

Abstract
Defects in the β subunit of rod cGMP phosphodiesterase 6 (PDE6β) are associated with autosomal recessive retinitis pigmentosa (RP), a childhood blinding disease with early retinal degeneration and vision loss. To date, there is no treatment for this pathology. The aim of this preclinical study was to test recombinant adeno-associated virus (AAV)-mediated gene addition therapy in the rod-cone dysplasia type 1 (rcd1) dog, a large animal model of naturally occurring PDE6β deficiency that strongly resembles the human pathology. A total of eight rcd1 dogs were injected subretinally with AAV2/5RK.cpde6β (n = 4) or AAV2/8RK.cpde6β (n = 4). In vivo and post-mortem morphological analysis showed a significant preservation of the retinal structure in transduced areas of both AAV2/5RK.cpde6β- and AAV2/8RK.cpde6β-treated retinas. Moreover, substantial rod-derived electroretinography (ERG) signals were recorded as soon as 1 month postinjection (35% of normal eyes) and remained stable for at least 18 months (the duration of the study) in treated eyes. Rod-responses were undetectable in untreated contralateral eyes. Most importantly, dim-light vision was restored in all treated rcd1 dogs. These results demonstrate for the first time that gene therapy effectively restores long-term retinal function and vision in a large animal model of autosomal recessive rod-cone dystrophy, and provide great promise for human treatment.
Introduction
Retinitis pigmentosa (RP), or rod-cone dystrophy, comprises a wide spectrum of incurable hereditary retinal dystrophies characterized by sequential degeneration of rod and cone photoreceptors.1 Early symptoms include night blindness and loss of peripheral vision due to progressive rod photoreceptors degeneration. This phase is followed by cone death and concomitant loss of central day vision. RP may be inherited as an autosomal recessive (50–60%), autosomal dominant (30–40%), X-linked (5–15%), or simplex/multiplex disease.2 To date, about 45 genes associated with RP etiology have been identified; most of them are primarily expressed in photoreceptor cells.3
Mutations in the gene encoding the β subunit of rod cGMP-phosphodiesterase 6 (PDE6β) are associated with one of the most prevalent forms of autosomal recessive RP, accounting for ~1–2% of all human RP cases.4,5,6,7,8,9,10,11,12 Rod PDE6 is localized on the disc membrane of rod outer segments where it plays a key role in the rod phototransduction cascade by controlling the level of cGMP and Ca2+ in the rod outer segments. Rod PDE6 is a heterotetrameric complex composed of two homologous catalytic subunits (PDE6α and PDE6β) and two copies of an inhibitory subunit (PDE6γ).13 As both PDE6α and PDE6β subunits are required for rod PDE6 activity, loss-of-function mutations in the pde6β gene result in an improper function of PDE6 holoenzyme and an accumulation of cGMP and Ca2+ in the rod outer segments. This ultimately leads to rod then cone photoreceptor death through apoptosis by non-well defined mechanisms.14,15,16
There is currently no cure for RP caused by PDE6β deficiency. Addition gene therapy is an attractive approach for PDE6β-RP.
Gene therapy approaches to delay photoreceptors degeneration have already been employed in three murine models of PDE6β-RP, the rodless 1 (rd1),17 the retinal degeneration 10 (rd10)18,19 and the PDE6βH620Q.20 Various vectors encoding murine or human pde6β gene have been tested, including adenovirus,21 adeno-associated virus (AAV),22,23,24,25 simian immunodeficiency virus,26 herpes simplex virus,27 and lentivirus.20,28 In most studies functional rescue was limited, despite significant improvement of retinal morphology with an increased number of outer nuclear layer (ONL) nuclei and outer segment length. One of the best functional results (37% preservation of rod electroretinography (ERG) responses and prolonged photoreceptor survival up to postnatal day (P) 35) was obtained in dark-reared rd10 mice, a model with a slower rate of degeneration than rd1 mice, treated at P14 using an AAV2/5smCBA.mpde6β vector.23 This suggest that difficulties in achieving sustained long-term functional rescue in murine models of PDE6β-deficiency may not be related to specific characteristics of the PDE6β subunit but to (i) excessively rapid loss of photoreceptors in these models and mainly to (ii) difficulties in achieving sufficient level of transgene expression in mutant photoreceptors before their irreversible degeneration. This hypothesis was reinforced by a recent successful gene addition therapy in the same mouse model after subretinal injection of a fast-acting AAV2/8(Y733F)smCBA.mpde6β vector.24 In this recent “proof-of-concept” study, the authors reported on a subsequent 58% preservation of rod ERG responses, rod-mediated vision-guided behavior and rod survival for up to 6 months.
Preclinical evaluation of recombinant AAV (rAAV)-mediated gene transfer in large animal models (i.e., dogs, cats, pigs, and nonhuman primates) is a key step for future clinical development.29 Indeed, the size and anatomy of the eye in large animals provide a more relevant model than rodents in terms of pathobiology and surgical approaches. Moreover, their longevity enables long-term evaluation of the treatment, which is crucial for pathologies such as PDE6β deficiency that typically progress over decades in humans.
The Rcd1 dog is a natural large animal model of progressive retinal degeneration. The Rcd1 dog carries a (2420G>A) nonsense mutation in the C-terminus of the PDE6β subunit30 leading to truncation and destabilization of the gene product and a nonfunctional PDE6 holoenzyme.31,32 Retinal development is normal in affected dogs until 13 days of age, at which point photoreceptor development is arrested. Rod degeneration then occurs gradually from 1 to 5 months of age, followed by cone loss within 1 to 2 years.33,34 This clinical course of generalized retinal atrophy, which strongly mimics retinal degeneration in humans, makes this PDE6β deficient dog a valuable large animal model for the evaluation of gene therapy.
In the present study, we investigated the long-term efficacy of early rAAV-mediated pde6β gene transfer in the rcd1 dog. Our findings demonstrate, for the first time, that gene transfer prevents long-term photoreceptor death and restores electrophysiological function and vision in this large animal model.
Results
Recombinant AAV2/5 and AAV2/8 vector design and subretinal administration to rcd1 dogs
We generated rAAV2/5 or rAAV2/8 vectors carrying the canine pde6β (cpde6β) gene under the control of the photoreceptor-specific human rhodopsin kinase (RK) promoter35,36 to drive rapid and strong transgene expression in photoreceptors (Figure 1).
Figure 1.

Schematic structure of recombinant adeno-associated virus (AAV) vectors. AAV2/5RK.cpde6β and AAV2/8RK.cpde6β vectors encode the canine pde6β cDNA under the control of a human RK promoter (–112 bp to +87 bp region of the proximal promoter36). BGHpA, bovine growth hormone polyadenylation signal; cpde6β, canine pde6β cDNA; ITR, inverted terminal repeats from AAV2; RK, human rhodopsin kinase promoter.
Rcd1 dogs display an early and severe rod-cone photoreceptor degeneration that starts in the central retina at P25, during postnatal differentiation of photoreceptor cells.33,34 This rapid onset of retinal degeneration in rcd1 dogs suggests that early therapeutic intervention is critical to prevent rod photoreceptor cell death in this model. To this end, we chose to treat rcd1 dogs at P20, before extensive rod photoreceptor loss.
A total of eight rcd1 dogs (A2–A9) were injected subretinally at the age of 20 days. Dogs A2 to A5 were treated with AAV2/5RK.cpde6β, whereas dogs A6 to A9 were injected with AAV2/8RK.cpde6β (Table 1). All injections were performed unilaterally in the right retina, except for dog A9 which received AAV2/8RK.cpde6β in both eyes due to the observation of significant vector reflux into the vitreous during the subretinal injection of the right eye. Subretinal blebs were restricted to the nasal superior retina and covered ~25% of the entire retinal surface, except in the right eye of dog A9 in which the bleb was much smaller.
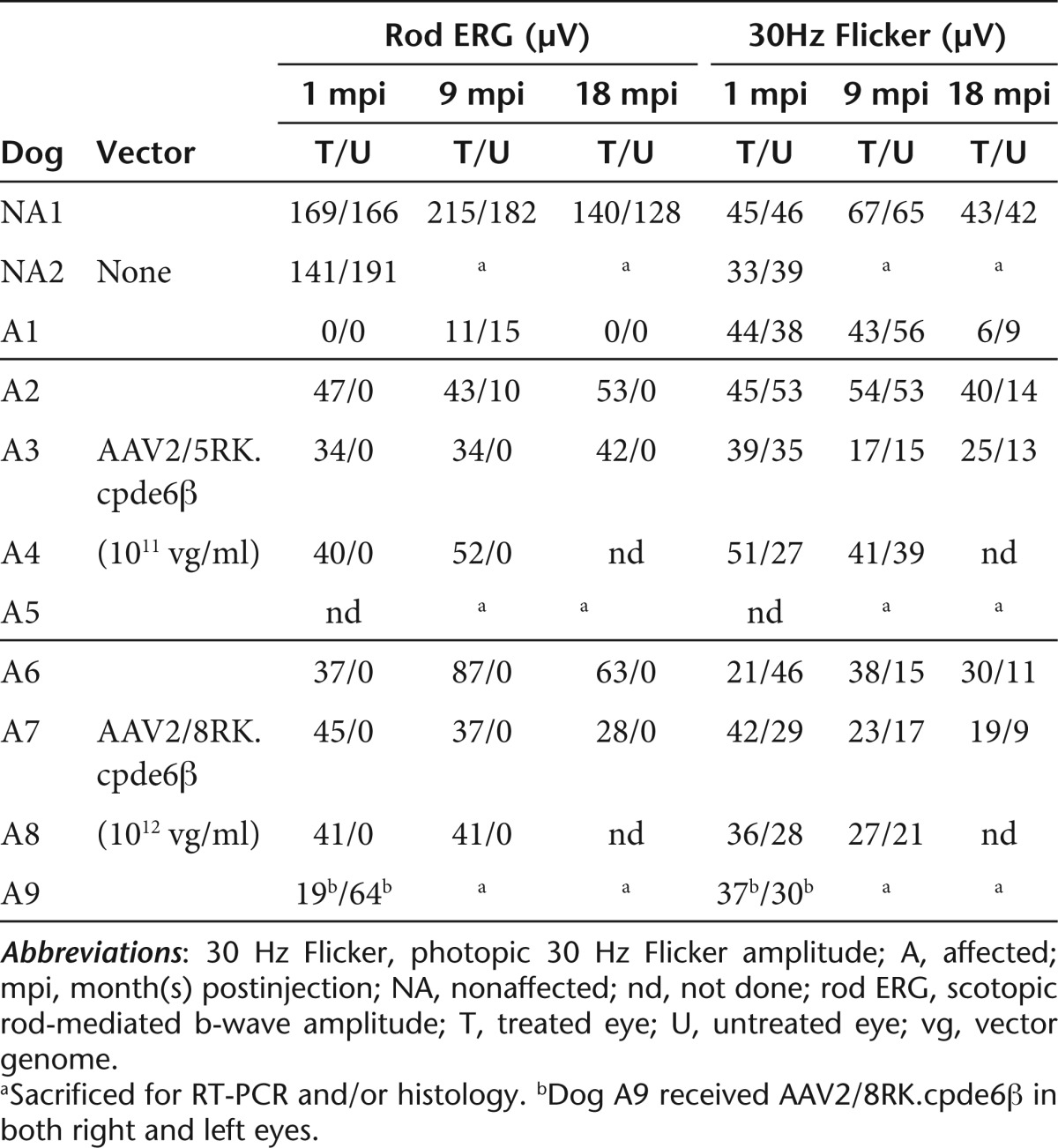
Table 1. List of dogs included in the study and ERG amplitudes.
In all treated eyes, no apparent surgically induced retinal damage was noted by fundus photography or optical coherence tomography (OCT) at 2 months postinjection (mpi), despite the small size of the eye and the immaturity of the retina at the time of the subretinal injection (data not shown).
Assessment of transgene expression in rcd1-treated retinas
To evaluate pde6β transgene expression after subretinal injection of AAV2/5RK.cpde6β and AAV2/8RK.cpde6β vectors, presence of pde6β transcripts was assessed by reverse transcription (RT)-PCR in treated retinas of dogs A4 and A8, at 4 mpi (Figure 2).
Figure 2.

Detection of transgene (wild-type) and endogenous (mutant) pde6β transcripts in A5- and A9-treated retinas at 4 mpi. (a) Reverse transcription-PCR (RT-PCR) amplification of retinal cDNA using 2056F- and 2561R-specific primers generates a 405 bp product encompassing a portion of exons 21 and 22 of transgene and native pde6β transcripts. Mismatched 2561R specifically creates a unique BtsCI restriction site in PCR products arising from pde6β transgene transcripts, dividing the 405 bp product into 385 and 20 bp fragments. This BtsCI restriction site is not present in PCR products derived from mutant RNA templates. Arrows indicate the positions of the 2056F and 2561R primers used in RT-PCR. (b) Agarose gel electrophoresis of RT-PCR products after complete BtsCI digestion. Lines 1 and 2 show products from Pde6β+/+ and Pde6β+/− (NA2) untreated control retinas, respectively. Lines 3 and 4 show products from untreated and AAV2/5RK.cpde6β-treated retinas of dog A5 at 4 mpi, respectively. Line 5 shows the AAV2/8RK.cpde6β-treated right retina of dog A9. +/+, Pde6β+/+ retina; AAV, adeno-associated virus; bp, base pairs; ladder, 1 kb molecular size ladder; T, treated retina; U, untreated retina.
As our preliminary RT-PCR analysis indicated that the rcd1 dogs expressed mutant (2420G>A) pde6β transcripts at the time of injection (P20) (data not shown), we used allele-specific RT-PCR to ensure discrimination between wild-type (transgene) and mutant 2420G>A (endogenous) pde6β transcripts in treated retinas.
Total retinal RNA was reverse-transcribed and subjected to PCR amplification designed to generate a 405 bp product encompassing a portion of exons 21 and 22 of both wild-type and mutant 2420G>A pde6β transcripts. A mismatched reverse primer specifically created a unique BtsCI restriction site in the PCR products arising from wild-type pde6β RNA templates, dividing the 405 bp product into 385 and 20 bp fragments (Figure 2a). This BtsCI restriction site was not created in the PCR products derived from mutant RNA templates. Thus, one could expect the complete digestion of RT-PCR products by BtsCI endonuclease to result in a 385-bp digestion product in RNA templates arising from Pde6β+/+ retinas, a 405 bp product in RNA templates arising from untreated Pde6β−/− retinas and both 385 and 405 bp products in RNA templates arising from Pde6β+/− and rAAV-treated Pde6β−/− retinas.
The results were as expected (Figure 2b). Transgene-derived 385 bp product was detected in both AAV2/5RK.cpde6β- and AAV2/8RK.cpde6β-treated retinas. On the contrary, this 385 bp product was never detected in the untreated rcd1 retina. These results clearly show that vector-encoded pde6β was expressed in both AAV2/5RK.cpde6β- and AAV2/8RK.cpde6β-treated retinas.
AAV2/5RK.cpde6β and AAV2/8RK.cpde6β preserve retinal structure
Fundus appearance and retinal thickness were evaluated in all dogs using color fundus photography and OCT. These noninvasive examinations were performed from 2 to 18 mpi for dogs A2, A3, A6, and A7, from 2 to 8 mpi for dogs A4 and A8, and from 2 to 3 mpi for dogs A5 and A9. Recordings on age-matched untreated Pde6β+/− (NA1) and Pde6β−/− (A1) dogs were performed as controls. All examinations were performed bilaterally to optimize the comparison between treated and untreated eyes.
Figure 3 shows the ophthalmoscopy results obtained for rAAV2/5-treated (A3), rAAV2/8-treated (A7) and age-matched Pde6β+/− control (NA1) dogs, at 4 and 18 mpi.
Figure 3.

In vivo assessment of retinal morphology in dogs A3 and A7 at 4 and 18 mpi. (a) Fundus photographs and retinal cross-sectional images obtained from control nonaffected, untreated dog NA1 at 18 months of age. (b) Fundus photographs and retinal cross-sectional images obtained from dog A3 treated with AAV2/5RK.cpde6β at 4 and 18 mpi. (c) Fundus photographs and retinal cross-sectional images obtained from A7 treated with AAV2/8RK.cpde6β at 4 and 18 mpi. Dark circles on fundus photographs schematically represent areas of the treated retinas exposed to recombinant adeno-associated virus (rAAV) vectors. Optical coherence tomography (OCT) scans were acquired on a horizontal line shown on the fundus images (dark-line). The localization and the size of the dark-lines represent the localization and the size of the OCT scans. Retinal thicknesses at the same location were measured using calibrated calipers and indicated on the OCT scan. mpi, months postinjection; µm, micrometers.
Fundus photography revealed a significant preservation of the retinal vasculature in both rAAV2/5- and rAAV2/8-treated eyes compared with an age-matched Pde6β+/− control. In contrast, severe retinal degeneration, hyper-reflective fundi and attenuated retinal vessels were observed in the untreated left eye of dogs A3 (Figure 3b, left) and A7 (Figure 3c, left) as early as 4 mpi. By 18 mpi, only few central retinal vessels were still visible in the untreated affected eyes and pallor of the optic nerve was observed (Figure 3b,c, right).
In all rAAV-treated eyes, this preservation of fundus appearance was associated with a preservation of central retinal thickness, as assessed by long-term OCT monitoring. At 4 and 18 mpi, the treated retina of dog A3 (224 and 207 µm) was 22 to 24% thicker than the contralateral untreated retina (174 and 157 µm) (Figure 3b). Meanwhile, the treated retina of dog A7 (240 µm) was 17–21% higher than the contralateral untreated retina (199 and 190 µm) (Figure 3c). At 18 mpi, the retinal thickness of A3 AAV2/5RK.cpde6β- and A7 AAV2/8RK.cpde6β-treated retinas was 207- and 240-µm thick, respectively, representing 83–97% of that observed in age-matched noninjected Pde6β+/− eyes (248 µm) (Figure 3a).
To more accurately define the therapeutic effect of rAAV-mediated gene transfer on retinal degeneration observed in vivo by OCT, we serially cryosectioned the entire treated retinas of dogs A5 and A9. The untreated left retinas of dogs A5 and NA2 were processed similarly and examined as controls.
The expression of the PDE6β subunit in treated rcd1 eyes was not evaluated as all commercially available antibodies against the mouse or human PDE6β subunit were tested and found to be nonspecific in dogs due to crossreactivity with the canine PDE6α subunit (data not shown). Rod photoreceptors were thus identified by staining for the entire rod PDE6 holoenzyme or GNAT1.
Figure 4 shows photographs of hematoxylin and eosin stained sections and images of rod PDE6 and GNAT1 immunofluorescence obtained from nasal superior retinas of dogs A5 and NA2.
Figure 4.

Post-mortem assessment of retinal morphology in dog A5 at 4 months postinjection. (a–c) Nasal retinal cryosections from the treated and untreated eyes of dog A5 at 4 months post subretinal delivery of rAAV2/5RK.cpde6β in the nasal superior retina. (a) Wide A5 retinal section displaying vector exposed and unexposed-areas. Retinal layers remained intact while the choroid has been partially detached from the retina during the embedding process. (b) Fundus photograph representing the A5 retina exposed to the rAAV2/5 vector (dark circle) and the localization of the wide retinal section (red line). (c) Nasal retinal cryosections from unaffected, untreated dog NA2 at 5 months of age and from the untreated and treated eyes of dog A5. Serial retinal cryosections were processed for hematoxylin and eosin coloration (top) and for immunohistochemistry using antibodies against rod PDE6 (middle) or GNAT1 (bottom). Primary antibodies were detected with Alexa 488-conjugated goat anti-rabbit IgG (green). Cell nuclei were counterstained with DAPI (blue). Bar = 10 µm. Vertical dark lines indicate ONL thickness (mean ± SEM, n = 10). GC, ganglion cells; INL, inner nuclear layer; IS, inner segments; ONL, outer nuclear layer; OS, outer segments; rAAV, recombinant adeno-associated virus; RPE, outer retinal pigment.
Low magnifications revealed a dramatic difference in retinal morphology between the vector-exposed and unexposed areas of the treated retina of dog A5 (Figure 4a,b). The vector-exposed region of the treated A5 retina retained a nearly normal morphology compared with the Pde6β+/− age-matched control retina (Figure 4c, top). The ONL from the treated A5 retina was 16–17 rows thick (39 ± 3 µm, n = 10), accounting for ~90% of the ONL thickness of the control retina (18–19 rows, 42 ± 2 µm, n = 10) (Figure 4c, top). Most importantly, the mean combined inner and outer segment thickness (22 ± 3 µm, n = 10) was indistinguishable from that of the Pde6β+/− control retina (22 ± 1 µm, n = 10).
In the vector-unexposed region of the A5 treated retina, however, a massive loss of photoreceptor cell nuclei was observed (Figure 4c, top) similarly to that seen in the noninjected contralateral retina. The ONL was reduced to 6–7 rows (20 ± 2 µm, n = 10) which represented only one-third to one-half of the number of rows observed in normal or in the vector-exposed retina respectively, and ~50% of normal or vector-exposed ONL thickness (Figure 4c, top). Moreover, photoreceptor inner and outer segments appeared to be shorter (16 ± 2 µm, n = 10), less dense and disorganized compared with the vector-exposed region and the nonaffected control retina.
Strong PDE6 labeling was found in outer segments in the vector-exposed region of the A5 treated retina (Figure 4c, middle), confirming the preservation of rod photoreceptor morphology after rAAV-mediated gene transfer. In contrast, only mild or undetectable PDE6 immunofluorescence was detected in the vector-unexposed region of the A5 treated retina and in the A5 untreated retina (Figure 4c, middle). A similar pattern of immunofluorescence was obtained using an antibody directed against GNAT1 (Figure 4c, bottom).
The analysis of the entire retina showed that the preserved morphology covered ~25% of the total retinal surface (data not shown) and was mostly restricted to the region exposed to the vector.
AAV2/5RK.cpde6β and AAV2/8RK.cpde6β rescue retinal function
Retinal function was evaluated by the same investigator (L.L.) in all dogs using simultaneous bilateral full-field flash ERG. ERGs were performed on all treated dogs at different timepoints following vector delivery, starting at 1 mpi, except for dog A5 for which retinal function was first assessed at 3 mpi. Follow-up was 18 mpi for dogs A2, A3, A6 and A7, 9 mpi for dogs A4 and A8, and 4 mpi for dogs A5 and A9. Recordings on age-matched untreated Pde6β+/− (NA1) and Pde6β−/− (A1) dogs were performed as controls (Table 1).
Representative ERG waveforms for dogs A1, A2, and A6 at 1, 9, and 18 mpi are shown in Figure 5. The kinetics of functional recovery in dogs A2 and A6 are presented in Figure 6.
Figure 5.

Bilateral full-field electroretinographic traces from dogs NA1, A1, A2, and A6 at 1, 9, and 18 months following subretinal injection. (a) Electroretinographic trace from control nonaffected, untreated dog NA1 at 18 months of age. (b) Electroretinographic traces from control affected, untreated dog A1 at 1 and 18 months of age. (c) Electroretinographic traces from dog A2 treated with AAV2/5RK.cpde6β. (d) Electroretinographic traces from dog A6 treated with AAV2/8RK.cpde6β. The top two recordings are low- and high-intensity dark-adapted responses, whereas the bottom two recordings show light-adapted responses (responses to single flash and 30 Hz flicker stimuli, respectively). AAV, adeno-associated virus; mpi, month(s) postinjection; T, treated eye; U, untreated eye.
Figure 6.

Kinetics of retinal function recovery in treated dogs A2 and A6. (a) Amplitudes of electroretinography (ERG) responses for control affected, untreated dog A1 from 1 to 18 months of age. (b) Amplitudes of ERG responses for dog A2 treated with AAV2/5RK.cpde6β from 1 to 18 mpi. (c) Amplitudes of ERG responses for dog A6 treated with AAV2/8RK.cpde6β from 1 to 18 mpi. The left and middle panels show scotopic rod and mixed cone-rod-mediated b-wave amplitudes, respectively. The right panel shows photopic 30 Hz flicker amplitude. Right eyes are shown in dark, left eyes in white. AAV, adeno-associated virus; mpi, month(s) postinjection.
We found that both AAV2/5RK.cpde6β and AAV2/8RK.cpde6β-subretinal delivery led to substantial restoration of rod function in all treated eyes as soon as 1 mpi (Table 1 and Figures 5c,d and 6b,c). In contrast, the rod responses were undetectable in the contralateral untreated eyes (Figures 5c,d and 6b,c) and in untreated control Pde6β−/− eyes (Table 1 and Figures 5b and 6a) at this timepoint.
Although the general trend of rod function recovery was similar in all rAAV2/5- or rAAV2/8-treated eyes, interindividual's variations were observed (Table 1) and might reflect small variations on the extent and/or localization of the vector bleb.
In all rAAV2/5- and rAAV2/8-treated dogs, the amplitude of rod function recovery increased slightly between 1 and 3 mpi and remained stable thereafter, up to 18 mpi, the longest period of observation (Figure 6b,c, left). In all treated dogs, restored rod function exhibited typical a- and b-wave components (Figure 5c,d).
At 18 mpi, the median b-wave amplitude recorded on AAV2/5RK.cpde6β- (47 ± 8 µV, n = 2) and AAV2/8RK.cpde6β- (46 ± 25 µV, n = 2) treated eyes represent 35% of that recorded on nonaffected eyes (134 ± 8 µV, n = 2) (Supplementary Figure S1). At 18 mpi, the maximal b-wave amplitudes were recorded on A2 AAV2/5RK.cpde6β- (53 µV, 38% of the normal rod function) and A6 AAV2/8RK.cpde6β- (63 µV, 45% of the normal rod function) treated retinas (Table 1 and Figure 6b,c, left).
The cone-mediated ERG (30 Hz Flicker) responses in treated and untreated eyes were similar in all AAV2/5RK.cpde6β- and AAV2/8RK.cpde6β-injected dogs over the first 8–12 month period postvector delivery (Figure 6b,c).
Interestingly, ERG max and 30 Hz flicker amplitudes remained stable in all treated eyes over the 12–18 mpi period while a progressive and consistent reduction of these responses was observed in all contralateral untreated eyes during this period (Table 1 and Figures 5c,d, and 6b,c).
At 18 mpi, the median amplitude of the 30 Hz flicker of AAV2/5RK.cpde6β- (33 ± 11 µV, n = 2) and AAV2/8RK.cpde6β- (25 ± 8 µV, n = 2) treated eyes represent 76 and 56% of that recorded on age-matched Pde6β+/− control eyes (43 ± 1µV, n = 2), respectively (Supplementary Figure S1). At 18 mpi, the maximal amplitudes of the 30 Hz flicker were recorded on A2 AAV2/5RK.cpde6β- (40 µV, 93% of the normal cone function) and A6 AAV2/8RK.cpde6β- (30 µV, 70% of the normal cone function) treated retinas (Table 1 and Figure 6b,c, left). They were approximately threefold higher than responses recovered for their contralateral untreated eyes (14 and 11µV).
Interestingly, the level of rod function restoration and cone function preservation obtained following delivery of rAAV2/5 was not significantly different from that achieved with the rAAV2/8 (Supplementary Figure S1).
AAV2/5RK.cpde6β and AAV2/8RK.cpde6β restore vision
Vision tests were performed on A2, A3, A6, and A7 treated Pde6β−/− dogs at 4, 8, 12, and 18 months postvector delivery (Table 2). Dim-light conditions were used to assess transmission of rescued rod activity to higher visual pathways and improvement of visually guided behavior. Bright light conditions were used to assess preservation of day vision in treated rcd1 dogs.
Table 2. Evolution of dim- and bright-light vision in PDE6β–/– treated dogs.
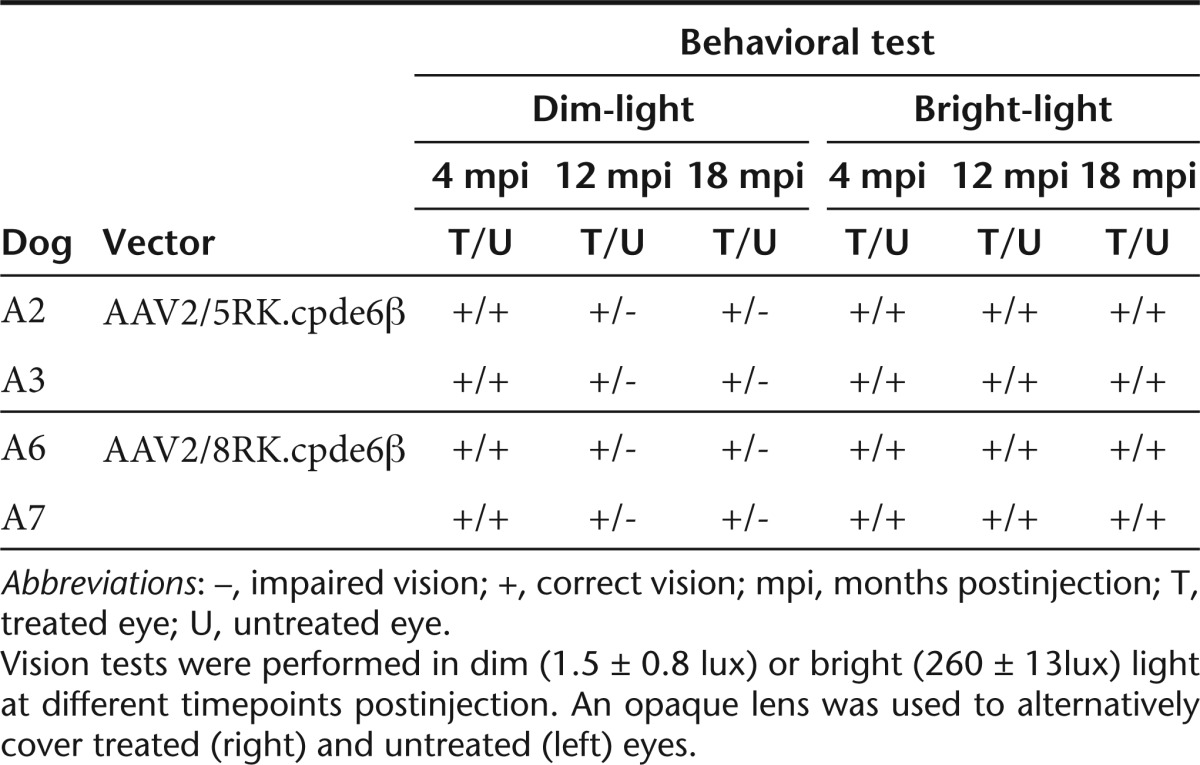
Under dim-light conditions at 4 and 8 mpi, none of the treated dogs showed behavioral signs of blindness in both eyes (Table 2). However, at the latest timepoints (12 and 18 mpi), all treated dogs avoided obstacles when their untreated eye was occluded while they showed difficulty to navigate around the obstacle panels when their treated eye was occluded. They moved very cautiously with their nose to the ground and tried to feel the panels with their forelegs. Several collisions with obstacles were also noted. These results were confirmed by a significant increase in transit time. For example, dog A6 completed the obstacle course in 7 seconds with the untreated eye covered and in 30 seconds with the treated eye covered (Supplementary Video S1, part I). Dog A6 took a comparable time to complete the obstacle course with the untreated eye covered than a control nonaffected Pde6β+/− dog with one eye covered (9 seconds, data not shown).
In bright light conditions, none of the treated dogs showed behavioral signs of blindness from 4 to 18 mpi regardless of which eye was covered (Table 2 and Supplementary Video S1, part II).
Discussion
In this study, a total of eight rcd1 dogs were subretinally injected with rAAV2/5 and rAAV2/8 vectors carrying the cpde6β cDNA under the control of the photoreceptor-specific RK promoter. The results demonstrated that rAAV-mediated cpde6β expression restored rod function and consequently, prevented photoreceptor death and concomitant vision loss in treated dogs for at least 18 mpi (the duration of the study).
Interestingly, rod-mediated ERG responses were undetectable in the rcd1 canine model from the earliest age measured37 (1 month of age, Table 1). At this age, the PDE6β subunit was undetectable by immunoblot and rod PDE6 function was absent, suggesting that the rcd1 dog carries a null mutation in the pde6β gene.30 In this matter, this canine model of PDE6β deficiency is similar to the rd1 murine model in which PDE6 function is also completely lacking,17 but different from the hypomorphic rd10 and Pde6βH620Q murine models in which photoreceptors develop and function almost normally before their degeneration.18,19,20
Here, we show that both AAV2/5RK.cpde6β and AAV2/8RK.cpde6β-gene transfer restored sustained and stable rod-mediated ERG responses in all treated rcd1 eyes as early as 1 month postinjection (Figure 5). From 1 to 18 mpi, median amplitudes of dark-adapted b-waves in rAAV-treated eyes account for 28–35% of those recorded in normal eyes (Supplementary Figure S1), consistent with the estimated area of retina directly exposed to the vectors (~25% of the total retinal surface).
This is the first demonstration that gene therapy can restore a rod function in a null animal model of PDE6β deficiency as all attempts to treat the rd1 mice by gene addition therapy have failed to provide functional rescue.21,26,27 In contrast, in the rd10 mouse, previous studies demonstrated that subretinal injection of AAV2/5smCBA.mpde6β led to the preservation of 37% of rod ERG responses up to P35,23 and that subretinal injection of AAV2/8(Y733F)smCBA.mpde6β led to a preservation of 58% of rod ERG responses for up to 6 months after treatment.24
This finding has obvious clinical significance because among the 21 causative-mutations already identified in patients with PDE6β-recessive RP,4,5,6,7,8,9,10,11,12 8 (38%) are nonsense mutations predicted to lead to a complete loss of PDE6 function.5,6,7,8,10 The most sustained rescue of rod function in null animal models of progressive photoreceptor defects reported to date was obtained in the Aipl1−/− mouse, in which both rod and cone functions are absent at birth. In this model, subretinal injection of AAV2/8RK.haipl1 at P10 led to a 50% restoration of wild-type scotopic ERG responses for at least 4 weeks postinjection.38 When scAAV2/8(Y733F)RK.haipl1 was used,39 this rescued rod function was more sustained persisting until P60 (the duration of the reported study).
In addition to this sustained restoration of rod function in treated rcd1 retinas, long-term preservation of cone-mediated ERG responses were demonstrated up to 18 mpi (Table 1 and Figures 5 and 6). This result strongly suggests that rAAV-mediated gene therapy inhibited or delayed the initiation of secondary cone death in treated rcd1 retinas. A similar beneficial effect of the primary rod treatment on the cone function has been previously obtained in rd10 mice treated with AAV2/8(Y733F)smCBA.mpde6β for at least 6 weeks postinjection24 (the duration of the reported study).
Several mechanisms have been proposed to explain the non-autonomous death of cones in rod-cone dystrophies: the loss of a rod-derived trophic support, the release of a toxic factor by dying rods, an increase in oxidative damage to cones once the rods have died or an imbalance in the metabolism of cones due to changes in retinal architecture.14,15,16 In all these models, primary loss of rods is responsible for secondary cone death, indicating that protection of cones after rAAV-mediated gene transfer might be correlated with a preservation of rod cells in treated rcd1 retinas.
Long-term preservation of central retinal thickness in treated rcd1 eyes was observed by OCT from 4 to 18 mpi (Figure 3). Our histology data at 4 mpi suggested that this reflects the retention of rod photoreceptor cells in treated retinas (Figure 4).
Interestingly, this preservation of rod photoreceptors was not widespread but mainly restricted to nasal vector-exposed areas of treated retinas. Similar results have been recently shown in RPGR canine models of X-linked RP following subretinal injection of AAV2/5IRBP.hrpgr or AAV2/5RK.hrpgr.40 This observation may have two notable clinical implications. First, it demonstrates that degenerating nontransduced photoreceptors have no major negative impact on transduced photoreceptor survival as previously suggested.41 Second, it surprisingly indicates that localized preservation of rod photoreceptor cells in the nasal superior retina (that did not comprise the cone-rich area centralis42) is sufficient to maintain the nearly total cone function and subsequently delay initiation of cone loss. We do not have yet a clear explanation of the apparent complete retention of cone function observed in the treated rcd1 retinas. It might be related to the physic and/or trophic interactions of preserved rods with other retinal cell populations.14,15,16 Further monitoring of the cone function for months or years in treated rcd1 dogs will be essential to confirm its stability overtime and injection of additional dogs will be required to identify the underlying molecular mechanism for this long-term preservation of cone function.
The effect of the rAAV-mediated gene therapy on the vision of treated rcd1 dogs was assessed by behavioral tests under dim (1.5 ± 0.8 lux) and bright (260 ± 13 lux) light conditions. It is important to note that, in these two conditions, it was impossible to discriminate rod- from cone-mediated vision because both types of photoreceptors were stimulated.
In dim light, all treated dogs displayed normal vision behavior using either their untreated and treated eyes up to 8 mpi (Table 2), probably due to the preservation of normal cone function in both eyes (Table 1 and Figures 5 and 6). Over the 8 to 18 mpi period, none of the treated dogs were still able to successfully negotiate the obstacle course using their untreated eye (Supplementary Video S1, part I) consistent with the progressive loss of cone function observed by ERG analysis at these late timepoints (Table 1 and Figures 5 and 6). In contrast, they maintained normal vision-elicited behavior using their treated eyes (Supplementary Video S1, part I), demonstrating that rAAV-mediated restoration of rod function and/or preservation of cone function preserve long-term night vision in treated rcd1 dogs.
In bright light, untreated dogs displayed vision over the 18-month period (Supplementary Video S1, part II) certainly due to the residual cone function in both eyes (Table 1 and Figures 5 and 6). It will be necessary to further monitor the vision of treated rcd1 dogs over several years to determine whether the preservation of cone function observed by ERG analysis will also lead to long-term preservation of day vision over time.
In this study, the efficacy of rAAV2/5 and rAAV2/8 vectors was comparable with both vectors providing similar rod-mediated ERG responses in treated rcd1 eyes (Table 1). This result was surprising as rAAV2/8 (1012 vg/ml) was injected at a tenfold higher titer than rAAV2/5 (1011 vg/ml) vector. One possible explanation might be the broader dispersion of rAAV2/8 in the canine retina after subretinal delivery.43 The higher potential of rAAV2/8 for widespread diffusion across synapses may reduce the global number of rAAV-delivered vector genomes in photoreceptors cells and subsequently the level of pde6β expression. Alternatively or in addition, it is possible that in both rAAV2/5- and rAAV2/8-treated eyes, the saturation threshold of pde6β expression level was reached.
Interestingly, both rAAV2/5 and rAAV2/8 vectors provided long-term therapy in the rcd1 dog. This result contradicts previous reports in mouse models of Pde6β,24 Aipl1,38 and Rpgrip144 deficiencies that have shown a strong functional advantage of rAAV2/8 and/or rAAV2/8(Y733F) vectors compared with the rAAV2/5 vector. This significant discrepancy in long-term rAAV2/5 efficiency between the rcd1 dog and these murine models of retinal dystrophies probably reflects the slower rate of retinal degeneration in the rcd1 dog.33,34 This result should be considered in the context of PDE6β-patients as they typically describe impaired or absent night vision in childhood, followed by a reduction of their visual field from young adulthood, which slowly progresses over decades.4,5,6
In this study, we used rAAV vectors carrying the cpde6β under the control of the human RK promoter which has been demonstrated to drive transgene expression in both rods and cones after subretinal delivery,35,36,40 whereas the β subunit of PDE6 is specifically expressed in rod photoreceptors.13 Recombinant AAV-mediated gene therapy in rcd1 retinas may thus led to undesirable ectopic expression of pde6β in transduced cones. It was unfortunately not possible to assess this pde6β expression in cones due to the limitations of pde6β immunostaining. However, based on the finding that rAAV-treated eyes exhibited no appreciable reduction of cone function over the 18-mpi period (Table 1), it is likely that this potential RK-mediated ectopic expression of pde6β has no toxic effects on the cone function, at least at this stage. These results have high clinical relevance because they suggest the potential safety of the human RK promoter for future treatment of pde6β defects.
In conclusion, this study demonstrated, for the first time, that rAAV-mediated gene transfer efficiently corrects the pde6β defect in a large animal model of pde6β deficiency, leading to (i) robust and stable restoration of rod function and (ii) concomitant long-term preservation of cone function and vision. As mutations in the pde6β gene are one of the leading causes of recessive RP4,5,6,7,8,9,10,11,12 and as retinal degeneration in the rcd1 dog is highly similar to the human disease, this preclinical study represent a major step towards the future development of this therapy in patients with PDE6β-deficiencies. In a larger sense, these PDE6β results offer great promise for the treatment of many other rapid recessive rod-cone dystrophies due to a rod-specific defect.
Materials and Methods
Plasmid construction and production of rAAV vectors. Recombinant AAV2/5.RK.cpde6β and AAV2/8.RK.cpde6β vectors were produced by triple transfection of 293 cells according to previously reported methods,45 using the SSV9RK.cpde6β vector plasmid. This construct carried the cpde6β cDNA (2,681 bp) directly under the control of the short human RK promoter (–112 bo to +87 bp region of the proximal promoter36) and the bovine growth hormone polyadenylation signal (BGHpA), flanked by two AAV2 inverted terminal repeat sequences.
For the SSV9RK.cpde6β construction, full-length cpde6β cDNA (NCBI RefSeq NM_001002934.1) was amplified by PCR from Pde6β+/+ canine retinal cDNA. Two primers designed to cover the entire sequence of the pde6β gene were used for this purpose. The forward primer encoded a HindIII end and the first 15 bp of the cpde6β gene (5′-AATTAAGCTTTAGACAGCCGGACAC-3′). The reverse primer (5′-CTTTATTCATAGTTGAGTTT-3′) encoded a 20 bp sequence of the cpde6β gene located 121 bp downstream of the stop codon. An endogenous EcoRV restriction site was present in the sequence of cpde6β 82 bp downstream of the stop codon. The PCR product (2,728 bp) was purified, digested by HindIII and EcoRV and cloned into a parental plasmid SSV9RK.crpgrip1, between the RK promoter and the bovine growth hormone polyadenylation site, after removal of the eGFP sequence (by HindIII and EcoRV digestion). The identity of the resulting SSV9RK.cpde6β construct was verified by sequencing.
Viral vector titers were determined by dot-blot and by quantitative real-time PCR and expressed as vector genomes (vg) per milliliter (vg/ml). The final vector titers of AAV2/5RK.cpde6β and AAV2/8RK.cpde6β were 1.1011 and 1.1012 vg/ml, respectively.
Animals. A total of nine affected Pde6β−/− and 1 Pde6β+/− control Irish Setter dogs were used in this study (Table 1). The first three Pde6β−/− individuals were kindly provided D.J. Maskell (University of Cambridge, Cambridge, UK). Animals were maintained at the Boisbonne Center (ONIRIS, Nantes-Atlantic College of Veterinary Medicine, Food Science and Engineering, Nantes, France) under a 12/12 hour light/dark cycle. All experiments involving animals were conducted in accordance with the Association for Research in Vision and Ophthalmology statement for the use of animals in ophthalmic and vision research.
Subretinal administration of rAAV vectors. Subretinal injections of AAV2/5RK.cpde6β and AAV2/8RK.cpde6β vectors were performed on 8 affected Pde6β−/− dogs at P20. For all dogs except A9, vector delivery was unilateral, leaving the contralateral eyes without injection as internal controls (Table 1). All subretinal injections were performed under general anesthesia induced by intramuscular injection of a mixture of diazepam (Hoffmann-La Roche, Basel, Switzerland) and ketamine (Rhone Merieux, Lyon, France) and maintained by inhalation of isofluorane gas. Pupils were fully dilated by topical administration of 0.3% atropine (Alcon Cusi SA, Barcelona, Spain), tropicamide (Novartis, Annonay, France) and phenylephrine hypochloride (Novartis).
Surgery was conducted using a transvitreal approach as previously described,46 without vitrectomy. Under microscopic control, 80–120 µl of vector solution were injected into the subretinal space. Immediately after the injection, the localization and the extent of the retinal surface directly exposed to the vector were recorded on schematic fundus drafts as it was not possible to obtain clear fundus photography of the dog retina until 2 months of age. The precise orientation of the vector bleb was further determined by fundus photography at 2 mpi, by the observation of brighter areas that delineate the border of the treated area (data not shown).
Post-surgical care included one topical administration of 0.3% atropine (Alcon Cusi SA) and two topical administrations of Ocryl lotion (Laboratoire TVM, Lempdes, France) and gentamicin dexamethasonum (Virbac France S.A., Carros, France) daily for 10 days post-surgery.
Histology and immunohistochemistry. At 4 mpi, A5 and A9 dogs were euthanized by intravenous injection of pentobarbital sodium (Vétoquinol, Lure, France). Eyes were enucleated and fixed for 2 hours in 4% paraformaldehyde in phosphate-buffered saline (PBS) solution before removal of the anterior chamber and the lens. Eyecups were embedded in optimal cutting temperature compound (OCT Cryomount; Microm Microtech, Francheville, France), and flash-frozen in a dry ice isopentane bath. Ten to fifteen micrometer cryosections were prepared.
For morphological examinations, sections were stained with hematoxylin and eosin before imaging by transmitter light microscopy (Nikon, Champigny sur Marne, France). Photoreceptor survival was assessed by counting the number of photoreceptor nuclei rows in one ONL column. Mean combined outer and inner segment thickness values of vector-exposed and unexposed areas of the retina were determined by averaging ten counts, performed on ten different slices.
For immunohistochemical studies, cryosections were air-dried, washed in PBS solution and incubated in blocking solution [20% normal goat serum (Invitrogen Life Technologies, Saint Aubin, France), 0.05% Triton X-100 in PBS] for 1 hour at room temperature. The bovine rod PDE6 (1:500) (Cytosignal, Irvine, CA) or GNAT1 (1:250) (Santa-Cruz Technology, Heidelberg, Germany) antibodies were incubated overnight at 4 °C in blocking solution. After three washes in 0.05% Triton X-100 in PBS solution, slides were incubated with the secondary antibody Alexa 488 goat anti-rabbit IgG conjugate (Life Technologies, Grand Island, NY) at 1:250 for 2 hours at room temperature. Slides were washed three times with 0.05% Triton X-100 in PBS solution and once with PBS solution before counterstaining the nuclei with DAPI diluted at 1:500. Slides were mounted in Prolong Gold anti-fade reagent (Life Technologies), observed with a fluorescence microscope (Nikon) and images captured with a digital camera (Nikon).
Specificity of the primary bovine rod PDE6 antibody for canine retina was demonstrated in the Pde6β+/+ retina by absence of co-labeling with peanut lectin agglutinin conjugated with fluorescein isothiocyanate (1:250; Vector Laboratories, Burlingame, CA) (data not shown).
RT-PCR
RNA extraction and retro-transcription. Total RNA was isolated from individual flash-frozen retinas or retinal cryosections of Pde6β+/+, Pde6β+/−, or Pde6β−/− dogs using TRIzol Reagent (Invitrogen Life Technologies). Rnase-free Dnase I (Ambion DNA-free kit; Invitrogen Life Technologies) was used according to the manufacturer's instructions to remove contaminating DNA before generation of cDNA by reverse-transcription. Five hundred nanograms of total RNA were reverse-transcribed using oligodT primers and M-MLV reverse transcriptase (Invitrogen Life Technologies) as per the manufacturer's instructions. Control assays without addition of reverse transcriptase were included and the products were used in the subsequent RT-PCR as negative controls.
Primers. Integrity of cDNA was determined by amplification of a 650 bp sequence of the β-actin gene using β-actinF (5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′) and β-actinR (5′-CTAGAAGCATTTGC GGTGGACGATGGAGGG-3′) primers.
For pde6β amplification, 2056F (5′-GAAGATCGTGGATGAGTCTAAGA-3′) and 2561R (5′-CTACTTATCATCAGTCAAGGCCA TC-3′) primers were designed to specifically create a BtsCI restriction site in RT-PCR products arising from wild-type RNA templates. The reverse primer was designed to neighbouring exon sequences to avoid amplification of genomic DNA (Figure 2). The forward 2056F primer was designed in a pde6β-specific sequence to avoid amplification of canine pde6α RNA.
To verify the specificity of the 2056F and 2561R primer set against pde6β, full-length canine pde6α cDNA (NCBI RefSeq NM_001003073.1) was amplified by PCR from Pde6α+/+ canine retinal cDNA. The forward primer encoded a BamHI end and an 18 bp sequence located 72 bp upstream of the start codon of the cpde6α gene (5′-ATTAAGGATCCTGACTC TGTCTTGC-3′). The reverse primer encoded an EcoRV end and a 15 bp sequence located 353 bp downstream of the stop codon of the cpde6α gene (5′- TATTGATATCTGGGTGATGAGGAGG -3′). The PCR product (3,061 bp) was purified, digested by BamHI and EcoRV and cloned into the plasmid SSV9RK.cpde6β previously constructed, between the RK promoter and the bovine growth hormone polyadenylation site after removal of the cpde6β cDNA (by BamHI and EcoRV digestion). The identity of the resulting SSV9RK.cpde6α construct was verified by sequencing.
Amplification and BstCI digestion. The amount of cDNA used in β-actin and pde6β allele-specific PCR was 40 ng. Both PCR were performed using GoTaq DNA polymerase (Promega France, Charbonnicres, France) and a Veriti Applied Biosystems thermocycler.
The β-actin reaction profile was as follows: an initial denaturation step at 95 °C for 5 minutes, followed by 40 cycles at 94 °C for 30 seconds, 55 °C for 30 seconds, 72 °C for 30 seconds and a final incubation step at 72 °C for 7 minutes. For pde6β, conditions of amplification were: an initial denaturation step at 95 °C for 5 minutes, followed by 10 cycles at 94 °C for 30 seconds, 60 °C for 30 seconds, 72 °C for 30 seconds, 30 cycles at 94 °C for 30 seconds, 56 °C for 30 seconds, 72 °C for 30 seconds and a final incubation step at 72 °C for 7 minutes.
Actin PCR products were resolved on a 1.5% agarose gel electrophoresis. Pde6β PCR products were purified by Nucleospin Extract II (Macherey-Nagel, Hoerdt, France) before digestion by BtsCI (New England Biolabs, Ipswich, MA) for 2 hours at 50 °C according to the manufacturer's instructions and analyzed by 3.5% agarose gel electrophoresis.
Fundus photography and OCT. Fundus photography and OCT were performed bilaterally on all treated dogs every 2 months after treatment. Before clinical examinations, the pupils were topically dilated as described above. Dogs were anesthetized by intravenous injection of xylazine (BayerHealth Care, Shawnee Mission, KS) and ketamine (Rhone Merieux).
Fundus photographs were taken with a Canon UVI retinal camera connected to a digital imaging system (Lhedioph Win Software; Lheritier, Saint-Ouen-l'Aumône, France).
Optical coherence tomography was performed by doing a 3-mm horizontal line scan in vector-exposed or vector unexposed areas of the retina (Stratus 3000; Carl Zeiss S.A.S; Le Pecq, France).
Electroretinography. Retinal function of treated Pde6β−/− dogs and age-matched untreated Pde6β−/− and Pde6β+/− controls was evaluated using bilateral full-field flash ERG. Initial ERG measurements were recorded at 4 weeks postinjection and every subsequent 2 months up to 4 (dogs A5 and A9), 9 (dogs A4 and A8), or 18 mpi (dogs A2, A3, A6, and A7), the latest timepoints evaluated in this study. For all dogs, each contralateral eye served as intra-individual noninjected control (except for dog A9 who received injection in both eyes).
Both pupils of animals were topically dilated as described above. Dogs were dark-adapted for 20 minutes and anesthetized following premedication by intravenous injection of thiopental sodium (Specia Laboratories, Paris, France) followed by isofluorane gas inhalation. Hydroxypropulmethylcellulose (Laboratoires Théa, Clermont-Ferrand, France) was applied to each eye to prevent corneal dehydration during recordings.
A computer-based system MonColor (Metrovision, Pérenchies, France) and contact lens electrodes (ERG-jet; Microcomponents SA, Grenchen, Switzerland) were used.
ERGs were recorded in a standardized fashion according to the International Society for Clinical Electrophysiology of Vision (ISCEV) recommendations,47 using a protocol described previously.48
Vision testing. Light and dim-light vision of selected treated Pde6β−/− dogs were evaluated at regular intervals from 7 to 18 mpi by recording ambulation of dogs through an obstacle avoidance course under 260 ± 13 lux and 1,5 ± 0.8 lux ambient light intensities, respectively. Details of the apparatus have been described previously.48 Animals were adapted to each light condition for 1 hour before beginning the test. An opaque lens was used to alternatively cover the treated or the untreated eye. Obstacle panel combinations were randomly determined for each test to prevent the dogs memorizing the positions of the obstacle panels. A Nightshot camera recorder (Sony DCR-PC110E PAL; Sony, New York, NY) was used for filming the dimmest light condition. All dim-light films were cleared before mounting with commercial Final Cut Pro 7 software (Apple, Cork, Ireland) using a standardized protocol.
SUPPLEMENTARY MATERIAL Figure S1. Median ERG responses of rAAV2/5- and rAAV2/8-treated eyes overtime. Video S1. Assessment of dim and bright light vision in dog A6 at 18 mpi.
Acknowledgments
We acknowledge K. Stieger (Justus-Liebig University, Giessen, Germany) for critical reading of this manuscript. We thank D.J. Maskell (University of Cambridge, Cambridge, UK) for the gift of the first rcd1 dogs and S. Khani (State University of New York, Buffalo, NY) for the gift of the human RK promoter. We also thank the Vector Core (www.vectors.univ-nantes.fr) for production of the rAAV vectors, the staff of the Boisbonne Center for animals care and Mireille Ledevin for retinal cryosections. This work was supported by the Agence Nationale pour la Recherche, the Association Française contre les Myopathies, the INSERM, the Fondation pour la Thérapie Génique en Pays de la Loire and the Ministère Français de l'Enseignement Supérieur et de la Recherche.
Supplementary Material
Median ERG responses of rAAV2/5- and rAAV2/8-treated eyes overtime.
Assessment of dim and bright light vision in dog A6 at 18 mpi.
REFERENCES
- Dryja TP. Retinitis pigmentosa and stationary night blindness. The Metabolic & Molecular Bases of Inherited Diseases. 2001. pp. 5903–5933.
- Hartong DT, Berson EL., and, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- RETNET at < https://sph.uth.tmc.edu/Retnet/ >. Last accessed 6 March2012
- McLaughlin ME, Sandberg MA, Berson EL., and, Dryja TP. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet. 1993;4:130–134. doi: 10.1038/ng0693-130. [DOI] [PubMed] [Google Scholar]
- McLaughlin ME, Ehrhart TL, Berson EL., and, Dryja TP. Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci USA. 1995;92:3249–3253. doi: 10.1073/pnas.92.8.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danciger M, Blaney J, Gao YQ, Zhao DY, Heckenlively JR, Jacobson SG.et al. (1995Mutations in the PDE6B gene in autosomal recessive retinitis pigmentosa Genomics 301–7. [DOI] [PubMed] [Google Scholar]
- Bayés M, Giordano M, Balcells S, Grinberg D, Vilageliu L, Martínez I.et al. (1995Homozygous tandem duplication within the gene encoding the beta-subunit of rod phosphodiesterase as a cause for autosomal recessive retinitis pigmentosa Hum Mutat 5228–234. [DOI] [PubMed] [Google Scholar]
- Danciger M, Heilbron V, Gao YQ, Zhao DY, Jacobson SG., and, Farber DB. A homozygous PDE6B mutation in a family with autosomal recessive retinitis pigmentosa. Mol Vis. 1996;2:10. [PubMed] [Google Scholar]
- Saga M, Mashima Y, Akeo K, Kudoh J, Oguchi Y., and, Shimizu N. A novel homozygous Ile535Asn mutation in the rod cGMP phosphodiesterase beta-subunit gene in two brothers of a Japanese family with autosomal recessive retinitis pigmentosa. Curr Eye Res. 1998;17:332–335. doi: 10.1076/ceyr.17.3.332.5214. [DOI] [PubMed] [Google Scholar]
- Hmani-Aifa M, Benzina Z, Zulfiqar F, Dhouib H, Shahzadi A, Ghorbel A.et al. (2009Identification of two new mutations in the GPR98 and the PDE6B genes segregating in a Tunisian family Eur J Hum Genet 17474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark GR, Crowe P, Muszynska D, O'Prey D, O'Neill J, Alexander S.et al. (2010Development of a diagnostic genetic test for simplex and autosomal recessive retinitis pigmentosa Ophthalmology 1172169–77.e3. [DOI] [PubMed] [Google Scholar]
- Ali S, Riazuddin SA, Shahzadi A, Nasir IA, Khan SN, Husnain T.et al. (2011Mutations in the ß-subunit of rod phosphodiesterase identified in consanguineous Pakistani families with autosomal recessive retinitis pigmentosa Mol Vis 171373–1380. [PMC free article] [PubMed] [Google Scholar]
- Ionita MA., and, Pittler SJ. Focus on molecules: rod cGMP phosphodiesterase type 6. Exp Eye Res. 2007;84:1–2. doi: 10.1016/j.exer.2005.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho-Pelluz J, Arango-Gonzalez B, Kustermann S, Romero FJ, van Veen T, Zrenner E.et al. (2008Photoreceptor cell death mechanisms in inherited retinal degeneration Mol Neurobiol 38253–269. [DOI] [PubMed] [Google Scholar]
- Wright AF, Chakarova CF, Abd El-Aziz MM., and, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet. 2010;11:273–284. doi: 10.1038/nrg2717. [DOI] [PubMed] [Google Scholar]
- Punzo C, Xiong W., and, Cepko CL. Loss of daylight vision in retinal degeneration: are oxidative stress and metabolic dysregulation to blame. J Biol Chem. 2012;287:1642–1648. doi: 10.1074/jbc.R111.304428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeler C. Retinal degeneration in the mouse is rodless retina. J Hered. 1966;57:47–50. doi: 10.1093/oxfordjournals.jhered.a107462. [DOI] [PubMed] [Google Scholar]
- Chang B, Hawes NL, Pardue MT, German AM, Hurd RE, Davisson MT.et al. (2007Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene Vision Res 47624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart AW, McKie L, Morgan JE, Gautier P, West K, Jackson IJ.et al. (2005Genotype-phenotype correlation of mouse pde6b mutations Invest Ophthalmol Vis Sci 463443–3450. [DOI] [PubMed] [Google Scholar]
- Davis RJ, Tosi J, Janisch KM, Kasanuki JM, Wang NK, Kong J.et al. (2008Functional rescue of degenerating photoreceptors in mice homozygous for a hypomorphic cGMP phosphodiesterase 6 b allele (Pde6bH620Q) Invest Ophthalmol Vis Sci 495067–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J, Tanabe T, Sun D, Zeng Y, Kjeldbye H, Gouras P.et al. (1996Photoreceptor cell rescue in retinal degeneration (rd) mice by in vivo gene therapy Nat Med 2649–654. [DOI] [PubMed] [Google Scholar]
- Jomary C, Vincent KA, Grist J, Neal MJ., and, Jones SE. Rescue of photoreceptor function by AAV-mediated gene transfer in a mouse model of inherited retinal degeneration. Gene Ther. 1997;4:683–690. doi: 10.1038/sj.gt.3300440. [DOI] [PubMed] [Google Scholar]
- Pang JJ, Boye SL, Kumar A, Dinculescu A, Deng W, Li J.et al. (2008AAV-mediated gene therapy for retinal degeneration in the rd10 mouse containing a recessive PDEbeta mutation Invest Ophthalmol Vis Sci 494278–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang JJ, Dai X, Boye SE, Barone I, Boye SL, Mao S.et al. (2011Long-term retinal function and structure rescue using capsid mutant AAV8 vector in the rd10 mouse, a model of recessive retinitis pigmentosa Mol Ther 19234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allocca M, Manfredi A, Iodice C, Di Vicino U., and, Auricchio A. AAV-mediated gene replacement, either alone or in combination with physical and pharmacological agents, results in partial and transient protection from photoreceptor degeneration associated with betaPDE deficiency. Invest Ophthalmol Vis Sci. 2011;52:5713–5719. doi: 10.1167/iovs.10-6269. [DOI] [PubMed] [Google Scholar]
- Kumar-Singh R., and, Farber DB. Encapsidated adenovirus mini-chromosome-mediated delivery of genes to the retina: application to the rescue of photoreceptor degeneration. Hum Mol Genet. 1998;7:1893–1900. doi: 10.1093/hmg/7.12.1893. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Miyoshi H, Verma IM., and, Gage FH. Rescue from photoreceptor degeneration in the rd mouse by human immunodeficiency virus vector-mediated gene transfer. J Virol. 1999;73:7812–7816. doi: 10.1128/jvi.73.9.7812-7816.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosi J, Sancho-Pelluz J, Davis RJ, Hsu CW, Wolpert KV, Sengillo JD.et al. (2011Lentivirus-mediated expression of cDNA and shRNA slows degeneration in retinitis pigmentosa Exp Biol Med (Maywood) 2361211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieger K, Lhériteau E, Lhéariteau E, Moullier P., and, Rolling F. AAV-mediated gene therapy for retinal disorders in large animal models. ILAR J. 2009;50:206–224. doi: 10.1093/ilar.50.2.206. [DOI] [PubMed] [Google Scholar]
- Suber ML, Pittler SJ, Qin N, Wright GC, Holcombe V, Lee RH.et al. (1993Irish setter dogs affected with rod/cone dysplasia contain a nonsense mutation in the rod cGMP phosphodiesterase beta-subunit gene Proc Natl Acad Sci USA 903968–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquirre G, Farber D, Lolley R, Fletcher RT., and, Chader GJ. Rod-cone dysplasia in Irish setters: a defect in cyclic GMP metabolism in visual cells. Science. 1978;201:1133–1134. doi: 10.1126/science.210508. [DOI] [PubMed] [Google Scholar]
- Liu YP, Krishna G, Aguirre G., and, Chader GJ. Involvement of cyclic GMP phosphodiesterase activator in an hereditary retinal degeneration. Nature. 1979;280:62–64. doi: 10.1038/280062a0. [DOI] [PubMed] [Google Scholar]
- Parry HB. Degenerations of the dog retina. II. Generalized progressive atrophy of hereditary origin. Br J Ophthalmol. 1953;37:487–502. doi: 10.1136/bjo.37.8.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre G, Farber D, Lolley R, O'Brien P, Alligood J, Fletcher RT.et al. (1982Retinal degeneration in the dog. III. Abnormal cyclic nucleotide metabolism in rod-cone dysplasia Exp Eye Res 35625–642. [DOI] [PubMed] [Google Scholar]
- Khani SC, Pawlyk BS, Bulgakov OV, Kasperek E, Young JE, Adamian M.et al. (2007AAV-mediated expression targeting of rod and cone photoreceptors with a human rhodopsin kinase promoter Invest Ophthalmol Vis Sci 483954–3961. [DOI] [PubMed] [Google Scholar]
- Beltran WA, Boye SL, Boye SE, Chiodo VA, Lewin AS, Hauswirth WW.et al. (2010rAAV2/5 gene-targeting to rods:dose-dependent efficiency and complications associated with different promoters Gene Ther 171162–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buyukmihci N, Aguirre G., and, Marshall J. Retinal degenerations in the dog. II. Development of the retina in rod-cone dysplasia. Exp Eye Res. 1980;30:575–591. doi: 10.1016/0014-4835(80)90041-x. [DOI] [PubMed] [Google Scholar]
- Sun X, Pawlyk B, Xu X, Liu X, Bulgakov OV, Adamian M.et al. (2010Gene therapy with a promoter targeting both rods and cones rescues retinal degeneration caused by AIPL1 mutations Gene Ther 17117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku CA, Chiodo VA, Boye SL, Goldberg AF, Li T, Hauswirth WW.et al. (2011Gene therapy using self-complementary Y733F capsid mutant AAV2/8 restores vision in a model of early onset Leber congenital amaurosis Hum Mol Genet 204569–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran WA, Cideciyan AV, Lewin AS, Iwabe S, Khanna H, Sumaroka A.et al. (2012Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa Proc Natl Acad Sci USA 1092132–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarra GM, Stephens C, de Alwis M, Bainbridge JW, Smith AJ, Thrasher AJ.et al. (2001Gene replacement therapy in the retinal degeneration slow (rds) mouse: the effect on retinal degeneration following partial transduction of the retina Hum Mol Genet 102353–2361. [DOI] [PubMed] [Google Scholar]
- Mowat FM, Petersen-Jones SM, Williamson H, Williams DL, Luthert PJ, Ali RR.et al. (2008Topographical characterization of cone photoreceptors and the area centralis of the canine retina Mol Vis 142518–2527. [PMC free article] [PubMed] [Google Scholar]
- Stieger K, Colle MA, Dubreil L, Mendes-Madeira A, Weber M, Le Meur G.et al. (2008Subretinal delivery of recombinant AAV serotype 8 vector in dogs results in gene transfer to neurons in the brain Mol Ther 16916–923. [DOI] [PubMed] [Google Scholar]
- Pawlyk BS, Bulgakov OV, Liu X, Xu X, Adamian M, Sun X.et al. (2010Replacement gene therapy with a human RPGRIP1 sequence slows photoreceptor degeneration in a murine model of Leber congenital amaurosis Hum Gene Ther 21993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JE, Rolling F, Li C, Conrath H, Xiao W, Xiao X.et al. (2002Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity J Virol 76791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Rabinowitz J, Provost N, Conrath H, Folliot S, Briot D.et al. (2003Recombinant adeno-associated virus serotype 4 mediates unique and exclusive long-term transduction of retinal pigmented epithelium in rat, dog, and nonhuman primate after subretinal delivery Mol Ther 7774–781. [DOI] [PubMed] [Google Scholar]
- Narfström K, Ekesten B, Rosolen SG, Spiess BM, Percicot CL., and, Ofri R, Committee for a Harmonized ERG Protocol, European College of Veterinary Ophthalmology Guidelines for clinical electroretinography in the dog. Doc Ophthalmol. 2002;105:83–92. doi: 10.1023/a:1020524305726. [DOI] [PubMed] [Google Scholar]
- Lhériteau E, Libeau L, Mendes-Madeira A, Deschamps JY, Weber M, Le Meur G.et al. (2010Regulation of retinal function but nonrescue of vision in RPE65-deficient dogs treated with doxycycline-regulatable AAV vectors Mol Ther 181085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Median ERG responses of rAAV2/5- and rAAV2/8-treated eyes overtime.
Assessment of dim and bright light vision in dog A6 at 18 mpi.