

Abstract
The pyrrole-imidazole alkaloids have fascinated chemists for decades because of their unique structures. The high nitrogen and halogen contents and the densely functionalized skeletons make their laboratory synthesis challenging. We describe herein an oxidative method for accessing the core skeletons of two classes of pyrrole-imidazole dimers. This synthetic strategy was inspired by the putative biosynthesis pathways and its development was facilitated by computational studies. Using this method, we have successfully prepared ageliferin, bromoageliferin, and dibromoageliferin in their natural enantiomeric form.
Introduction
The pyrrole-imidazole alkaloids, often referred to as the oroidin family of alkaloids, are a group of structurally diverse natural products formed through oxidation, cyclization, and oligomerization of oroidin and its debrominated congeners (1).1 For example, ageliferins (2),2 massadines (3),3 and benzosceptrins (4)4 are the [4+2], [3+2], and [2+2] dimerization products of 1, respectively (figure 1). While cycloadditions of 1 are generally believed to be involved in the biosynthesis of 2–4,5 attempts to dimerize 1 in the test tube failed to deliver these alkaloids because of the mismatched electronic properties.6 Recently, Molinski, Romo, and co-workers used cell-free extracts of the pyrrole-imidazole alkaloid-producing sponges to induce the dimerization of hymenidine (1b) in vitro giving the [2+2] dimer benzosceptrin (4).7 They further proposed that the dimerization of 1 is promoted by a single-electron transfer (SET). We have been interested in studying the biomimetic synthesis of the pyrrole-imidazole alkaloids and proposed that the dimerization of 1 proceeds through a radical mechanism.8 Agreeing to the hypothesis put forth by Molinski and Romo, we believed that a SET oxidation of 1 would give radical cation 1•+ that is highly active toward cycloadditions. We have further shown that both the [4+2] and [3+2] dimerization products could be obtained from an oxidative radical cyclization reaction in model systems. We describe herein the design of this biomimetic synthetic strategy and its application to the asymmetric synthesis of ageliferins (2).
Figure 1.

Structures of selected pyrrole-imidazole alkaloids.
The unique structures of the dimeric pyrrole-imidazole alkaloids have, over several decades, inspired chemists to develop numerous synthetic strategies and methods. These include work from the groups of Overman,9 Carreira,10 Romo,11 Lovely,12 Ohta,13 Austin,14 Baran,15 Birman,16 Harran,17 Gin,18 Gleason,19 Namba-Williams-Nishizawa20 and us.8 Notably, the synthesis of the [2+2] dimer has been accomplished by Baran and coworkers15a and Birman and co-workers,16 the [3+2] dimers by Baran and co-workers,15c–h and the [4+2] dimers by Baran and co-workers15b and us.8c It should also be noted that Ohta and co-workers have used a biomimetic Diels–Alder approach to achieve the racemic synthesis of 12,12’-dimethyl-2a,13 and Romo and co-workers11 and Lovely and co-workers12 have independently developed a biomimetic skeletal rearrangement approach to synthesize the core skeleton of 3. Complementary to other reported approaches, our biomimetic approach comprises a manganese(III)-promoted oxidation of a β-ketoester and a radical tandem cyclization to yield the skeleton of the [4+2] or [3+2] dimer with control.8a Its development was guided by computational studies. The realization of this intramolecular radical tandem cyclization reaction suggests that the SET-mediated oroidin dimerization is chemically viable.
Results and Discussion
Research Design
We hypothesized that the biogenic dimerization of 1 proceeds through a radical mechanism as shown in Scheme 1. Activation of 1 by a SET oxidation gives radical cation 1•+, which undergoes a [4+2] (path a) or a [3+2] (path b) cycloaddition with 1 to give 5 or 6. Subsequent SET and further transformations yield 2 and 3. This cycloaddition reaction can be either a concerted or a stepwise process.
Scheme 1.

A divergent biomimetic approach for the synthesis of the [4+2] and [3+2] pyrrole-imidazole dimers.
To test this hypothesis, we designed a model system to study if the radical cycloaddition of 1 and 1•+ is viable. We envisioned that the reaction efficiency could be improved significantly by tethering 1 and 1•+, and replacing the olefin group with an enol. We thus chose 7 as the model system for 1. We anticipated that a SET oxidation of 7 would give radical cation 7•+, which resembles 1•+/1. After losing a proton, radical 7• could undergo a 5-exo followed by a 6-endo cyclization to afford the ageliferin core skeleton 8, or a 5-exo followed by another 5-exo cyclization to afford the massadine core skeleton 9.
Model Studies
We sought to initiate the radical cascade of 7• by oxidizing 7 with a Mn(III) salt.21 The first intramolecular Mn(III)-promoted radical cyclization of a β-dicarbonyl compound was reported by Corey and Kang in 1984 as part of their gingkolide synthesis project.22 Later, Snider and co-workers achieved asymmetric Mn(III) radical addition reactions by using chiral auxiliaries,23 and Yang and co-workers found that chelating lanthanide triflates could enhance both the diastereoselectivity and the reaction rate.24,25
Our work began with studying the oxidation of 10 (X=H or Cl). The α-chlorine atom was incorporated to prevent over-oxidation. To our disappointment, treating 10 with various Mn(III) and Ce(IV) salts led only to complete decomposition (Scheme 2). We found that the imidazolinone group was destroyed but the olefinic group remained intact, indicating that the oxidation occurred without radical cyclization. We suspected that radical 10• adopts the anti conformation as indicated in Scheme 2 to minimize the dipole moment. A 1,5-hydrogen abstraction of the benzylic proton occurred and destroyed the imidazolinone group. This anti conformation could possibly be destabilized by introducing an α-alkyl group (e.g., 11•). Indeed, we found that oxidation of 11 with Mn(OAc)3 proceeded cleanly to afford 12 in 63% yield. The ageliferin skeleton was obtained as the only product.8a
Scheme 2.

Realization of the Mn(III)-promoted oxidative 5-exo/6-endo cyclization.
With the working conditions for the biomimetic [4+2] dimerization reaction in hand, we next studied the asymmetric synthesis of the ageliferin core skeleton using an allylic stereocenter as the controlling element (Scheme 3).26 We found that oxidation of 13 gave 15 and 16 in a 2:1 ratio, indicating that the two faces of the olefin are slightly differentiated. We then tested if the C9-C10 relative stereochemistry could be controlled by the olefin geometry to afford the nagelamide skeleton.27 Surprisingly, we found that oxidation of 14 gave the same anti products 15 and 16 in a 2:3 ratio, suggesting that the two cyclizations proceeded stepwisely and the 6-endo cyclization was slower than the epimerization of the C10 radical. The relative configurations of 15 and 16 were determined by 1H NMR experiments. This assignment is supported by the following experiments. Removal of the TIPS group of 15 and 16 followed by oxidation of the resulting alcohol provided ketones having all identical physical properties except the direction of optical rotation.8a
Scheme 3.

Model studies of the Mn(III)-promoted oxidative cyclization reaction.
We proposed that the stereochemical information of the (Z)-olefin could be retained by constraining it in a cyclic system. Pleasingly, we found that oxidation of 17 yielded 18 with the desired syn C9-C10 configuration corresponding to that of nagelamides. The diastereoselectivity of the reaction was also improved as 18 was the only observed product.
The proposed mechanism for the oxidation of 11 is shown in Scheme 4. It has been reported that the cis-products are kinetically favored whereas the trans-products are thermodynamically favored in the Mn(III)-promoted oxidative cyclization of allylic β-ketoesters.28 We therefore believe that the cis configuration at the 5,6-ring junction in 12 resulted from trapping of the kinetic cyclization product of 11• (i.e., 19). The loss of the olefinic stereochemical information in the oxidation of 14 suggests that epimerization of the radical center of 19 is faster than the second cyclization. While radical 19 could be first oxidized to a cation and then cyclized to 12, we consider the ionic cyclization less likely because of the mismatched polarization of acylimidazolinone. Also, addition of Cu(OAc)2, which is known to accelerate the oxidation of secondary radicals by 350 times,29 had no effect on this reaction.
Scheme 4.

The proposed mechanism of the Mn(III)-promoted oxidative 5-exo/6-endo radical cyclization.
The observed regioselectivity for the cyclizations of 11 is rather unusual. The 5-exo cyclization is normally kinetically favored over the 6-endo cyclization for alkyl radicals while the opposite is true for α-acyl radicals. Based on the seminal computational studies by Houk and co-workers, the planarity of α-acyl radicals is maintained in the transition states, causing the reversed regioselectivity.30 We obtained 12 from 11 through a 5-exo cyclization of an α-acyl radical (11•→19) and a 6-endo cyclization of an alkyl radical (19→21). We are particularly interested in the mechanism of the second radical cyclization step because it determines the formation of the [4+2] versus the [3+2] dimer skeleton. This cyclization could proceed through a direct 6-endo cyclization (19→21), a 5-exo cyclization followed by a 1,2-shift (19→20→21), or a reversible 5-exo cyclization of 19 giving 21 as a thermodynamic product (20⇆19→21). We therefore analyzed all these three pathways with DFT calculations to gain insights into this cyclization reaction (Figure 2).
Figure 2.

The energy diagram for the 6-endo and the 5-exo cyclizations of 22, and the 1,2-shift of 23.
Curran, Snider, and co-workers previously showed that Mn is not bound to the substrate during the radical cyclization.31 The computational studies are therefore less complicated because the transition metal is not involved. We further simplified the computational work by replacing the benzyl and phenyl groups of 19–21 with a methyl group and examined the reactions of radicals 22–24. Because the stereochemical information of C11’ in 20 and C15’ in 21 is lost in the product 12, we calculated the energy levels for both diastereomeric pathways.
We first performed the calculations with Spartan using three different methods: UHF/6-31G*, UB3LYP/6-31G**//UHF/6-31G*and UB3LYP/6-31G**.30c,32 The geometries obtained from the UB3LYP/6-31G** calculations were further refined with Gaussian using the UB3LYP functional and the 6-311G** basis set. The corresponding energy diagram is shown in Figure 2. The 6-endo cyclization was found to be both kinetically and thermodynamically favored by all these methods; however, the HF/DFT hybrid method significantly underestimated the activation energies of the 6-endo cyclization pathway.33 According to the UB3LYP/6-311G** calculations, the activation barrier for the 6-endo cyclization is 5.7 or 4.1 kcal/mol lower than the 5-exo cyclization for 22, depending on the configuration of the newly formed stereocenters. The activation barrier for the 1,2-shift is forbiddingly high (44.7 or 50.8 kcal/mol).
We note that the 6-endo cyclization product 24 is 16.7 or 11.4 kcal/mol more stable than the 5-exo cyclization product 23. These results agree with the reported ESR studies on the captodative stabilization of radicals,34 which predict 24 to be 16.8 kcal/mol more stable than 23. The synergetic stabilization of the amino and carbonyl groups results in a 10.8 kcal/mol captodative effect. We further examined the atomic spin densities for the transition states that lead to 23 and 24 and found that the captodative stabilization also greatly contributes to the kinetic preference for the 6-endo cyclization33 whereas the steric effects may also play a role.35
Based on these results, we predicted that installation of an electron-withdrawing group to the 15’-position would introduce captodative stabilization to the 5-exo cyclization pathway and results in a switch of the pathway selectivity. Computational studies indicated that the introduction of a chloro (25) or a cyano group (26) would lower the activation barrier of the 5-exo cyclization pathway without affecting the 6-endo cyclization pathway (Figure 3). We subsequently validated this hypothesis experimentally and identified the stereochemistry of 20 and 21. We therefore focus herein the computational results corresponding to the observed stereochemistry. Our calculations suggest that the 6-endo cyclization pathway is favored for 22 (Oimid) by 4.1 kcal/mol, but the 5-exo cyclization pathways is favored for 25 (Oimid-Cl) and 26 (Oimid-CN) by 3.7 and 6.2 kcal/mol, respectively. The 6-endo cyclization product of 25 is also predicted to undergo a barrierless elimination of the chlorine atom to afford the imidazolinone directly.
Figure 3.

The energy diagram for the 6-endo versus 5-exo cyclization for 22, 25 and 26.
We were pleased to find that the experimental results are fully in line with the computational predictions (Scheme 5). Oxidation of the chloro-substituted 27 afforded the 5-exo cyclization product 28 and the mono-cyclized product 29 in a 1:1.2 ratio. The structure of 28 has been confirmed by single crystal X-ray analysis.8a Replacing the phenyl group of 27 with a methyl group gave similar results. Oxidation of 30 gave 31 and 32 in a 1:1.4 ratio. We reasoned that trans-29 and trans-32 were formed because the second cyclization was hindered by the chlorine atom, rendering the retro-cyclization of the first 5-exo cyclization competitive. A trans-lactone was thus formed, further slowing down the second 5-exo cyclization because of torsional strains.
Scheme 5.

Switching the reaction pathway through controlling the captodative stabilization of radicals.
The computational studies suggested that stronger captodative stabilization could be obtained for the 5-exo cyclization pathway with a cyano-substituted imidazolinone. Indeed, oxidation of 33 gave the 5-exo cyclization product 34 as the only product.35 This reaction delivered the massadine core skeleton directly through a 5-exo/5-exo radical tandem cyclization but with an undesired spiro configuration. This observed stereo-chemistry supports the proposed conformation of 11• and 19 shown in Scheme 4.
Because the oxidation of 33 gave the massadine core skeleton with an undesired spiro configuration, we briefly explored the potential of constructing the massadine core skeleton through oxidative ring-contraction of the ageliferin core skeleton, a method that was also studied by the groups of Romo,11 Lovely,12 and Baran5l (Scheme 6). Unfortunately, the oxidation of 12 with mCPBA proceeded slowly from the undesired β-face, giving also 28. The reaction rate was low due to the presence of a pseudo-axial phenyl group on the β-face while the concaved α-face of 12 is not accessible.
Scheme 6.

Conversion of the ageliferin core to the massadine core by an oxidative ring-contraction reaction.
Synthesis of Ageliferins
We sought to accomplish the synthesis of ageliferins (2) using the biomimetic radical cyclization reaction described above.36 We initially planned to use imidazolinone as the aminoimidazole surrogate. However, we were not able to convert imidazolinone to aminoimidazole at various stages. Activating imidazolinone with thionation, methylation, sulfonation, or phosphoration followed by attacking with a nitrogen nucleophile all failed. Model studies suggested that the activation of imidazolinone occurred predominantly on the nitrogen instead of the oxygen atom. We next planned to use thioimidazolinone as the aminoimidazolinone surrogate but found it too labile to be introduced early. We therefore decided to explore the feasibility of using azidoimidazole as a protected aminoimidazole.
We first calculated the activation barrier (UB3LYP/6-311G**) of the Mn(III)-promoted oxidative cyclization for imidazole 35 and azidoimidazole 36 to evaluate if the aromaticity of imidazole would prevent the cyclization to occur. Encouragingly, the reaction was predicted to proceed as easily as compared to imidazolinone 22 (Figure 4). Consistent with the computational predictions, our model studies showed that oxidative cyclization of imidazole 37 and azidoimidazole 39 gave the ageliferin core skeletons 38 and 40 in 40% and 42% yield, respectively (Scheme 7). The mono-cyclized product was also obtained in ca. 15% yield in both reactions.
Figure 4.

Comparing the activation barriers of the radical cyclization of imidazolinone 22, imidazole 35, and azidoimidazole 36.
Scheme 7.

Mn(III)-promoted oxidative cyclization of imidazole substrates.
We have also tested if the biomimetic oxidative ring-contraction reaction could be realized with 40. While oxidation of azidoimidazole 40 occurred without ring-contraction, reducing the azido group restored the migratory reactivity. Treating 40 with PPh3 gave a stable phosphine imide that reacted with mCPBA to give 41 (Scheme 8). Similar to the oxidation of 12, the oxidant approached from the β-face to afford the undesired spiro configuration. Removal of the lactone group and reduction of the azido group by PPh3 gave 42. Subsequent oxidation with mCPBA afforded 43, which also bears the undesired spiro configuration. The structures of 41 and 43 were confirmed by single crystal X-ray analyses.37
Scheme 8.

The biomimetic oxidative ring-contraction reactions.
Returning to the synthesis of ageliferins, we started our work by coupling azidoimidazole 44 with allylic ester 45 and prepared 46 (Scheme 9).8b Oxidative cyclization of azidoimidazole 46 proceeded uneventfully to give the ageliferin core skeleton 47. The best results were obtained when the oxidation was carried out with Mn(OAc)3 in acetic acid at 50—60 °C or with Mn(picolinate)3 in methanol at 90 °C. Purification of 47 was found challenging and reverse-phase silica gel was used to minimize material loss. While 47 can be obtained as a 2.5—3:1 mixture of the C9’ diastereomers in 18—25% isolated yield, we found that using crude 47 directly for the following steps gave a higher overall yield of 49.
Scheme 9.

Synthesis of ageliferins (2).
The stereochemical outcomes of the oxidation of 46 are in sharp contrast to those of the oxidation of 13. The existence of 47 as a mixture of C9’ diastereomers suggests that, for 46, the second, 6-endo radical cyclization was relatively slow and the first, 5-exo cyclization became reversible. Since the oxidative cyclization of the azidoimidazole analog of 13 did not yield a mixture of C9’ diastereomers,33 we concluded that the slowing of the 6-endo cyclization is because of the increased steric effects imposed by the C10 side-chain instead of the strong aromaticity of azidoimidazole.
Decarboxylation of the crude mixture of 47 yielded a single diastereomer. In contrast to 42, the cis product was found to be thermodynamically more stable. We further found that the cistrans ratio is sensitive to the size of the C10 side-chain, which may gear the C9 side-chain and increase the equatorial-equatorial eclipsing of the C9 and C9’ side-chains for the trans isomer.
Converting the revealed hydroxyl group to an amino group was surprisingly difficult. Eventually we installed this critical nitrogen atom through activating the hydroxyl group by mesylation. Converting 48 to an iodide followed by displacing the iodide by an azide afforded 49 in 11—13% yield over 9 steps. Introducing the azido group by a Mitsunobu reaction or reacting 48 directly with an azide resulted mainly in elimination.
With the N7 and N7’ nitrogen functional groups in place, we next sought to introduce the two pyrrole side-chains. A Staudinger reduction was used to reveal the N7 amine of 49. We found that only the alkyl triphenylphosphine imide was hydrolyzed under the neutral conditions. The imidazolyl phosphine imide was moderately stable even under acidic conditions, rendering it a suitable protecting group for amino-imidazole.38 The acetonide and Boc protecting groups could be selectively hydrolyzed to give 50. The excess triphenylphosphine and triphenylphosphine oxide from the Staudinger reduction were easily removed by washing 50 with diethyl ether.
For the synthesis of ageliferin (2a) and dibromoageliferin (2c), we introduced the pyrrole groups to the less hindered N7 and N7’ amines of 50 selectively. With a careful control of the reaction time, 51 was obtained as the major products in 66% yield. The N7’-mono-acylated product, obtained as the minor product in ca. 20% yield, could be recycled to improve the overall yield of 51. For the synthesis of bromoageliferin (2b), the dibromopyrrole side-chain was first introduced to the N7 amine after the Staudinger reduction of 49. Next, the N7’ amine was released and the bromopyrrole group was introduced to give 51b.
To construct the second aminoimidazole group, we first installed a guanidine group on 51 and oxidized the hydroxyl group by 2-iodoxybenzoic acid (IBX). The resulting aldehyde cyclized smoothly with the guanidine to yield 52. When the reaction was conducted at small scale, this cyclization was slow but can be facilitated by adding 0.5 equivalent of trifluoroacetic acid. The cyclization product 52 was directly purified by HPLC without workup to prevent decomposition induced by the IBX oxidation byproduct.
To complete the synthesis, the C9’ configuration of 52 was corrected through a slow equilibration under acidic conditions (60–72 h, d.r.>10:1). The C10’ carbonyl group was then removed by sequential reductions to afford 53. The BOM protecting group was cleaved by treating 53 with boron trichloride. The resulting chloromethyl group was hydrolyzed with ammonia in acetonitrile-water. Using the methanolic ammonia solution for this hydrolysis resulted in the conversion of the chloromethyl group to a methoxymethyl (MOM) group. Finally, the triphenylphosphine imide group was hydrolyzed by hydro-chloric acid at 60 °C to afford the ageliferins (2).
Summary
Through computational and experimental studies, we demonstrated that the biogenic dimerization of oroidin (1a) may proceed through a radical mechanism. Activation of 1 by SET oxidation is likely used by enzymes to promote the otherwise difficult cycloaddition reactions. We further designed a biomimetic synthesis of the [4+2] dimer of 1 using a manganese(III)-promoted radical tandem cyclization reaction. Our synthetic strategy allowed for a flexible introduction of the pyrrole side-chains with different bromine patterns, leading to the asymmetric synthesis of ageliferin (2a), bromoageliferin (2b), and dibromoageliferin (2c). We are currently investigating the biological functions of these natural products.
Supplementary Material
ACKNOWLEDGMENT
Financial Support was provided by NIH (NIGMS R01-GM079554, R01-GM079554-S1, NCCAM R01-AT006889), the Welch Foundation (I-1596), and UT Southwestern. C.C. is a Southwestern Medical Foundation Scholar in Biomedical Research. We thank Prof. Joseph Ready for the access to the Gaussian 03 suite of programs. The calculations with Gaussian 09 were supported in part by the National Science Foundation through TeraGrid resources provided by National Center for Supercomputing Applications (NCSA) under CHE090129. The X-ray analysis of 41 and 43 was performed by Dr. Muhammed Yousufuddin (University of Texas at Arlington) and Dr. Joseph Ziller (University of California, Irvine), respectively.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental procedures, characterization data, and computational details. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.a Hoffmann H, Lindel T. Synthesis. 2003;35:1753–1783. [Google Scholar]; b Jacquot DEN, Lindel T. Curr. Org. Chem. 2005;9:1551–1565. [Google Scholar]; c Du H, He Y, Sivappa R, Lovely CJ. Synlett. 2006:965–992. [Google Scholar]; d Köck M, Grube A, Seiple IB, Baran PS. Angew. Chem. Int. Ed. 2007;46:6586–6594. doi: 10.1002/anie.200701798. [DOI] [PubMed] [Google Scholar]; e Weinreb SM. Nat. Prod. Rep. 2007;24:931–948. doi: 10.1039/b700206h. [DOI] [PubMed] [Google Scholar]; f Arndt H-D, Riedrich M. Angew. Chem. Int. Ed. 2008;47:4785–4788. doi: 10.1002/anie.200801793. [DOI] [PubMed] [Google Scholar]; g Heasley B. Eur. J. Org. Chem. 2009:1477–1489. [Google Scholar]; h Forte B, Malgesini B, Piutti C, Quartieri F, Scolaro A, Papeo G. Mar. Drugs. 2009;7:705–753. doi: 10.3390/md7040705. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Gaich T, Baran PS. J. Org. Chem. 2010;75:4657–4673. doi: 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]; j Al-Mourabit A, Zancanella MA, Tilvi S, Romo D. Nat. Prod. Rep. 2011;28:1229–1260. doi: 10.1039/c0np00013b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a Rinehart KL. Pure Appl. Chem. 1989;61:525–528. [Google Scholar]; b Keifer PA, Schwartz RE, Koker MES, Robert G, Hughes J, Rittschof D, Rinehart KL. J. Org. Chem. 1991;56:2965–2975. [Google Scholar]; c Kobayashi J, Tsuda H, Murayama T, Nakamura H, Ohizumi Y, Ishibashi M, Iwamura M. Tetrahedron. 1990;46:5579–5586. [Google Scholar]
- 3.a Nishimura S, Matsunaga S, Shibazaki M, Suzuki K, Furihata K, van Soest RWM, Fusetani N. Org. Lett. 2003;5:2255–2257. doi: 10.1021/ol034564u. [DOI] [PubMed] [Google Scholar]; b Grube A, Immel S, Baran PS, Köck M. Angew. Chem. Int. Ed. 2007;46:6721–6724. 8107. doi: 10.1002/anie.200701935. [DOI] [PubMed] [Google Scholar]; c Zhang H, Khalil Z, Conte MM, Plisson F, Capon RJ. Tetrahedron Lett. 2012;53:3784–3787. [Google Scholar]
- 4.a Walker RP, Faulkner DJ, Engen DV, Clardy J. J. Am. Chem. Soc. 1981;103:6772–6773. [Google Scholar]; b Appenzeller J, Tilvi S, Martin M-T, Gallard J-F, El-bitar H, Dau ETH, Debitus C, Laurent D, Moriou C, Al-Mourabit A. Org. Lett. 2009;11:4874–4877. doi: 10.1021/ol901946h. [DOI] [PubMed] [Google Scholar]; c Tilvi S, Moriou C, Martin M-T, Gallard J-F, Sorres J, Patel K, Petek S, Debitus C, Ermolenko L, Al-Mourabit A. J. Nat. Prod. 2010;73:720–723. doi: 10.1021/np900539j. [DOI] [PubMed] [Google Scholar]; d Kubota T, Araki A, Yasuda T, Tsuda M, Fromont J, Aoyama K, Mikami Y, Wälchli MR, Kobayashi J. i. Tetrahedron Lett. 2009;52:7268–7270. [Google Scholar]
- 5.a Ref. 4a. b Ref 2b. Olofson A, Yakushijin K, Horne DA. J. Org. Chem. 1997;62:7918–7919. doi: 10.1021/jo9715682.Kinnel RB, Gehrken H-P, Swali R, Skoropowski G, Scheuer PJ. J. Org. Chem. 1998;63:3281–3286.Al Mourabit A, Potier P. Eur. J. Org. Chem. 2001:237–243.Wang Y-G, Morinaka BI, Reyes JCP, Wolff JJ, Romo D, Molinski TF. J. Nat. Prod. 2010;73:428–434. doi: 10.1021/np900638e.Ma Z, Lu J, Wang X, Chen C. Chem. Commun. 2011;47:427–429. doi: 10.1039/c0cc02214d.Feldman KS, Nuriye AY, Li J. J. Org. Chem. 2011;76:5042–5060. doi: 10.1021/jo200740b. For an alternative hypothesis, see: Baran PS, O'Malley DP, Zografos AL. Angew. Chem. Int. Ed. 2004;43:2674–2677. doi: 10.1002/anie.200453937.Baran PS, Li K, O'Malley DP, Mitsos C. Angew. Chem. Int. Ed. 2006;45:249–252. doi: 10.1002/anie.200503374.Northrop BH, O'Malley DP, Zografos AL, Baran PS, Houk KN. Angew. Chem. Int. Ed. 2006;45:4126–4130. doi: 10.1002/anie.200600514.O'Malley DP, Li K, Maue M, Zografos AL, Baran PS. J. Am. Chem. Soc. 2007;129:4762–4775. doi: 10.1021/ja069035a.
- 6.a Ref. 4a. Pöverlein C, Breckle G, Lindel T. Org. Lett. 2006;8:819–821. doi: 10.1021/ol0526219.
- 7.a Stout EP, Wang Y-G, Romo D, Molinski TF. Angew. Chem. Int. Ed. 2012;51:4877–4881. doi: 10.1002/anie.201108119. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Stout EP, Morinaka BI, Wang Y-G, Romo D, Molinski TF. J. Nat. Prod. 2012;75:527–530. doi: 10.1021/np300051k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan X, Chen C. Angew. Chem. Int. Ed. 2006;45:4345–4348. doi: 10.1002/anie.200601208. b Ref. 5g. Wang X, Lu ZMJ, Tan X, Chen C. J. Am. Chem. Soc. 2011;133:15350–15353. doi: 10.1021/ja207386q.Lu J, Tan X, Chen C. J. Am. Chem. Soc. 2007;129:7768–7769. doi: 10.1021/ja072844p.
- 9.a Overman LE, Rogers BN, Tellew JE, Trenkle WC. J. Am. Chem. Soc. 1997;119:7159–7160. [Google Scholar]; b Lanman BA, Overman LE, Paulini R, White NS. J. Am. Chem. Soc. 2007;129:12896–12900. doi: 10.1021/ja074939x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a Starr JT, Koch G, Carreira EM. J. Am. Chem. Soc. 2000;122:8793–8794. [Google Scholar]; b Chinigo GM, Carreira ABEM. Org. Lett. 2011;13:78–81. doi: 10.1021/ol102577q. [DOI] [PubMed] [Google Scholar]; c Breder A, Chinigo GM, Waltman AW, Carreira EM. Chem. Eur. J. 2011;17:12405–12416. doi: 10.1002/chem.201101862. [DOI] [PubMed] [Google Scholar]
- 11.a Dilley AS, Romo D. Org. Lett. 2001;3:1535–1538. doi: 10.1021/ol015864j. [DOI] [PubMed] [Google Scholar]; b Dransfield PJ, Dilley AS, Wang S, Romo D. Tetrahedron. 2006;62:5223–5247. [Google Scholar]; c Wang S, Dilley AS, Poullennec KG, Romo D. Tetrahedron. 2006;62:7155–7161. [Google Scholar]; d Zancanella MA, Romo D. Org. Lett. 2008;10:3685–3688. doi: 10.1021/ol801289b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a Lovely CJ, Du H, Dias HVR. Org. Lett. 2001;3:1319–1322. doi: 10.1021/ol015687m. [DOI] [PubMed] [Google Scholar]; b Lovely CJ, Du H, He Y, Dias HVR. Org. Lett. 2004;6:735–738. doi: 10.1021/ol036403w. [DOI] [PubMed] [Google Scholar]; c Sivappa R, Hernandez NM, He Y, Lovely CJ. Org. Lett. 2007;9:3861–3864. doi: 10.1021/ol0711568. [DOI] [PubMed] [Google Scholar]
- 13.a Kawasaki I, Sakaguchi N, Fukushima N, Fujioka N, Nikaido F, Yamashita M, Ohta S. Tetrahedron Lett. 2002;43:4377–4380. [Google Scholar]; b Kawasaki I, Sakaguchi N, Khadeer A, Yamashita M, Ohta S. Tetrahedron. 2006;62:10182–10192. [Google Scholar]
- 14.Koenig SG, Miller SM, Leonard KA, Löwe RS, Chen BC, Austin DJ. Org. Lett. 2003;5:2203–2206. doi: 10.1021/ol0344063. [DOI] [PubMed] [Google Scholar]
- 15.Baran PS, Zografos AL, O'Malley DP. J. Am. Chem. Soc. 2004;126:3726–3727. doi: 10.1021/ja049648s. b Ref. 5i-l. Yamaguchi J, Seiple IB, Young IS, O'Malley DP, Maue M, Baran PS. Angew. Chem. Int. Ed. 2008;47:3578–3580. doi: 10.1002/anie.200705913.O'Malley DP, Yamaguchi J, Young IS, Seiple IB, Baran PS. Angew. Chem. Int. Ed. 2008;47:3581–3583. doi: 10.1002/anie.200801138.Su S, Seiple IB, Young IS, Baran PS. J. Am. Chem. Soc. 2008;130:16490–16491. doi: 10.1021/ja8074852.Seiple IB, Su S, Young IS, Lewis CA, Yamaguchi J, Baran PS. Angew. Chem. Int. Ed. 2010;49:1095–1098. doi: 10.1002/anie.200907112.Su S, Rodriguez RA, Baran PS. J. Am. Chem. Soc. 2011;133:13922–13925. doi: 10.1021/ja206191g.Seiple IB, Su S, Young IS, Nakamura A, Yamaguchi J, Jørgensen L, Rodriguez RA, O'Malley DP, Gaich T, Köck M, Baran PS. J. Am. Chem. Soc. 2011;133:14710–14726. doi: 10.1021/ja2047232.
- 16.Birman VB, Jiang X-T. Org. Lett. 2004;6:2369–2371. doi: 10.1021/ol049283g. [DOI] [PubMed] [Google Scholar]
- 17.a Garrido-Hernandez H, Nakadai M, Vimolratana M, Li Q, Doundoulakis T, Harran PG. Angew. Chem. Int. Ed. 2005;44:765–769. doi: 10.1002/anie.200462069. [DOI] [PubMed] [Google Scholar]; b Li Q, Hurley P, Ding H, Roberts AG, Akella R, Harran PG. J. Org. Chem. 2009;74:5909–5919. doi: 10.1021/jo900755r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ding H, Roberts AG, Harran PG. Angew. Chem. Int. Ed. 2012;51:4340–4343. doi: 10.1002/anie.201200205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bultman MS, Ma J, Gin DY. Angew. Chem. Int. Ed. 2008;47:6821–6824. doi: 10.1002/anie.200801969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a Hudon J, Cernak TA, Ashenhurst JA, Gleason JL. Angew. Chem. Int. Ed. 2008;47:8885–8888. doi: 10.1002/anie.200803344. [DOI] [PubMed] [Google Scholar]; b Cernak TA, Gleason JL. J. Org. Chem. 2008;73:102–110. doi: 10.1021/jo701866g. [DOI] [PubMed] [Google Scholar]
- 20.a Namba K, Kaihara Y, Yamamoto H, Imagawa H, Tanino K, Williams RM, Nishizawa M. Chem. Eur. J. 2009;15:6560–6563. doi: 10.1002/chem.200900622. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Namba K, Inai M, Sundermeier U, Greshock TJ, Williams RM. Tetrahedron Lett. 2010;51:6557–6559. doi: 10.1016/j.tetlet.2010.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a Snider BB. Chem. Rev. 1996;96:339–363. doi: 10.1021/cr950026m. [DOI] [PubMed] [Google Scholar]; b Snider BB. Tetrahedron. 2009;65:10738–10744. doi: 10.1016/j.tet.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Melikyan GG. Org. React. 1997;49:427–675. [Google Scholar]
- 22.Corey EJ, Kang M.-c. J. Am. Chem. Soc. 1984;106:5384–5385. [Google Scholar]
- 23.Zhang Q, Mohan RM, Cook L, Kazanis S, Peisach D, Foxman BM, Snider BB. J. Org. Chem. 1993;58:7640–7651. [Google Scholar]
- 24.Yang D, Ye X-Y, Xu M, Pang K-W, Cheung K-K. J. Am. Chem. Soc. 2000;122:1658–1663. [Google Scholar]
- 25.Renaud P, Gerster M. Angew. Chem. Int. Ed. 1998;37:2562–2579. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2562::AID-ANIE2562>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 26.Hoffmann RW. Chem. Rev. 1989;89:1841–1860. [Google Scholar]
- 27.Endo T, Tsuda M, Okada T, Mitsuhashi S, Shima H, Kikuchi K, Mikami Y, Fromont J, Kobayashi J. J. Nat. Prod. 2004;67:1262–1267. doi: 10.1021/np034077n. [DOI] [PubMed] [Google Scholar]
- 28.Sung K, Wang YY. J. Org. Chem. 2003;68:2771–2778. doi: 10.1021/jo026681t. [DOI] [PubMed] [Google Scholar]
- 29.Heiba EI, Dessau RM. J. Am. Chem. Soc. 1971;93:524–527. [Google Scholar]
- 30.a Spellmeyer DC, Houk KN. J. Org. Chem. 1987;52:959–974. [Google Scholar]; b Broeker JL, Houk KN. J. Org. Chem. 1991;56:3651–3655. [Google Scholar]; c Leach AG, Wang R, Wohlhieter GE, Khan SI, Jung ME, Houk KN. J. Am. Chem. Soc. 2003;125:4271–4278. doi: 10.1021/ja029342q. [DOI] [PubMed] [Google Scholar]
- 31.Curran DP, Morgan TM, Schwartz CE, Snider BB, Dombroskit MA. J. Am. Chem. Soc. 1991;113:6607–6617. [Google Scholar]
- 32.Wong MW, Radom L. J. Phys. Chem. 1995;99:8582–8588. [Google Scholar]
- 33.See Supporting Information for details.
- 34.Welle FM, Beckhaus H-D, Rüchardt C. J. Org. Chem. 1997;62:552–558. doi: 10.1021/jo961703v. [DOI] [PubMed] [Google Scholar]
-
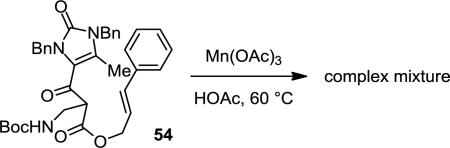
35.We believe that the steric effects are less important in determining the 5-exo/6-endo selectivity of the second cyclization. Replacing the cyano group of 33 with a methyl group did not result in a switch of the cyclization pathway. Instead, a complex mixture of reaction products was obtained from the oxidation of 54.
- 36.We previously reported the synthesis of ent-2a using ent-45 (Ref. 8c). The natural enantiomer of 2a is now synthesized for biological studies.
- 37.Based on the crystal structures of 41 and 43, the triphenylphosphine imide of aminoimidazole exists in the indicated form with no distortion and no stabilization from the N14′ nitrogen atom. The typical crystalinic N=P, N(sp2)–P, and N(sp3)–P bond length is 1.597(0.018), 1.651(0.024), and 1.683(0.005) Å, respectively. The N=P bond length is 1.599 and 1.596 Å, and the C–N=P bond angel is 123.1° and 124.1° in the crystals of 41 and 43, respectively.
- 38.The use of triphenylphosphine imide as a protecting group has been reported: Liu ST, Liu CY. J. Org. Chem. 1992;57:6079–6080.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.