammation in the gut. In general, even if dysbiosis of the gut microbiota was not involved in initiating colitis, it could still play a role in maintaining the inflammatory pathology.
Colon cancer and the alpha-bug hypothesis
Infection-driven chronic inflammation can promote carcinogenesis in the affected tissues or organs. For instance, chronic infection with Helicobacter pylori is a major cause of gastric cancer and chronic hepatitis B or C virus infection can lead to liver cancer47. In these and certain other types of cancer, e.g., cervical cancer (human papillomavirus), Burkitt’s lymphoma (Epstein-Barr virus), and urinary bladder cancer (Schistosoma hematobium), the oncogenic risk is defined by a specific pathogenic organism. By contrast, colon cancer has not been linked to a single microorganism, although the colonic microbiota is required for induction of chronic colitis and colon cancer, as shown in experiments utilizing germ-free mice48. Some studies suggest that the triggering of colon cancer may involve the action of multiple members of the colonic microbiota in combination with risk factors associated with diet and host genetics49,50. Recently, it has been proposed that certain pro-oncogenic bacteria, which are dubbed “alpha-bugs” and possess unique virulence traits, may trigger colon cancer by co-opting and collaborating with the colonic microbiota51 (Figure 3).
Figure 3. The “alpha-bug” hypothesis in colon cancer.

Nonenterotoxigenic strains of B. fragilis (NTBF) are usually symbionts and those expressing polysaccharide A (PSA) have been shown to inhibit IL-17- and Th17-mediated immune responses. On the other hand, enterotoxigenic strains of B. fragilis (ETBF) activate Stat3 signalling in the colon leading to IL-17- and Th17-dependent inflammation, which is required for colonic hyperplasia and tumor formation in the multiple intestinal neoplasia (Min) mouse model. Although ETBF secretes a pro-oncogenic toxin (BFT), the participation of the colonic microbiota is necessary for colon carcinogenesis. According to the “alpha-bug” hypothesis, ETBF remodels the colonic microbiota and co-opts it in a collaborative manner to induce colon cancer in combination with disease modifiers and host genetics. It is currently unclear exactly how ETBF influences and interacts with the colonic microbiota to promote carcinogenesis. Moreover, it is uncertain whether the microbiota is modulated by Th17 inflammation or, conversely, contributes to its induction (hypothetical connections indicated by dashed arrows).
The alpha-bug hypothesis is based on studies with Bacteroides fragilis52. Although a constituent of the normal intestinal microbiota, B. fragilis may cause serious disease as an opportunistic pathogen. It is a frequent anaerobic isolate from clinical specimens (> 80% of the infections by Bacteroides spp) despite accounting for ≈ 0.3% of all the Bacteroides spp in the colon and comprising < 1 to 2% of the cultured fecal microbiota53,54. A subgroup of strains that produce one of three isotypes of a 20-kDa zinc-dependent metalloprotease toxin (B. fragilis toxin, BFT; also known as fragilysin) comprise the enterotoxigenic B. fragilis (ETBF) which is associated with inflammatory diarrheal disease and relapses of IBD54. More recently, ETBF was shown to induce the formation of colon tumors in multiple intestinal neoplasia (Min) mice52.
The capacity of ETBF to induce T helper type 17 (Th17)-dependent inflammatory responses is crucial for pathogenesis in the Min model, as IL-17 blockade or CD4+ T cell depletion inhibits ETBF-induced colitis, colonic hyperplasia, and tumor formation52. Interestingly, in the colons of Min mice, ETBF selectively activates signal transducer and activator of transcription 3 (Stat-3), which is essential for Th17 cell differentiation and, moreover, is a key regulator of oncogenesis55. In stark contrast, nontoxigenic B. fragilis (NTBF) does not activate Stat3 or induce Th17 responses and is avirulent in this model, despite sharing the ability of ETBF to chronically colonize Min mice52. These differential effects of ETBF and NTBF suggest that the capacity of the former to secrete BFT may be crucial for carcinogenesis, although it should be noted that NTBF expresses polysaccharide A, a symbiosis factor that suppresses Th17 proinflammatory responses in the gut56,57(Figure 3).
Nevertheless, BFT appears to be, in its own right, a pro-oncogenic and pro-inflammatory bacterial toxin. Indeed, BFT indirectly stimulates cleavage of E-cadherin, and thereby disrupts intercellular adhesion and compromises the barrier function of the epithelium. Cleavage of E-cadherin by BFT also triggers β-catenin nuclear signalling leading to c-Myc expression and persistent proliferation of human colonic epithelial cells (HT29/C1)58. In colonic epithelial cells, moreover, BFT activates NF-κB-dependent expression of chemokines that stimulate neutrophil transepithelial migration59. These activities may play a contributory role in IBD and the oncogenic transformation in the colon.
Antibiotic treatment of ETBF-colonized Min mice can change the carcinogenesis rate51. Because this treatment modifies the colonic microbiota without interfering with ETBF colonization, it may be implied that the interactions of ETBF with the microbiota can modulate the outcomes of colon carcinogenesis51. It is uncertain at present exactly how ETBF impacts on, or interacts with, the colonic microbiota to promote carcinogenesis. One possibility is that by inducing Th17-mediated inflammation, ETBF could modify the intraluminal environment in a manner that alters the colonic microbiota and its oncogenic potential. Additionally, these inflammatory environmental changes might selectively suppress the growth of cancer-protective bacterial species. Alternatively, the microbiota might be a crucial contributory factor for ETBF-induced Th17 responses (Figure 3). It is also possible that changes to the local microenvironment resulting from the growing tumour may cause a new selective pressure and further alterations to the microbial community, which can influence the outcome of colorectal cancer progression60.
The postulated “alpha” role of ETBF in animal models and its presence in low abundance (<1 to 2% of the colonic microbiota)51 suggest that it could be a keystone pathogen in colon tumorigenesis. Although B. fragilis has been epidemiologically associated with colon cancer in humans61, it is not known at present whether this bacterium can remodel the human gut microbiota in ways that promote inflammation and colonic epithelial cell transformation, as supported by the “alpha-bug” hypothesis. Nevertheless, recent genomic analysis of the microbiome of human colorectal cancers revealed a significant enrichment of Fusobacterium species and a depletion of the Bacteroidetes and Firmicutes, most notably the Clostridia, relative to normal colon tissue62,63.
It is currently unclear whether all ETBF strains can induce experimental colon tumors or whether the disease is BFT isotype-specific. From a translational viewpoint, if ETFB is a keystone pathogen in human colon oncogenesis, it may be possible to develop new diagnostic tools (perhaps BFT isotype-specific) to identify individuals at high risk51. Such a translational approach may additionally have to include other potential “alpha bugs”, whose specific virulence traits and modes of action might endow them with similar leading roles in colon tumorigenesis, e.g., attaching and effacing E. coli64,65. In this regard, E. coli strains possessing the pks genomic island (responsible for expression of colibactin, a polyketide-peptide genotoxin) cause DNA damage to enterocytes in vivo followed by cell division with incomplete DNA repair, thereby potentially contributing to colorectal cancer development66.
Major functions by minor members
Methanogens are archaea that constitute a minor component of the gut microbiota. However, their ability to reduce small organic compounds (e.g., carbon dioxide, acetic acid, formic acid, or methanol) into methane in the presence of H2 has significant consequences67. The removal from the gut of excess H2 through methanogenesis prevents the inhibition of bacterial nicotinamide adenine dinucleotide (NADH) dehydrogenase, thereby leading to an increased yield of ATP from bacterial metabolism and a greater harvest of energy from the diet. Methanogenesis is not the only microbial mechanism to remove excess H2 from the gut, as this can also be mediated through the reduction of sulphate to sulphide by sulphate-reducing bacteria. However, methanogens appear to outcompete sulphate-reducing bacteria for H2 in the human colon68. In comparison to methanogens that do not colonize the intestine, the gut-dwelling Methanobrevibacter smithii shows significant enrichment for genes involved in the utilization of CO2, H2 and formate for methanogenesis. It also encodes genes that are probably involved in acetate assimilation and the use of methanol and ethanol69. These features suggest that M. smithii can remove a variety of bacterial metabolic end-products, which may be conducive for syntrophic metabolism with diverse gut microorganisms. M. smithii could thus be important in stabilizing gut microbial communities and appears to be a good example of a “low-abundance microbe with abundant functions”70. Moreover, M. smithii displays an enriched repertoire for glucosyltransferase genes (relative to other sequenced non-gut methanogens), which endows it with great flexibility in decorating its surface with glycans. These, strikingly, mimic those present in the gut mucosa and may thus serve to prevent the activation of host immune responses and inflammation69.
However, the ability of M. smithii to improve the efficiency of bacterial fermentation of dietary polysaccharides may also have adverse effects. M. smithii was shown to promote host adiposity in experiments in gnotobiotic mice co-colonized with this archeaon and Bacteroides thetaiotaomicron71. Moreover, the presence of M. smithii enhances the bacterial digestion of dietary glycans by influencing the transcriptional profile of B. thetaiotaomicron; the most robust response was the induction of several fructofuranosidases, resulting in increased utilization of otherwise inaccessible fructans. The substitution of M. smithii with the sulfate-reducing bacterium Desulfovibrio piger, in a similar co-colonization experiment, did not significantly affect B. thetaiotaomicron’s transcriptome or host adiposity71.
In this context, M. smithii could be regarded as a keystone pathogen as it is a relatively minor constituent of the gut microbiome and can direct bacterial metabolism in ways that promote host adiposity. Whether M. smithii remains at relatively low colonization levels also in obesity needs to be explored further. One study showed that the Methanobacteriales count is increased in the gut of obese individuals relative to normal-weight or post-gastric-bypass individuals, although the Methanobacteriales are still at least 4 log10 units less abundant than total bacteria72. Another study found only a modest, and statistically non-significant, increase in the numbers M. smithii in obese as compared to lean individuals73. Interestingly, the numbers of M. smithii are significantly elevated in anorexic patients, possibly representing an adaptive response for maximal exploitation of the limited caloric diet of these individuals73. On the basis of the implicating evidence from experimental animals, discussed above, and the genomic and metabolic features of M. smithii in the human and mouse gut, this organism has been proposed as a potential key therapeutic target for reducing energy harvest in obese individuals and thereby treating this disease69.
Conclusions and perspectives
The advent of new molecular approaches to characterize the human microbiome has dramatically changed our appreciation of microbial diversity, although we are still far from understanding the complex host-microbial and inter-microbial interactions that either promote health or lead to disease. For at least some polymicrobial inflammatory diseases, the “keystone pathogen” hypothesis may shed light on the mechanisms governing the structure of microbial communities and how they instigate disease. There is now a substantial body of literature in support of a role for “keystone” pathogens that provoke inflammation by remodeling the microbiota. This can occur through direct effects on the microbiota (e.g., altered transcriptional profile), indirect effects resulting from host modulation (e.g., manipulation of host signalling leading to impaired immunosurveillance) or, in principle, by both mechanisms.
One intriguing issue which requires further research is why the presence of a keystone pathogen (e.g., P. gingivalis in the human periodontium or ETBF in the human colon) does not always lead to the conversion of a symbiotic microbiota to a dysbiotic one: P. gingivalis, for example, can frequently be detected at low levels in the “normal” periodontal microbiota of healthy individuals. This may, of course, be related to the strain and virulence diversity within the population structure of the relevant pathogen and this warrants more detailed molecular and functional characterization of the key virulence factors that mediate the pathogen’s keystone effects. Alternatively, there may be individuals who, by virtue of the composition of their commensal microbiota or their intrinsic immune/inflammatory status, can resist or tolerate the conversion of the microbiota from a symbiotic to a dysbiotic state. In this case, disease modifiers (genetic or environmental) could play a significant role in susceptibility or resistance to a given disease. The identification of such modifiers remains a formidable challenge and could include the presence of protective members of the microbiota able to counteract the influence of the keystone pathogen, hyporesponsive or lack-of-function polymorphisms that mitigate inflammation, or polymorphisms that counteract microbial immune evasion.
The analysis of the current literature in this Opinion suggests that the keystone pathogen concept is a plausible hypothesis. Bacteria may not be the only organisms capable of manipulating the commensal microbiota to cause disease. Recently, viruses have been shown to co-opt the intestinal commensal microbiota to promote viral pathogenesis74,75. Further research is needed to identify keystone pathogens that fulfill the criteria of low relative abundance and community-wide impact, which involves host modulation and drives disease pathogenesis. Thus, a keystone pathogen is an agent that remodels the commensal microbiota into a dysbiotic state by causing disruption of host homeostasis. A keystone pathogen does not rely on already disrupted host homeostasis to cause disease, as proposed for “pathobionts” which are not necessarily low-abundance species and promote chronic inflammatory pathology only in hosts with specific genetic or environmental alterations (e.g., immunocompromised hosts)76,77. Identifying microbial species that act as stabilizing elements of symbiotic microbial communities (Box 1) is equally important. Finally, perhaps the greatest challenge for the future will be the translation of the experimental animal findings to human medicine for the development of new diagnostic tools and treatment modalities targeting keystone pathogens in complex dysbiotic diseases.
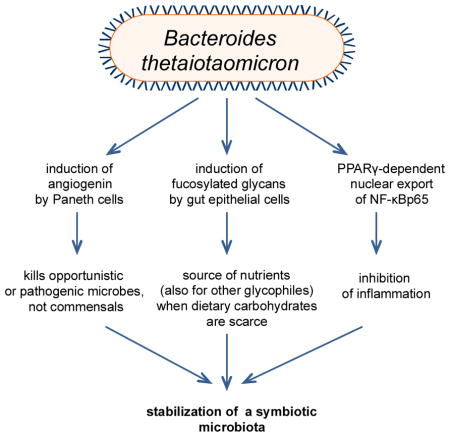
Box 1. B. thetaiotaomicron: a key stabilizing element of symbiotic microbial ecology.
Bacteroides thetaiotaomicron is an anaerobic symbiont in the distal intestine with an unusually large repertoire of genes involved in the acquisition and metabolism of polysaccharides79. This glycobiome enables B. thetaiotaomicron to turn to host polysaccharides when dietary polysaccharides become limited. B. thetaiotaomicron not only hydrolyzes host-derived glycans but proactively determines the type of glycans produced by gut epithelial cells80,81. The induction of host-derived glycans by B. thetaiotaomicron may serve an adaptive function, creating a habitable niche for itself that other glycophiles could exploit, thereby contributing to ecosystem stability and functional diversity. Another mechanism by which B. thetaiotaomicron may stabilize microbial ecology toward a healthy host-microbiota relationship involves its ability to induce the antimicrobial peptide angiogenin, which kills opportunistic or pathogenic organisms but not B. thetaiotaomicron or other commensals82. Moreover, B. thetaiotaomicron inhibits proinflammatory gene transcription through PPARγ-dependent nuclear export of NF-κBp65 (ref. 83), thereby potentially resisting inflammatory changes that could destabilize the symbiotic microbiota. These unique features have prompted the characterization of B. thetaiotaomicron as a keystone species84. In diametric opposition to a “keystone pathogen”, a low-abundance symbiont with a community-wide impact that promotes a homeostatic relationship with the host could be considered as a “keystone symbiont”. In this regard, B. thetaiotaomicron does not fulfill the low-abundance criterion and its keystone status falls under the loose definition of the term85,86. Implicit in the keystone metaphor, however, is not only the principle of high impact on the surrounding components of the community, but also the minority principle (the keystone is only one of many stones in an arch).
Acknowledgments
Work in the authors’ laboratories is supported by grants from the National Institutes of Health (DE015254, DE018292, DE021580, and DE021685 to G.H.; DE18274 and DE012768 to R.P.D.) and the Medical Research Council (UK) (G0900408 to M.A.C.).
Footnotes
Competing Interests Statement
The authors have no competing interests as defined by Nature Publishing Group, or other interests that might be perceived to influence the interpretation of the article.
References
- 1.Paine RT. A note on trophic complexity and community stability. Am Nat. 1969;103:91–93. [Google Scholar]
- 2.Power ME, et al. Challenges in the quest for keystones. BioScience. 1996;46:609–620. [Google Scholar]
- 3.Estes JA, Palmisano JF. Sea otters: their role in structuring nearshore communities. Science. 1974;185:1058–60. doi: 10.1126/science.185.4156.1058. [DOI] [PubMed] [Google Scholar]
- 4.Paine RT. Food Web Complexity and Species Diversity. Am Nat. 1966;100:65–75. [Google Scholar]
- 5.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 6.Nell S, Suerbaum S, Josenhans C. The impact of the microbiota on the pathogenesis of IBD: lessons from mouse infection models. Nat Rev Microbiol. 2010;8:564–77. doi: 10.1038/nrmicro2403. [DOI] [PubMed] [Google Scholar]
- 7.Littman DR, Pamer EG. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe. 2011;10:311–23. doi: 10.1016/j.chom.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol. 2008;8:411–20. doi: 10.1038/nri2316. [DOI] [PubMed] [Google Scholar]
- 9.Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010;8:481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 10.Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stecher B, et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007;5:2177–89. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–20. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- 13.Genco RJ, Van Dyke TE. Prevention: Reducing the risk of CVD in patients with periodontitis. Nat Rev Cardiol. 2010;7:479–80. doi: 10.1038/nrcardio.2010.120. [DOI] [PubMed] [Google Scholar]
- 14.Lundberg K, Wegner N, Yucel-Lindberg T, Venables PJ. Periodontitis in RA-the citrullinated enolase connection. Nat Rev Rheumatol. 2010;6:727–730. doi: 10.1038/nrrheum.2010.139. [DOI] [PubMed] [Google Scholar]
- 15.Lalla E, Papapanou PN. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol. 2011;7:738–48. doi: 10.1038/nrendo.2011.106. [DOI] [PubMed] [Google Scholar]
- 16.Gaffen SL, Hajishengallis G. A new inflammatory cytokine on the block: re-thinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and IL-17. J Dent Res. 2008;87:817–28. doi: 10.1177/154405910808700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eskan MA, et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol. 2012;13:465–473. doi: 10.1038/ni.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore WE, et al. Bacteriology of severe periodontitis in young adult humans. Infect Immun. 1982;38:1137–48. doi: 10.1128/iai.38.3.1137-1148.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Socransky SS. Microbiology of periodontal disease -- present status and future considerations. J Periodontol. 1977;48:497–504. doi: 10.1902/jop.1977.48.9.497. [DOI] [PubMed] [Google Scholar]
- 20.Holt SC, Ebersole J, Felton J, Brunsvold M, Kornman KS. Implantation of Bacteroides gingivalis in nonhuman primates initiates progression of periodontitis. Science. 1988;239:55–7. doi: 10.1126/science.3336774. [DOI] [PubMed] [Google Scholar]
- 21.Holt SC, Ebersole JL. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the “red complex”, a prototype polybacterial pathogenic consortium in periodontitis. Periodontol 2000. 2005;38:72–122. doi: 10.1111/j.1600-0757.2005.00113.x. [DOI] [PubMed] [Google Scholar]
- 22.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–44. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 23.Hajishengallis G, Lambris JD. Microbial manipulation of receptor crosstalk in innate immunity. Nat Rev Immunol. 2011;11:187–200. doi: 10.1038/nri2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Darveau RP. The oral microbial consortium’s interaction with the periodontal innate defense system. DNA Cell Biol. 2009;28:389–395. doi: 10.1089/dna.2009.0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hajishengallis G, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011;10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang M, et al. Microbial hijacking of complement-toll-like receptor crosstalk. Sci Signal. 2010;3:ra11. doi: 10.1126/scisignal.2000697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang S, et al. The C5a receptor impairs IL-12-dependent clearance of Porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol. 2011;186:869–877. doi: 10.4049/jimmunol.1003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darveau RP, Belton CM, Reife RA, Lamont RJ. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun. 1998;66:1660–5. doi: 10.1128/iai.66.4.1660-1665.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madianos PN, Papapanou PN, Sandros J. Porphyromonas gingivalis infection of oral epithelium inhibits neutrophil transepithelial migration. Infect Immun. 1997;65:3983–90. doi: 10.1128/iai.65.10.3983-3990.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bainbridge B, et al. Role of Porphyromonas gingivalis phosphoserine phosphatase enzyme SerB in inflammation, immune response, and induction of alveolar bone resorption in rats. Infect Immun. 2010;78:4560–9. doi: 10.1128/IAI.00703-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frias-Lopez J, Duran-Pinedo A. Effect of periodontal pathogens on the metatranscriptome of a healthy multispecies biofilm model. J Bacteriol. 2012;194:2082–2095. doi: 10.1128/JB.06328-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hasturk H, et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol. 2007;179:7021–9. doi: 10.4049/jimmunol.179.10.7021. [DOI] [PubMed] [Google Scholar]
- 33.Page RC, et al. Immunization of Macaca fascicularis against experimental periodontitis using a vaccine containing cysteine proteases purified from Porphyromonas gingivalis. Oral Microbiol Immunol. 2007;22:162–8. doi: 10.1111/j.1399-302X.2007.00337.x. [DOI] [PubMed] [Google Scholar]
- 34.Kumar PS, et al. Changes in periodontal health status are associated with bacterial community shifts as assessed by quantitative 16S cloning and sequencing. J Clin Microbiol. 2006;44:3665–73. doi: 10.1128/JCM.00317-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doungudomdacha S, Rawlinson A, Douglas CW. Enumeration of Porphyromonas gingivalis, Prevotella intermedia and Actinobacillus actinomycetemcomitans in subgingival plaque samples by a quantitative-competitive PCR method. J Med Microbiol. 2000;49:861–74. doi: 10.1099/0022-1317-49-10-861. [DOI] [PubMed] [Google Scholar]
- 36.Chaves ES, Jeffcoat MK, Ryerson CC, Snyder B. Persistent bacterial colonization of Porphyromonas gingivalis, Prevotella intermedia, and Actinobacillus actinomycetemcomitans in periodontitis and its association with alveolar bone loss after 6 months of therapy. J Clin Periodontol. 2000;27:897–903. doi: 10.1034/j.1600-051x.2000.027012897.x. [DOI] [PubMed] [Google Scholar]
- 37.Moore WE, et al. The microflora of periodontal sites showing active destructive progression. J Clin Periodontol. 1991;18:729–39. doi: 10.1111/j.1600-051x.1991.tb00064.x. [DOI] [PubMed] [Google Scholar]
- 38.Frank DN, Pace NR. Gastrointestinal microbiology enters the metagenomics era. Curr Opin Gastroenterol. 2008;24:4–10. doi: 10.1097/MOG.0b013e3282f2b0e8. [DOI] [PubMed] [Google Scholar]
- 39.Chassaing B, Darfeuille-Michaud A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1720–28. doi: 10.1053/j.gastro.2011.01.054. [DOI] [PubMed] [Google Scholar]
- 40.Garrett WS, et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131:33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garrett WS, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lupp C, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2:119–29. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 43.Hoffmann C, et al. Community-wide response of the gut microbiota to enteropathogenic Citrobacter rodentium infection revealed by deep sequencing. Infect Immun. 2009;77:4668–78. doi: 10.1128/IAI.00493-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bry L, Brigl M, Brenner MB. CD4+-T-cell effector functions and costimulatory requirements essential for surviving mucosal infection with Citrobacter rodentium. . Infect Immun. 2006;74:673–81. doi: 10.1128/IAI.74.1.673-681.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elinav E, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–57. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vijay-Kumar M, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–31. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rogers AB, Fox JG. Inflammation and Cancer. I. Rodent models of infectious gastrointestinal and liver cancer. Am J Physiol Gastrointest Liver Physiol. 2004;286:G361–6. doi: 10.1152/ajpgi.00499.2003. [DOI] [PubMed] [Google Scholar]
- 48.Uronis JM, et al. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One. 2009;4:e6026. doi: 10.1371/journal.pone.0006026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Azcarate-Peril MA, Sikes M, Bruno-Barcena JM. The intestinal microbiota, gastrointestinal environment and colorectal cancer: a putative role for probiotics in prevention of colorectal cancer? Am J Physiol Gastrointest Liver Physiol. 2011;301:G401–24. doi: 10.1152/ajpgi.00110.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hope ME, Hold GL, Kain R, El-Omar EM. Sporadic colorectal cancer--role of the commensal microbiota. FEMS Microbiol Lett. 2005;244:1–7. doi: 10.1016/j.femsle.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 51.Sears CL, Pardoll DM. Perspective: alpha-bugs, their microbial partners, and the link to colon cancer. J Infect Dis. 2011;203:306–11. doi: 10.1093/jinfdis/jiq061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu S, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–22. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holton J. Enterotoxigenic Bacteroides fragilis. Curr Infect Dis Rep. 2008;10:99–104. doi: 10.1007/s11908-008-0018-7. [DOI] [PubMed] [Google Scholar]
- 54.Sears CL. Enterotoxigenic Bacteroides fragilis: a rogue among symbiotes. Clin Microbiol Rev. 2009;22:349–369. doi: 10.1128/CMR.00053-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 56.Round JL, et al. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–7. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–5. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 58.Wu S, Morin PJ, Maouyo D, Sears CL. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology. 2003;124:392–400. doi: 10.1053/gast.2003.50047. [DOI] [PubMed] [Google Scholar]
- 59.Kim JM, et al. Nuclear factor-κB activation pathway in intestinal epithelial cells is a major regulator of chemokine gene expression and neutrophil migration induced by Bacteroides fragilis enterotoxin. Clin Exp Immunol. 2002;130:59–66. doi: 10.1046/j.1365-2249.2002.01921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver–passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol. 2012;10 doi: 10.1038/nrmicro2819. in press. [DOI] [PubMed] [Google Scholar]
- 61.Toprak NU, et al. A possible role of Bacteroides fragilis enterotoxin in the aetiology of colorectal cancer. Clin Microbiol Infect. 2006;12:782–6. doi: 10.1111/j.1469-0691.2006.01494.x. [DOI] [PubMed] [Google Scholar]
- 62.Kostic AD, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292–8. doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Castellarin M, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maddocks OD, Short AJ, Donnenberg MS, Bader S, Harrison DJ. Attaching and effacing Escherichia coli downregulate DNA mismatch repair protein in vitro and are associated with colorectal adenocarcinomas in humans. PLoS One. 2009;4:e5517. doi: 10.1371/journal.pone.0005517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martin HM, et al. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology. 2004;127:80–93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- 66.Cuevas-Ramos G, et al. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci U S A. 2010;107:11537–42. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Walker A. Say hello to our little friends. Nat Rev Microbiol. 2007;5:572–3. doi: 10.1038/nrmicro1720. [DOI] [PubMed] [Google Scholar]
- 68.Strocchi A, Furne J, Ellis C, Levitt MD. Methanogens outcompete sulphate reducing bacteria for H2 in the human colon. Gut. 1994;35:1098–101. doi: 10.1136/gut.35.8.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Samuel BS, et al. Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc Natl Acad Sci U S A. 2007;104:10643–8. doi: 10.1073/pnas.0704189104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arumugam M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–80. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Samuel BS, Gordon JI. A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism. Proc Natl Acad Sci U S A. 2006;103:10011–6. doi: 10.1073/pnas.0602187103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang H, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci U S A. 2009;106:2365–70. doi: 10.1073/pnas.0812600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Armougom F, Henry M, Vialettes B, Raccah D, Raoult D. Monitoring bacterial community of human gut microbiota reveals an increase in Lactobacillus in obese patients and Methanogens in anorexic patients. PLoS One. 2009;4:e7125. doi: 10.1371/journal.pone.0007125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kane M, et al. Successful transmission of a retrovirus depends on the commensal microbiota. Science. 2011;334:245–249. doi: 10.1126/science.1210718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kuss SK, et al. Intestinal Microbiota promote enteric virus replication and systemic pathogenesis. Science. 2011;334:249–252. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chow J, Tang H, Mazmanian SK. Pathobionts of the gastrointestinal microbiota and inflammatory disease. Curr Opin Immunol. 2011;23:473–80. doi: 10.1016/j.coi.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chow J, Mazmanian SK. A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe. 2010;7:265–76. doi: 10.1016/j.chom.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Honda K. Porphyromonas gingivalis sinks teeth into the oral microbiota and periodontal disease. Cell Host Microbe. 2011;10:423–5. doi: 10.1016/j.chom.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 79.Xu J, et al. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science. 2003;299:2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- 80.Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr. 2002;22:283–307. doi: 10.1146/annurev.nutr.22.011602.092259. [DOI] [PubMed] [Google Scholar]
- 81.Sonnenburg JL, et al. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science. 2005;307:1955–9. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- 82.Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat Immunol. 2003;4:269–73. doi: 10.1038/ni888. [DOI] [PubMed] [Google Scholar]
- 83.Kelly D, et al. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-g and RelA. Nat Immunol. 2004;5:104–12. doi: 10.1038/ni1018. [DOI] [PubMed] [Google Scholar]
- 84.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–20. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 85.Davic RD. Linking keystone species and functional groups: A new operational definition of the keystone species concept - Response. Conserv Ecol. 2003;7:r11. [Google Scholar]
- 86.Simberloff D, editor. Community and ecosystem impacts of single-species extinctions. Princeton University Press; Princeton, New Jersey: 2003. [Google Scholar]