

Abstract
Toxoplasma gondii(T. gondii) is an apicomplexan parasite that can cause eye disease, brain disease, and death, especially in congenitally infected and immune-compromised people. Novel medicines effective against both active and latent forms of the parasite are greatly needed. The current study focused on the discovery of such medicines by exploring a family of potential inhibitors whose anti-apicomplexan activity has not been previously reported. Initial screening efforts revealed that niclosamide, a drug approved for anthelmintic use, possessed promising activity in vitro against T. gondii. This observation inspired the evaluation of the activity of a series of salicylanilides and derivatives. Several inhibitors with activities in the nanomolar range with no appreciable in vitro toxicity to human cells were identified. An initial structure-activity relationship was explored. Four compounds were selected for evaluation in an in vivo model of infection, and two derivatives with potentially enhanced pharmacological parameters demonstrated the best activity profiles.
Introduction
The apicomplexan family of parasites, which includes members such as Plasmodium, Babesia and Toxoplasma, are protozoa of great medical and economic significance1. T. gondii is one of the most successful parasites on earth, infecting all warm-blooded animals and one-third to one-half of the human population. This parasite can cause disease, toxoplasmosis, with eye and neurological damage, systemic illness, and death. Toxoplasmosis can be especially devastating in those infected congenitally, immune-compromised persons, or those with post-natally acquired infection.
Though the only definitive hosts of this obligate, intracellular parasite are members of the Felidae (cat) family, in humans and other intermediate hosts T. gondii exists in two life stages: the rapidly proliferating tachyzoite form, and the latent, encysted bradyzoite form, which remains in the body for the duration of the lifetime of the host, maintaining the risk of recurrence2. There are currently no effective treatments against the bradyzoite form, and those medicines which target the tachyzoite form (pyrimethamine and sulfadiazine are the most effective) can be associated with toxicity and hypersensitivity3. Novel, nontoxic anti-Toxoplasma agents are greatly needed.
Niclosamide (5-chloro-N-(2-chloro-4-nitrophenyl)-2-hydrobenzamide, 4) is a well-established FDA-approved anthelminthic drug4 whose activity in the tapeworm is thought to involve the uncoupling of oxidative phosphorylation5. It is not toxic at high concentrations when administered orally6. Earlier unpublished studies by our group had shown that niclosamide was a potential inhibitor of T. gondii (MIC50 250–200 nM). Although niclosamide has the disadvantage of low solubility and low bioavailability7,8, its promising activity against T. gondii inspired the preparation and testing of a series of salicylanilides and derivatives in the anticipation of potentially improving potency and physicochemical and pharmacological properties. These were evaluated for activity against T. gondii tachyzoites and for toxicity towards host cells in vitro. Experiments were conducted to determine whether the observed activity was due to static or cidal effects. The most promising inhibitors that emerged from this study were the carbamate derivatives 14a and 14b, which possess an ionizable moiety appended to the salicylanilide core.
As an apicomplexan parasite, T. gondii is often used as a model organism to study other members of this Order, such as Plasmodium, Babesia and Eimeria. Because the inhibitory activity of the subject compounds against other apicomplexans is of great interest, selected compounds were also tested for efficacy against both drug-sensitive and drug-resistant strains of Plasmodium falciparum, the causative agent of the most virulent form of malaria in humans, and were found to be effective as described herein.
Chemistry
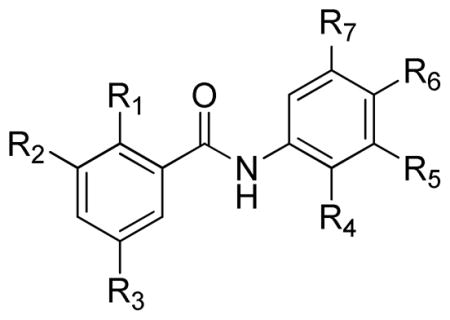
Commercially available salicylic acids 1 were coupled with commercially available anilines 2 in hot xylenes in the presence of PCl3 to furnish salicylanilides 39 (Scheme 1). R1 – R6 are defined in Table 1.
Scheme 1.

Preparation of 3a – 3 ae.
Table 1.
Activity of Salicylanilides against T. gondii
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | R7 | MIC50 | MIC90 |
| 3a | OH | H | Cl | H | CH3 | H | H | >1 μM | >1 μM |
| 3b | OH | H | Cl | H | Br | H | H | 750-500 nM | 1 μM–750 nM |
| 3c | OH | H | Cl | H | CH2CH3 | H | H | 500-250 nM | 750-500 nM |
| 3d | OH | H | Cl | H | C≡CH | H | H | >1 μM | >1 μM |
| 3e | OH | H | Cl | H | CH=CH2 | H | H | 500-250 nM | 1 μM–750 nM |
| 3f | OH | H | Cl | H | CF3 | H | H | 500-250 nM | 500-250 nM |
| 3g | OH | H | Cl | H | CN | H | H | >1 μM | >1 μM |
| 3h | OH | H | Cl | H | F | H | H | 570-500 nM | 1 μM–750 nM |
| 3i | OH | H | Cl | H | C(CH3)3 | H | H | 16-8 nM | 31-16 nM |
| 3j | OH | H | Cl | H | CF3 | H | CF3 | 31-16 nM | 250-125 nM |
| 3k | OH | H | H | Cl | H | NO2 | H | >1 μM | >1 μM |
| 3l | OH | H | Cl | H | CH2Ph | H | H | 1 μM–750 nM | 1 μM–750 nM |
| 3m | OH | H | CH3 | Cl | H | NO2 | H | 500-250 nM | 500-250 nM |
| 3n | OH | H | Cl | H | Cl | H | Cl | 500-250 nM | 500-250 nM |
| 3o | OH | H | Cl | H | F | H | F | >1 μM | >1 μM |
| 3p | OH | H | Cl | F | H | Cl | H | 750-500 nM | 750-500 nM |
| 3q | OH | H | Cl | OCH3 | OCH3 | OCH3 | H | >1 μM | >1 μM |
| 3r | OH | H | Cl | Cl | H | H | CN | 750-500 nM | 1 μM–750 nM |
| 3s | OH | H | Cl | H | CH3 | H | CH3 | >1 μM | >1 μM |
| 3t | OH | H | Cl | H | C≡CH | F | H | >1 μM | >1 μM |
| 3u | OH | H | Cl | H | OCH2CH3 | H | H | >1 μM | >1 μM |
| 3v | OH | H | Cl | H | OCH3 | OCH3 | H | >1 μM | >1 μM |
| 3w | OH | H | Cl | H | OCH(CH3)2 | H | H | >1 μM | >1 μM |
| 3x | OH | H | Cl | H | OCH3 | H | CH3 | >1 μM | >1 μM |
| 3y | OH | H | Cl | H | OPh | H | H | >1 μM | >1 μM |
| 3z | OH | H | Cl | H | OCH(CF3)2 | H | H | >1 μM | >1 μM |
| 3aa | OH | I | I | H | Cl | H | H | >1 μM | >1 μM |
| 3ab | OH | H | Cl | H | OCH2CH2O | H | >1 μM | >1 μM | |
| 3ac | OH | H | F | H | OCH2CH3 | H | H | >1 μM | >1 μM |
| 4 | OH | H | Cl | Cl | H | NO2 | H | 250-200 nM | 250-200 nM |
| 5 | OH | H | Cl | Cl | H | NH2 | H | >1 μM | >1 μM |
| 7a | PhC(=O)O | H | Cl | Cl | H | NO2 | H | 125-61 nM | 250-125 nM |
| 7b |

|
H | Cl | Cl | H | NO2 | H | >1 μM | >1 μM |
| 7c |
|
H | Cl | Cl | H | NO2 | H | >1 μM | >1 μM |
| 7d |

|
H | Cl | Cl | H | NO2 | H | 750-500 nM | 1 μM–750 nM |
| 10a | CH3O | H | F | H | CH3 | H | H | >1 μM | >1 μM |
| 10b | CH3O | H | F | H | OCH3 | H | H | >1 μM | >1 μM |
| 14a |

|
H | Cl | Cl | H | NO2 | H | 31-16 nM | 250-125 nM |
| 14b |

|
H | Cl | H | CF3 | H | CF3 | 250-125 nM | 250-125 nM |
Reduction of niclosamide 4 with Zn dust in methanol and acetic acid followed by salt formation gave amino salicylanilide hydrochloride 5 (Scheme 2).
Scheme 2.

Simple ester or carbamate derivatives of 4 were obtained through treatment of 4 with various carbonyl chlorides 6 to provide acylated derivatives 7 (Scheme 3).
Scheme 3.

The fluorine-containing salicylanilide methyl ethers 10 were synthesized by HATU-mediated condensation of 5-fluoro-2-methoxybenzoic acid 8 with nitroanilines 9 (Scheme 4).
Scheme 4.

Sarcosine tert-butyl ester hydrochloride 11 was transformed into the free base and treated with phosgene in toluene to provide tert-butyl 2-((chlorocarbonyl)(methyl) amino)acetate 12. We found that triphosgene and a solution of phosgene in toluene are essentially equivalent for this transformation. 12 reacted with 4 smoothly in warm pyridine under DMAP catalysis to furnish the expected carbamate ester. Sequential removal of the ester function by treatment with trifluoroacetic acid, condensation of the resulting carboxylic acid 13a with tert-butyl carbazate using EDCI, and treatment of the resulting protected acid hydrazide with HCl in dioxane furnished the sarcosine hydrazide hydrochloride 14a. In a similar fashion, salicylanilide 3j was converted to the corresponding sarcosine hydrazide hydrochloride 14b (Scheme 5).
Scheme 5.

In Vitro Bioassays
Parasite proliferation was monitored using stably transfected type I RH-YFP parasites, which constitutively express Yellow Fluorescent Protein. Proliferation also was tested using a [3H]-Uracil incorporation assay, as Uracil is incorporated into nucleic acids of T. gondii tachyzoites, but not mammalian cells, as they divide. Complimentary challenge assays ensured that the observed fluorescence data was due to parasite inhibition and not to quenched fluorescence. Pyrimethamine and sulfadiazine were used as positive controls, and DMSO at a concentration of 0.1% was used as the negative control. Because T. gondii is an obligate parasite, compounds that are toxic to host cells will appear to inhibit parasite growth. Therefore, all test compounds were simultaneously evaluated for efficacy and toxicity against human cells.
Selected compounds exhibiting the ability to inhibit tachyzoite growth were evaluated to determine whether their activity was due to a static or cidal effect.
Compound activity against P. falciparum, a causative agent of malaria, was assessed using the Malaria SYBR Green I - Based Fluorescence (MSF) Assay. The assay is a microtiter plate drug sensitivity assay that uses the presence of malarial DNA as a measure of parasitic proliferation in the presence of antimalarial drugs or experimental compounds based on modifications of previously described methods by Plouffe et al10 and Johnson et al11. As the intercalation of SYBR Green I dye and its resulting fluorescence is relative to parasite growth, a test compound that inhibits the growth of the parasite will result in a lower fluorescence.
Selected compounds were examined for activity against two strains of P. falciparum: D6 (CDC/Sierra Leone), a drug-sensitive strain readily killed by chloroquine, and TM91-C235, a multi-drug resistant strain resistant to chloroquine.
In Vivo Bioassays
Selected compounds were evaluated for in vivo efficacy against T. gondii and P. falciparum. To assess toxic effects when administered orally, selected compounds were administered orally to mice daily for nine days at a dose of 100mg/kg. At the end of the ten days, the animals were evaluated for toxic effects.
Compounds 14a and 14b were tested for efficacy against T. gondii in the oocyst stage following per oral challenge in mice. Mice were infected by oral gavage with ME49 or TgGoatUS4 oocysts. Mice were treated with either 100mg/kg or 25mg/kg of test substance. Diluent was DMSO (1.0%)/PEG 400 (5.0%)/0.5% CMC (94.0%) where PEG 400 is polyethylene glycol, average MW 400, 0.5% CMC = carboxymethylcellulose 0.5% in water.
Results and Discussion
A limited medicinal chemistry effort was undertaken to probe the effects of the variation of ring substituents and the derivatization of the phenolic oxygen of the core salicylanilide structure. Salicylanilides 3a – 3ae, 4, 5, and derivatives 7a–7d, 10a, 10b, 14 a, and 14b were tested for in vitro efficacy against T. gondii tachyzoites. It was decided that only those compounds possessing MIC50 ≤ 1 μM would be considered active against T. gondii. The efficacy and corresponding cellular toxicity data appear in Table 1. Of the 39 compounds assayed, 16 (41%) had MIC50 ≤ 1 μM, 12 (31%) had MIC50 ≤ 500 nM, 6 (15%) had MIC50 ≤ 250 nM, and 4 (10%) had MIC50 ≤ 125 nM.
Initially the phenolic functionality was maintained, and two alterations to the A (salicyl) ring were made while retaining the 2′-chloro-4′-nitro B (anilide) ring. Replacement of the 5-chloro of 4 with methyl (3m) decreased potency, while replacement with H (3k) eliminated activity altogether. Compounds with a 4-fluoro substituent (3ac, 10a, and 10b) or 3, 5-diiodo substitution (3aa) demonstrated no activity. The decision was made to proceed with the study of 4-chloro Aring analogs with a variety of B ring substituents.
Altering the electron-withdrawing character of the B ring substituents of 4 had a profound effect on activity. When a nitro group was replaced with an amino group at position 4′, (5) all activity was lost. Likewise, compounds possessing O-alkyl electron-donating substituents at 2′ or 4′ (3q, 3v, 3ab) were devoid of activity. It was surprising to note that replacement of the 2′ chloro with the more electronegative fluoro substituent, while simultaneously replacing the 4′ nitro with the less powerful electron withdrawing chloro group (3p), removed all activity.
A series of 3′ monosubstituted compounds were examined. Various activities were observed, and it is clear that the nature of the substituent at this position has a profound effect. In this series, the activity range shows tBu (3i) ≫ Et (3c) ≈ CH2CH2 (3e) ≈ CF3 (3f) ≈ F (3h)> Br (3b) ≈ CH2Ph (3l). All other 3′ substituents resulted in compounds with no activity. Clearly the introduction of 3′-alkoxy or aryloxy substitution resulted in no increase in activity and actually may even be detrimental. When compared to 3i, the best in the 3′-monosubstituted series, both electronegative (halo, CF3) and modestly electron-donating (alkyl) substitutions provided moderate activity.
The activity of a few of the 3′-monosubstituted salicylanilides prompted the evaluation of four compounds with substituents at both the 3′ and 5′ positions. Compounds 3o (3′,5′-difluoro) and 3s (3′,5′-dimethyl) were inactive, and 3n (3′,5′-dichloro) showed slight activity. Interestingly, the 3′, 5′-bis(trifluoromethyl) derivative 3j displayed promising activity.
A cursory study of the effect of capping the phenol of 4 via acylation was undertaken. The acylated derivatives 7a–d were prepared and tested. The carbamates 7b, 7c, and 7d, impart altered polarity and hydrogen bonding capabilities compared to 4. 7d showed modest activity, but 7c and 7d were devoid of activity. We were surprised to learn that benzoate ester 7a showed an apparent increase in potency over 4. In other studies, we found that 7a and other carboxylic esters of 4 were hydrolytically labile when incubated in a mixture of THF and buffer (pH 7.4 or pH 8.5), returning 4 at rates dependent on the nature of the ester moiety (data not shown). Any differential activity of 7a over 4 therefore may be due to altered solubility and permeability parameters which 7a may possess, and that eventual liberation of active 4 may be responsible for enhancing the observed activity. The intrinsic activity of intact 7a cannot yet be ruled out, and this interesting phenomenon is currently under study. The carbamates 7b, 7c, and 7d are much more stable against hydrolysis when incubated in a mixture of THF and buffer (pH 7.4 or pH 8.5) (data not shown), and are not expected to yield free 4 during bioassay. The fact that these derivatives have no activity suggests that either the increased steric demand of the carbamate groups, or the capping of the phenolic oxygen, renders these compounds inactive.
Two 5-fluorosalicylanilide methyl ethers (10a and 10b) were synthesized, tested, and proved to be inactive. This series of salicylanilide ethers was not further pursued.
In an attempt to improve the activity of compound 4 and one of the most promising salicylanilide analogs 3j, we prepared ionizable derivatives of each compound. The acid hydrazide salts 14a and 14b were designed to possess enhanced solubility and bioavailability relative to the parent structures. We were delighted to learn that 14a and 14b possess compelling in vitro and at least minimal in vivo activity against a highly virulent challenge. It is not yet clear whether the ionized derivatives have innate activity, or whether these compounds provide, by virtue of increased solubility and permeability, a more effective delivery of the parent salicylanilides. It is possible that the sarcosine hydrazide moiety of these derivatives provides an initial enhancement of concentration by virtue of increased solubility, and ultimately cleaves to liberate the active salicylanilide. Research is currently underway to elucidate this mode of enhancement.
The limited number of compounds studied allows a number of observations that suggest a primitive structure activity relationship. It is appreciated that salicylanilides have been studied for decades, and that it is known that the electronic environment of one aromatic ring strongly influences that of the other. The evaluation of effects on activity of rationally modifying a given substituent while holding the others constant was limited by the scope of the research project and the relative availability of starting materials. The main observation that emerged is that there is a strong interplay between all of the substituents, and that a complex relationship of interaction across the amide bond exists. For example, whereas the activity of 4 (MIC50 = 250–200 nM, R6 = nitro) is destroyed in 5 (R6 = amino), 3i (MIC50 = 16-8 nM, R6 = H, R5 = t-butyl), which possesses neither the strong electron-withdrawing group of 4 or the strong electron-donating group of 5. Further research to elaborate on the observations and trends presented below is planned. The compounds studied are represented by generic structure 15.

Observations of effects of substituents on activity and possible trends in Table 1 are summarized in BOX 1:
BOX 1.
| Observed Substituent Effects | Trends | |
|---|---|---|
| R1 |
|
Appears that R1-tolerated substituents may be involved in intra- or intermolecular H-bond donors or acceptors. They may also impart electronic effects. The contributions of R1 may be muted by effects of other substituents. |
| R3 |
|
Appears that R3 substituents play a largely electronic role, but that the contributions of R3 to activity may be muted by effects of other substituents. |
| R4 |
|
Appears that R4 substituents play a largely electronic role, but that the contributions of R4 to activity may be muted by effects of other substituents. |
| R5 | When R1 = OH and R2 = R3 = R4 = H, the following observations are made:
|
Appears that a combination of steric and electronic effects of R5 substituents contribute to activity. |
| R6 |
|
Appears that the electron acceptor effects of R6, the electron donor effects of R1, and the nature of R3 contribute to the activity when all three substituents are present. Also appears that the effects of R6 are not necessary for activity when the appropriate R5 substituent is present. |
| R7 | When R1 = OH and R2 = R3 = R4 = H, the following observations are made:
|
Appears that combined electronic effects of R5 and R7 substituents contribute to activity. |
These screening efforts revealed that six of the compounds were the most effective inhibitors. Of these, 3i, 3j, 7a, 14a, and 14b were selected for further in vitro evaluation. Serial dilutions of these compounds to give additional test concentrations were made and studied to identify inhibitory IC50 and IC90 values. Graphical presentation of parasite inhibition is in Figure 1, while graphical display of toxicity to HFF cells is in Figure 2. The measured IC50 and IC90 ranges and the corresponding toxicity data are compared in Table 2.
Figure 1.

Inhibition of T. gondii RH-YFP fluorescence in the presence of Compounds 3i, 3j, 7a, 14a, 14b. FIBS, host fibroblasts alone, not infected; P/S, infected control treated with pyrimethamine and sulfadiazine in combination; RH-YFP, untreated infected fibroblast control; 0.1% DMSO (vehicle) infected fibroblast control; [nM], concentration of inhibitor dissolved in 0.1% DMSO. Ordinate axis: Relative Fluorescence Units.
Figure 2.

Effect of Selected Compounds on Survival of HFF Cells. Ordinate: Optical Density.
Table 2.
Comparison of IC50 and IC90 Values of Lead Compounds Characterized Further In vitro
| Compound | IC50, nM | IC90, nM | Toxicity, nMa |
|---|---|---|---|
| 3i | 16-8 | 31-16 | >1000 |
| 3j | 31-16 | 250-125 | >1000 |
| 7a | 125-61 | 250-125 | >1000 |
| 14a | 31-16 | 250-125 | >1000 |
| 14b | 250-125 | 250-125 | >1000 |
The HFF used as the host cells in the parasite assay also were used for the toxicity assay
Since the ideal antiparasitic agent would have cidal activity, it is of interest whether potential antiparasitic drugs exhibit a static (inhibition of growth and/or replication) or cidal (lethal) effect. In order to determine whether leading compounds in this study inhibited parasite proliferation by either a cidal or static mechanism, four were selected (3i, 3j, 7a, and 14a) and applied at four to eight times MIC50 to parasites. In this assay, RH-YFP tachyzoites were treated with each compound at 1 μM under various dosing conditions:
Condition A: Parasites were treated for four days, then compound was removed
Condition B: Parasites were treated for ten days, then compound was removed
Condition C: Compound was refreshed at four days then removed at ten days
Condition D: Compound was maintained for the duration of the experiment
The four and ten day time points were taken to reveal the impact of extended exposure of the parasites to the test substance. Compounds were refreshed at four days to examine whether compound degradation could contribute to an observed static effect. Parasite growth was assessed at days 11, 17, and 25. The growth data, as a function of RH-YFP fluoresence, is expressed in Figure 3.
Figure 3.


Effect of various dosing conditions on prolonged survival of RH-YFP tachyzoites, measured by inhibition of RH-YFP fluorescence. Abscissa: 4 days, Condition A; 10 days, renewed at 4, Condition B; 10 days, Condition C; All time, Condition D. Ordinate: Relative Flourescence Units.
Treatment with 3i under Condition A reveals that the parasite burden is roughly equivalent to untreated controls at day 11. At day 17, Condition C dosing of 3i also shows renewed growth. Application of 3i under Condition D demonstrates inhibition. These data suggest that 3i is parasitostatic with 4 and 10 days of exposure to the compounds in vitro. In contrast, compounds 3j and 7a inhibited growth under all Conditions employed in this experiment. No parasite growth observed after the removal of these compounds, even at day 25, suggesting that their activity is parasitocidal. Compounds 3j and 7a demonstrated a cidal effect after four days of treatment, while 14a demonstrated a cidal effect after ten days of treatment, comparable to treatment with the combination of pyrimethamine and sulfadiazine.
The effect of selected compounds on other apicomplexan parasites was also explored. Compounds 3i, 3j, 7a, and 14a were examined for activity against two strains of P. falciparum, a causative agent of malaria. One of these strains, D6 (CDC/Sierra Leone), is drug-sensitive and readily killed by chloroquine, while the second strain, TM91-C235, is multi-drug resistant and shows resistance to chloroquine. The activity of these compounds was assessed using the Malaria SYBR Green I - Based Fluorescence (MSF) Assay. This microtiter plate drug sensitivity assay uses the presence of malarial DNA as a measure of parasitic proliferation. As shown in Table 3, all compounds demonstrated activity against both P. falciparum strains, with 7a the most effective (D6 chloroquine sensitive IC50 = 770 nM) and TM91-C235 chloroquine resistant IC50 = 690 nM). Compounds 7a, 3j, and 14a were equally effective against the chloroquine-sensitive D6 and the multi-drug resistant Thai strain, TM91-C235, while compound 3i had a two-fold higher IC50 against TM91-C235 (D6 IC50 = 3100 nM and TM91-C235 IC50 = >6500 nM). The lack of cross-resistance in compounds 7a, 3j, and 14a is an encouraging finding for a novel scaffold and a valuable lead quality compound attribute given the rapid development of drug resistance against many antimalarials in the field. This initial finding is the basis for future research directed to the development of agents effective against P. falciparum.
Table 3.
Inhibition of P. falciparum D6 and C235 by selected compounds.
| Compound | D6 IC50 (nM) | D6 R2 | C235 IC50 (nM) | C235 R2 |
|---|---|---|---|---|
| Chloroquine | 7 nM | - | 89 nM | - |
| 3i | 3100 nM | 0.93 | >6000 nM | 0.67 |
| 3j | 1400 nM | 0.96 | 1300 nM | 0.97 |
| 7a | 770 nM | 0.97 | 690 nM | 0.97 |
| 14a | 2700 nM | 0.95 | 2700 nM | 0.87 |
The in vitro data for T. gondii prompted the selection of 3i, 3j, 7a, 14a, and 14b for evaluation in mouse models of T. gondii infection. Initial difficulties were encountered with the formulation and preliminary safety studies of 3i, 3j, and 7a, presumably due to limited aqueous solubility. Compound 3i, although slightly more active as a static agent in vitro, was not further derivatized herein because it was not cidal and because of its poorer solubility. It was anticipated that 14a and 14b, by virtue of their polar, ionizable appended functionality, may possess improved physicochemical profiles. Consequently, 14a and 14b were chosen for evaluation in a mouse model of T. gondii oocyst infection.
To explore whether 14a or 14b exerted toxic effects when dosed orally, each compound was administered by gavage to mice daily for nine days at a dose of 100mg/kg. At the beginning, throughout and at the end of the ten days, all mice were alive and appeared healthy using well-being criteria including sleek fur, normal activity pattern and movement, suggesting that neither compound had any observable toxic effect upon oral administration at the dosage studied. Thus, compounds 14a and 14b were tested for efficacy in a mouse model of T. gondii oocyst infection. The oocyst form of the parasite is excreted by cats and is often the form by which people and other animals become infected. This oocyst infection in mice is highly virulent and often fatal making even limited survival following such a virulent challenge of importance. Mice were infected by oral gavage with ME49 or TgGoatUS4 oocysts. Mice were treated with either a high dose (100mg/kg) or low dose (25mg/kg) of 14a or 14b 1mL suspension via oral gavage, or were not treated. All uninfected mice dosed with compound alone remained asymptomatic, whereas all mice inoculated orally with oocysts of either strain died of acute toxoplasmosis 8-9 days post infection, and tachyzoites were found in smears of their mesenteric lymph nodes. Treatment with 14a and 14b increased survival by 1 day (Table 4, Figure 4).
Table 4.
Efficacy of compounds 14a and 14b in B7 mice infected with T. gondii Me-49 or Tg-Goat-US4 oocysts.
| Compound | Dose | Challenge | # Mice | Day of Death |
|---|---|---|---|---|
| 14a | 100 mg/kg | None | 5 | None |
| 14a | 100 mg/kg | Me-49 | 5 | 8,9,9,9,9 |
| 14a | 25 mg/kg | None | 5 | None |
| 14a | 25 mg/kg | Me-49 | 5 | 8,9,9,9,9 |
| None | None | Me-49 | 5 | 8,8,8,8,8 |
| 14b | 100 mg/kg | None | 5 | None |
| 14b | 100 mg/kg | TgGoatUS4 | 5 | 8,9,9,9,9 |
| 14b | 25 mg/kg | None | 5 | None |
| 14b | 25 mg/kg | TgGoatUS4 | 5 | 8,9,9,9,9 |
| None | None | TgGoatUS4 | 5 | 8,8,8,8,8 |
Figure 4.

Effect of Compound 14a (left panel) on survival in a mouse model following challenge with T. gondii oocysts of the Me-49 strain. On day 8, p<0.014. N= 5 per group. Doses 25 and 100 mg/kg (dose 25/100) are shown with a single line as both have the same results. Effect of Compound 14b (right panel) on survival in a mouse model following challenge with T. gondii oocysts of the TgGoatUS4 strain. On day 8, p<0.014. N=5 per group. Doses 25 and 100 mg/kg (dose 25/100) are shown with a single line as both have the same results.
Kaplan Meier curves in Figure 4 permit direct comparison of the detailed data set in Table 4.
This is a highly virulent oocyst challenge lethal to all mouse strains tested so far. Although the protection is of modest biological significance adding only a day of increased survival, it is protection against an otherwise lethal infection and was consistent across doses and strains of parasites and replicate experiments. These dosages of compounds also were not harmful.
In an attempt to discover the molecular target of selected compounds, insertional mutagenesis experiments were performed. The goal of this study was the identification of one or more genes which, when disrupted, confer resistance to the parasite, thus potentially identifying the gene product which, upon interaction with the active compound, inhibits the growth of the parasite. THdhxgTRP tachyzoites were successfully transfected with pLK47 vector plasmid to create parasites with random gene mutations. No parasite growth was observed after prolonged incubation in the presence of 3i, 3j, 7a or 14b (Data not shown). The value of this approach to elucidate molecular targets or target pathways of T. gondii inhibitors was recently demonstrated. In an unrelated study conducted in our laboratory, this methodology has successfully identified the T. gondii trafficking pathway inhibited by a series of N-benzoyl-2-hydroxybenzamides12. This insertional mutagenesis approach was used successfully in parallel studies (reference 12) which served as controls. One interpretation of the data reported herein is that the molecular target of the active inhibitors of the study may be essential. Specifically, the fact that it was not possible to identify the molecular target with an insertional mutagenesis strategy which has been successful in other work with other targets, indicates that it is likely that when this target is mutated it is lethal for the parasites (i.e., that the target is essential). The cidal activity of the lead compounds also indicates this is the case and that it is not possible to identify the target by prolonged culture of the parasite in the presence of the concentration of the inhibitor we utilized. Future approaches to target and interacting molecule identification might also include prolonged growth at subinhibitory concentrations to try to identify a resistant mutant followed by sequencing of that mutant. Expression profiling by determining transcriptome of treated parasites early after their treatment to identify the signature of pathways perturbed by the compounds using The Connectivity map might also be useful. Also, proteomics with a similar approach, and trying pull down experiments with candidate proteins from T.gondii that are homologues of known targets of niclosamide, determining compound effects on these targets, and whether overexpression of the targets could rescue inhibition of parasite growth are other approaches to target identification which could be used in the future.
As T. gondii and P. falciparum are both members of the same phylum Apicomplexa, it is gratifying that the activity profile of the inhibitors is similar for both parasites. It will be of interest in the future to characterize utility of these compounds as agents to treat toxoplasmosis and malaria, and perhaps diseases caused by other apicomplexan parasites as well. When molecular targets is/are identified in the future it will be of interest to determine whether selectivity or broader utility for multiple apicomplexan parasites with similar targets will be the strengths of this new family of inhibitors. Discoveries of safe and highly cidal leads as potent as the compounds we have created for diseases such as toxoplasmosis and malaria that are worldwide scourges and harm many children are rare.
Conclusions
Herein, the inhibitory properties of the anti-helminthic drug niclosamide inspired the synthesis and evaluation of a series of salicylanilides. The initial in vitro screen yielded five promising agents, compounds 3i, 3j, 7a, 14a, and 14b, which were active at low nanomolar concentrations and were not toxic to human host cells. Compound 3i had a static effect on parasite proliferation, which resumed after drug pressure was removed, while the activity of compounds 3j, 7a, and 14a was cidal. Compounds 14a and 14b compounds were effective at prolonging very slightly survival of mice infected with T. gondii oocysts, and showed no signs of toxicity. Diluent alone had no effect. It will next be important to explore the activity of these compounds against the latent, encysted bradyzoite life stage.
3i, 3j, 7a, 14a were examined for activity against two drug-resistant strains of P. falciparum, the causative agent of the most virulent form of malaria in humans. All compounds demonstrated activity against P. falciparum, with 7a as the most effective.
The next steps in the exploration of this series will include detailed investigation of structure-activity relationships, optimizing potency and pharmacodynamic properties, and the identification of the molecular target of this family of compounds. The failure to create drug-resistant insertional mutants in this study suggests the possibility that the target of the compounds studied may be essential. This is a promising characteristic for novel medicines, given the risk of emergence of drug-resistant strains. Identification of the molecular target of these compounds would enhance the optimization of activity through structural modification.
Experimental Section
Chemistry
Synthesis of Potential Inhibitors
Unless otherwise stated, all solvents and reagents were used as received from vendors. 1H NMR spectra were measured at either 400 MHz (Varian) or 500 MHz (Varian Inova AS500) in DMSO-d6 or CDCl3. HPLC-MS analyses were carried out with a Shimadzu LCMS 2020 using a Phenomenex CHO-8463 C18 column (50 × 3.0 mm) with a gradient of 10% acetonitrile: 90% water (0.1% formic acid) to 100% acetonitrile (0.1% formic acid) over five minutes. Retention times (TR) are reported in minutes (min). Mass spectra (ESI) are reported in positive (m/z+) and/or negative (m/z−) mode. The calculated exact mass is denoted as EM. Unless otherwise stated, all compounds were obtained at ≥ 95% purity (HPLC-MS). Niclosamide (Compound 4) was purchased from Sigma-Aldrich (St. Louis, MO). Reactants and reagents were used as received from the vendor except as noted.
General Method of Salicylanilide Synthesis9
A suspension of the salicylic acid derivative and aniline in xylenes (0.1 to 0.5 M) was warmed to reflux, and then a solution of phosphorous trichloride in CH2Cl2 or xylenes was introduced dropwise. When the reaction was complete, as determined by TLC or HPLC-MS, the reaction mixture was rapidly transferred while hot by pipette, cannulation, or decanting to a beaker and allowed to cool under rapid stirring. This action removed tarry residue which may accumulate on the reaction vessel walls during the reaction. Typically the product crystallized from the reaction solvent as it cooled, or was induced to crystallize upon the slow addition of hexanes when the temperature of the reaction solvent reached 75 to 80° C.
N-(3-methylphenyl)-5-chloro-2-hydroxybenzamide (3a)
A boiling solution of 5-chlorosalicylic acid (0.72 g, 4.17 mmol) in xylenes (10 mL) was treated dropwise with a 2.0 M solution of PCl3 in CH2Cl2 (0.83 mL, 1.67 mmol). After 2 h, the reaction solution was transferred via pipette to a beaker and was allowed to cool to room temperature under rapid stirring. The product separated as off-white crystals. The crude product was recrystallized from EtOAc. 1H NMR (400 MHz, DMSO-d6) δ 2.291 (s, 3H), 6.942 (d, J = 7.2 Hz, 1H), 6.988 (d, J = 8.8Hz, 1H), 7.227 (M, 1H), 7.425–7.508 (M, 3H), 7.943 (d, J = 2.24 Hz, 1H), 10.322 (s, 1H). HPLC TR 2.70 min; m/z+ 261.95 [M+H] +; m/z− 259.85, [M-H]−, (EM 261.06).
N-(3-bromophenyl)-5-chloro-2-hydroxybenzamide (3b)
Using the method described for compound 3a, 5-chlorosalicylic acid (0.57 g, 3.30 mmol) reacted with 3-bromoaniline (0.36 mL, 3.30 mmol) and 2M PCl3 in CH2Cl2 (0.66 mL, 1.32 mmol) in xylenes (8 mL). The crude product was recrystallized from EtOAc/hexanes. 1H NMR (500 MHz, DMSO-d6) δ 7.024 (m, 2H), 7.354 (m, 3H), 7.474(m, 1H), 7.660 (m, 1H), 7.895 (m, 1H), 8.056 (s, 1H), 10.482 (s, 1H). HPLC TR 2.84 min; m/z+ 327.85 [M+H]+; m/z− 325.75 [M-H]−, (EM 324.95).
N-(3-ethylphenyl)-5-chloro-2-hydroxybenzamide (3c)
Using the method described for compound 3a, 5-chlorosalicylic acid (0.63 g, 3.65 mmol) reacted with 3-ethylaniline (0.45 mL, 3.65 mmol) and 2M PCl3 in CH2Cl2 (0.73 mL, 1.45 mmol) in xylenes (9 mL). The crude product was recrystallized from EtOAc/hexanes. 1H NMR (400 MHz, DMSO-d6) δ 1.198 (t, J = 7.6 Hz, 3H), 2.618 (q, J = 7.6Hz, 2H), 7.012 (m, 2H) 7.254 (m, 1H), 7.457 (m, 3H), 7.983 (m, 1H), 10.357 (s, 1H). HPLC TR 2.86 min; m/z+ 275.95 [M+H] +; m/z− 273.80 [M-H]−; (EM 275.07).
N-(3-ethynylphenyl)-5-chloro-2-hydroxybenzamide (3d)
Using the method described for compound 3a, 5-chlorosalicylic acid (0.75 g, 4.35 mmol) reacted with 3-ethynyllaniline (0.45 mL, 4.35 mmol) and 2M PCl3 in CH2Cl2 (0.87 mL, 1.74 mmol) in xylenes (19 mL). The crude product was recrystallized from 2-methyl-1-propanol. 1H NMR (400MHz, DMSO-d6) δ 4.191 (s, 1H), 6.997 (d, J = 8.8Hz, 1H), 7.231 (d, J = 7.6Hz, 1H), 7.366 (dd, J = 8.0, 8.0 Hz, 1H), 7.442 (dd, J = 8.8, 2.8 Hz, 1H), 7.692 (m, 1H), 7.889 (m, 2H), 10.423 (s, 1H). HPLC TR 2.67 min; m/z+ 271.90 [M+H]+; m/z− 269.80 [M-H]−; (EM 271.04).
N-(3-vinylphenyl)-5-chloro-2-hydroxybenzamide (3e)
Using the method described for compound 3a, 5-chlorosalicylic acid (0.56 g, 3.24 mmol) reacted with 3-vinyllaniline (0.37 mL, 3.24 mmol) and 2M PCl3 in CH2Cl2 (0.65 mL, 1.30 mmol) in xylenes (10 mL). The crude product was recrystallized from EtOH. 1H NMR (400 MHz, DMSO-d6) δ 5.305 (dd, J = 0.8, 11.6Hz, 1H), 5.825 (dd J = 0.8, 17.6Hz, 1H), 6.750 (dd, J = 11.6, 17.6Hz, 1H) 7.024 (d, J = 8.8Hz, 1H), 7.272 (d, J=7.6Hz, 1H), 7.360 (dd, J = 7.6, 8.0 Hz, 1H), 7.478 (dd, J = 2.8, 8.8Hz, 1H), 7.618 (d, J = 8.0 Hz, 1H), 7.804 (s, 1H), 7.972 (d, J = 2.8, 1H), 10.416 (s, 1H). HPLC TR 2.77 min; m/z+ 273.90 [M+H]+; m/z− 271.80 [M-H]−; (EM 273.06).
N-(3-(triflouromethyl)phenyl)-5-chloro-2-hydroxybenzamide (3f)
Using the method described for compound 3a, 5-chlorosalicylic acid (2.19 g, 12.69 mmol) reacted with 3-trifluoromethylaniline (1.58 mL, 12.69 mmol) and 2M PCl3 in CH2Cl2 (2.54 mL, 5.08 mmol) in xylenes (32 mL). The crude product was recrystallized from EtOH. 1H NMR (500 MHz, DMSO-d6) δ 7.035 (d, J=9.0 Hz, 1H), 7.488 (m, 2H), 7.619 (dd, J=8.0, 8.0Hz, 1H), 7.934 (m, 2H), 8.209 (s, 1H), 10.624 (s, 1H). HPLC TR 2.843 min; m/z+ 315.90 [M+H]+; m/z− 628.80 [2M-H]−, 314.80 [M-H]−, (EM 315.03).
N-(3-cyanophenyl)-5-chloro-2-hydroxybenzamide (3g)
Using the method described for compound 3a, 5-chlorosalicylic acid (2.06 g, 11.94 mmol) reacted with 3-amino benzonitrile (1.41 g, 11.94 mmol) and 2M PCl3 in CH2Cl2 (2.39 mL, 4.78 mmol) in xylenes (30 mL). The crude product was recrystallized from EtOH. 1H NMR (500 MHz, DMSO-d6) δ 7.012 (dd, J = 1.4, 8.5Hz, 1H), 7.459 (dd, J = 1.8, 8.5Hz, 1H), 7.580 (m, 2H), 7.856 (m, 1H), 7.916 (m, 1H), 8.182 (s, 1H), 10.588 (s, 1H). HPLC TR 2.479 min.; m/z+ = 313.95 [M+CH3CN+H]+, 272.90 [M+H]+; m/z− = 542.95 [2M-H]−, 270.85[M-H]−, (EM 272.04).
N-(3-fluorophenyl)-5-chloro-2-hydroxybenzamide (3h)
Using the method described for compound 3a, 5-chlorosalicylic acid (2.42 g, 14.02 mmol) reacted with 3-amino benzonitrile (1.35 mL, 14.02 mmol) and 2M PCl3 in CH2Cl2 (2.80 mL, 5.61 mmol) in xylenes (30 mL). The crude product was recrystallized from 2-methyl-1-propanol. 1H NMR (500 MHz, DMSO-d6) δ 6.995 (m, 2H), 7.433 (m, 3H), 7.706 (m, 1H), 7.897 (d, J = 2.8 Hz, 1H), 10.519 (s, 1H), 11.641 (s, 1H). HPLC TR 2.639 min; m/z+ 265.90 [M+H]+; m/z− 263.85 (EM 265.03).
N-(3-tert-butylphenyl)-5-chloro-2-hydroxybenzamide (3i)
Using the method described for compound 3a, 5-chlorosalicylic acid (2.04 g, 11.82 mmol) reacted with 3-tert-butylaniline (1.76 g, 11.82 mmol) and 2M PCl3 in CH2Cl2 (2.336 mL, 4.73 mmol) in xylenes (30 mL). At completion of reaction, the hot xylenes solvent was decanted, cooled to room temperature, and then diluted with hexanes (30 mL). This was stored at 4°C for 30 hours during which time an off-white crystalline solid separated. The product was recrystallized from EtOAc/hexanes to give a mixture of the title compound (89.9% and an unidentified impurity (10.1%). 1H NMR of the major component (400 MHz, DMSO-d6) δ 1.280 (S, 9H), 7.172 (dq, J=8.0, 0.8Hz, 1H), 7.283 (dd, J=8.0, 0.8Hz, 1H), 7.455 (dd, J=8.8, 2.6 Hz−, 1H), 7.560 (dd, J=8.0, 0.8Hz, 1H), 7.679 (M, 1H), 7.982 (d, J=2.6Hz, 1H), 10.345 (S, 1H), 11.903 (S, 1H). HPLC TR 3.095 min; m/z+ 303.95 [M+H]+; m/z− = 301.85[M-H]−; (EM 303.10).
N-(3,5-bis(triflouromethyl)phenyl-5-chloro-2-hydroxybenzamide (3j)
Using the method described for compound 3a, 5-chlorosalicylic acid (0.94g, 5.48 mmol) reacted with 3,5-bis(trifluoromethyl)aniline (0.85 mL, 5.48 mmol) and 2M PCl3 in CH2Cl2 (1.10 mL, 2.19 mmol) in xylenes (15 mL). At completion of reaction, the hot xylenes solvent was decanted, cooled to room temperature, and then diluted with hexanes (50 mL). This was stirred at room temperature for 14 hours, during which time pure product separated as white crystals. 1H NMR (400 MHz, DMSO-d6) δ 7.048 (d, J=8.7Hz, 1H), 7.493 (dd, J=9.0, 2.7Hz, 1H), 7.845 (M, 2H), 8.449 (S, 2H), 10.851 (S, 1H), 11.427 (S, 1H). HPLC TR 3.118 min; m/z− 381.80; (EM 383.01).
N-(2-chloro-4-nitrophenyl)-2-hydroxybenzamide (3k)
Using the method described for compound 3a, salicylic acid (1.03 g, 7.46 mmol) reacted with 2-chloro-5-nitroaniline (1.29 g, 7.46 mmol) and 2M PCl3 in CH2Cl2 (1.50 mL, 2.98 mmol) in xylenes (20 mL). The crude product was recrystallized from EtOH. 1H NMR (400 MHz, DMSO-d6) δ 7.055 (M, 1H), 7.495 (M, 1H) 8.053 (dd, J=8.0, 1.5Hz, 1H) 8.283 (dd, J=9.0, 2.5Hz, 1H), 8.415 (d, J=2.5Hz, 1H), 8.854 (d, J=9.0Hz, 1H). HPLC TR 2.482 min; m/z+ 292.95 [M+H]+; m/z− 290.80 [M-H]+; (EM 292.03).
N-(3-benzyl phenyl)-5-chloro-2-hydroxybenzamide (3l)
Using the method described for compound 3a, 5-chlorosalicylic acid (0.81g, 4.69 mmol) reacted with 3-benzylaniline (0.86 g, 4.69 mmol) and 2M PCl3 in CH2Cl2 (0.94 mL, 1.88 mmol) in xylenes (15 mL). The crude product was recrystallized from toluene. 1H NMR (500 MHz, DMSO-d6) δ 3.951 (S, 2H), 7.017 (m, 2H), 7.248 (m, 6H), 7.460 (dd, J=8.5, 2.5Hz, 1H), 7.550 (m, 2H), 7.948 (d, J=2.0Hz, 1H) 10.363 (s, 1H), 11.846 (s, 1H), HPLC TR 3.017 min; m/z+ 337.95 [M+H]+; m/z− 335.85; (EM 337.09).
N-(2-chloro-5-nitrophenyl)-5-methyl-2-hydroxybenzamide (3m)
Using the method described for compound 3a, 5-methylsalicylic acid (0.77g, 5.06 mmol) reacted with 2-chloro-4-nitroaniline (0.87 g, 5.06 mmol) and 2M PCl3 in CH2Cl2 (1.01 mL0, 2.02 mmol)in xylenes (18 mL). The desired product (64.5% pure) was found to contain 5.5% of an unidentified contaminant after collection upon cooling of the reaction solvent. 1H NMR (500 MHz, DMSO-d6) δ 2.261 (s, 3H), 6.949 (d, J=8.5Hz, 1H), 7.272 (dd, J=8.5, 2.0Hz, 1H), 7.812 (d, J=2.0Hz, 1H), 8.257 (dd, J=9.0, 2.5, 1H), 8.384 (d, J=2.5, 1H), 8.824 (d, J=9.0Hz, 1H). HPLC TR 2.628 min; m/z+ 306.95 [M+1]+; m/z− 304.85 [M-H]−; (EM 306.04).
N-(2,4-dichlorophenyl)-5-chloro-2-hydroxybenzamide (3n)
A suspension of 5-chlorosalicylic acid (1.73 g, 10.0 mmol) and 2,4-dichloraniline (1.62 g, 10.0 mmol) in xylenes (50 mL) was heated to reflux and a solution of PCl3 (0.35 mL, 4.0 mmol) in xylenes (5.0 mL) was introduced in a dropwise manner. After 90 minutes, the reaction mixture was transferred to a beaker via pipette and was allowed to cool to room temperature under rapid stirring. The crude product was recrystallized from EtOAc. 1H NMR (500 MHz, DMSO-d6) δ 7.075 (dd, J=8.8, 1.2Hz, 1H), 7.473 (m, 2H), 7.693 (dd, J=2.4, 1.2Hz, 1H), 7.982 (dd, J=2.8, 1.2Hz, 1H), 8.457 (dd, J=8.8, 1.2Hz, 1H), 10.925 (s, 1H), 12.241 (s, 1H). HPLC TR 2.889 min; m/z+ 315.85 [M+H]+; m/z− 313.70 [M-H]−; (EM-314.96).
N-(2,4-difluorophenyl)-5-chloro-2-hydroxybenzamide (3o)
Using the method described for compound 3n, 5-chlorosalicylic acid (1.73 g, 10.0 mmol) and 2,4-difluoroaniline (1.29 g, 10.0 mmol) reacted in refluxing xylenes (25 mL) in the presence of PCl3 (0.35 mL, 4.0 mmol). The crude product was recrystallized from EtOH. 1H NMR (500 MHz, DMSO-d6) δ 7.045 (d, J=9.0Hz, 1H), 7.143 (M, 1H), 7.410 (M, 1H), 7.498 (dd, J=9.0, 2.5Hz, 1H), 7.973 (d, J=2.5Hz, 1H), 8.116 (M, 1H), 10.624 (s, 1H), 12.139 (s, 1H.) HPLC TR 2.523 min; m/z+ 283.90 [M+H]+; m/z− 281.75 [M-H]−; (EM=283.02).
5-chloro-N-(4-chloro-2-fluorophenyl)-2-hydroxybenzamide (3p)
Using the method described for compound 3n, 5-chlorosalicylic acid (1.73 g, 10.0 mmol) and 2,4-difluoroaniline (1.46 g, 10.0 mmol) reacted in refluxing xylenes (25 mL) in the presence of PCl3 (0.35 mL, 4.0 mmol). The crude product was recrystallized from EtOAc. 1H NMR (500 MHz, DMSO-d6) δ 7.057 (d, J=8.8Hz, 1H), 7.335 (dd, J=8.8, 1.2Hz), 7.508 (dd, J=8.8, 2.8Hz, 1H), 7.577 (dd, J=10.4, 2.0Hz, 1H), 7.959 (d, J=2.8Hz, 1H), 8.245 (dd, J=8.8, 8.8Hz), 10.704 (s, 1H), 12.166 (s, 1H). HPLC TR 2.756 min; m/z+ 299.90 [M+H]+; m/z− 297.75 [M-H]−; (EM=298.99).
5-chloro-2-hydroxy-N-(3,4,5-trimethoxyphenyl)benzamide (3q)
Using the method described for compound 3n, 5-chlorosalicylic acid (1.73 g, 10.0 mmol) and 3,4,5-trimethoxaniline (1.83 g, 10.0 mmol) reacted in refluxing xylenes (20 mL) in the presence of PCl3 (0.35 mL, 4.0 mmol). The crude product was dissolved in 100 mL boiling toluene: heptane (1: 1 v/v) and the resulting solution was decanted from a dark insoluble residue. White crystalline product separated upon cooling. 1H NMR (500 MHz, DMSO-d6) δ 11.88 – 11.84 (s, 1H), 10.33 – 10.29 (s, 1H), 7.99 – 7.95 (d, J = 2.7 Hz, 1H), 7.51 – 7.45 (dd, J = 8.8, 2.6 Hz, 1H), 7.15 – 7.11 (s, 2H), 7.05 – 6.99 (d, J = 8.8 Hz, 1H), 3.81 – 3.77 (s, 6H), 3.68 – 3.64 (s, 3H), 3.35 – 3.31 (s, 3H). HPLC TR 2.400 min; m/z+ 337.95 [M+H]+; m/z− 335.85 [M-H]−; (EM=337.07).
5-chloro-N-(2-chloro-5-cyanophenyl)-2-hydroxybenzamide (3r)
Using the method described for compound 3n, 5-chlorosalicylic acid (0.86 g, 5.0 mmol) and 3-amino-4-chlorobenzonitrile (0.76 g, 5.0 mmol) reacted in refluxing xylenes (10 mL) in the presence of PCl3 (0.18 mL, 2.0 mmol). The crude product was recrystallized from EtOH/water to provide a tan colored solid. 1H NMR (500 MHz, DMSO-d6) δ 7.053 (d, J=8.5Hz), 7.345 (dd, J=8.5, 3.0Hz), 7.407 (dd, J=8.5, 2.5Hz), 7.598 (d, J=8.5Hz), 8.079 (d, J=3.0Hz), 8.979 (s, J=2.5Hz), 11.105 (s, 1H), 11.700 (s, 1H.) HPLC TR 2.551 min; m/z+ 306.90 [M+H]+; m/z− = 304.75 [M-H]−; (EM=306.00).
5-chloro-N-(3,5-dimethylphenyl)-2-hydroxybenzamide (3s)
Using the method described for compound 3a, 5-methylsalicylic acid (1.04g, 6.03 mmol) reacted with 3,5-dimethylaniline and 2M PCl3 in CH2Cl2 (1.21 mL, 2.42 mmol) and 2M in refluxing xylenes (15 mL). Pure white crystalline product resulted. 1H NMR (500 MHz, DMSO-d6) δ 2.260 (s, 6H), 6.780 (s, 1H), 7.015 (M, 2H), 7.315 (s, 2H), 7.451 (dd, J=9.0, 2.5Hz, 1H), 7.965 (d, J=2.5Hz, 1H), 10.265 (s, 1H). HPLC TR 2.848 min; m/z+ 275.95, m/z− 273.80; (EM=275.07).
5-chloro-N-(3-cyano-4-fluorophenyl)-2-hydroxybenzamide (3t)
Using the method described for compound 3n, 5-chlorosalicylic acid (1.73 g, 10.0 mmol) and 3-ethynyl-4-fluoroaniline (1.36 g, 10.0 mmol) reacted in refluxing xylenes (25 mL) in the presence of PCl3 (0.35 mL, 4.0 mmol). The crude product was recrystallized from EtOAc/hexanes. 1H NMR (500 MHz, DMSO-d6) δ 11.57 (s, 1H), 10.64 (s, 1H), 8.24 (m, 1H), 8.03 (m, 1H), 7.87 (m, 1H), 7.64 (m, 1H), 7.48 (m, 1H), 7.04 (m, 1H). HPLC TR 2.546 min; m/z+ 290.85 [M+H]+; m/z− 288.80 [M-H]−; (EM 289.03).
5-chloro-N-(3-ethoxyphenyl)-2-hydroxybenzamide (3u)
Using the method described for compound 3n, 5-chlorosalicylic acid (1.73 g, 10.0 mmol) and 3-ethoxyaniline (1.37 g, 10.0 mmol) reacted in refluxing xylenes (25 mL) in the presence of PCl3 (0.35 mL, 4.0 mmol). The crude product was purified by column chromatography (silica gel, 35 EtOAc: 65 hexanes).) 1H NMR (500 MHz, DMSO-d6) δ 12.03 – 11.61 (s, 1H), 10.62 – 10.19 (s, 1H), 8.09 – 7.86 (d, J = 2.7 Hz, 1H), 7.61 – 6.51 (m, 6H), 4.47 – 3.72 (q, J = 7.0 Hz, 2H), 1.69 – 0.97 (t, J = 7.0 Hz, 3H). HPLC TR 2.729 min; m/z+ 307.90 [M+H]+; m/z− 305.85 [M-H]−; (EM=307.06).
5-chloro-N-(3,4-dimethoxyphenyl)-2-hydroxybenzamide (3v)
Using the method described for compound 3n, 5-chlorosalicylic acid (1.73 g, 10.0 mmol) and 3,4-dimethoxyaniline (1.53 g, 10.0 mmol) reacted in refluxing xylenes (25 mL) in the presence of PCl3 (0.35 mL, 4.0 mmol). The crude product was purified by column chromatography (silica gel, 35 EtOAc: 65 hexanes) then recrystallized from EtOAc. 1H NMR (500 MHz, DMSO-d6) δ 12.04 – 12.00 (s, 1H), 10.33 – 10.29 (s, 1H), 8.03 – 7.99 (d, J = 2.6 Hz, 1H), 7.51 – 7.45 (dd, J = 8.8, 2.7 Hz, 1H), 7.41 – 7.37 (d, J = 2.4 Hz, 1H), 7.28 – 7.22 (dd, J = 8.7, 2.4 Hz, 1H), 7.04 (d, J = 8.8 Hz, 1H), 6.94 (d, J = 8.8 Hz, 1H). NMR HPLC TR 2.359 min; m/z+ 307.90 [M+H]+; m/z− 305.85 [M-H]−; (EM=307.06).
5-chloro-2-hydroxy-N-(3-isopropoxyphenyl)benzamide (3w)
Using the method described for compound 3n, 5-chlorosalicylic acid (1.73 g, 10.0 mmol) and 3-isopropoxyaniline (1.51 g, 10.0 mmol) reacted in refluxing xylenes (25 mL) in the presence of PCl3 (0.35 mL, 4.0 mmol). The crude product was purified by column chromatography (silica gel, 1 EtOAc: 1 hexanes). 1H NMR (500 MHz, DMSO-d6) δ 12.23 – 11.64 (s, 1H), 10.87 – 10.01 (s, 1H), 7.96 – 7.92 (d, J = 2.7 Hz, 1H), 7.50 – 7.43 (dd, J = 8.8, 2.7 Hz, 1H), 7.39 – 7.34 (t, J = 2.2 Hz, 1H), 7.27 – 7.18 (m, 2H), 7.03 – 6.98 (d, J = 8.8 Hz, 1H), 6.72 – 6.67 (ddd, J = 7.8, 2.5, 1.4 Hz, 1H), 4.71 – 4.43 (hept, J = 6.0 Hz, 1H), 1.34 – 1.13 (d, J = 6.0 Hz, 6H). HPLC TR 2.843 min; m/z+ 305.95 [M+H]+; m/z− 303.85 [M-H]−; (EM 305.08).
5-chloro-2-hydroxy-N-(5-methoxy-2-methylphenyl)benzamide (3x)
Using the method described for compound 3n, 5-chlorosalicylic acid (0.645 g, 3.74 mmol) and 5-methoxy-2-methylaniline (0.513 g, 3.74 mmol) reacted in refluxing xylenes (15 mL) in the presence of PCl3 (0.13 mL, 1.50 mmol). The crude product was recrystallized from EtOH/H2O. 1H NMR (400 MHz, DMSO-d6) δ 3.331 (s, 1H), 3.742 (s, 1H), 6.708 (dd, J=8.4, 1.2, Hz), 7.053 (d, J=8.4Hz, 1H), 7.175 (d, J=8.4Hz, 1H), 7.188 (m, 1H), J=7.617 (s, 1H), 8.018 (s, 1H). HPLC TR 2.510 min; m/z+ 291.90 [M+H]+; m/z− 289.85 [M-H]−; (EM 291.07).
5-chloro-2-hydroxy-N-(3-phenoxyphenyl)benzamide (3y)
Using the method described for compound 3n, 5-chlorosalicylic acid (0.919 g, 5.33 mmol) and 3-phenoxyaniline (0.987 g, 5.33 mmol) reacted in refluxing xylenes (20 mL) in the presence of PCl3 (0.19 mL, 2.13 mmol). The crude product was recrystallized from EtOAc/hexanes. 1H NMR (500 MHz, CDCL3) δ 6.858 (m, 1H), 6.982 (d, J=8.8Hz, 1H), 7.057 (d, J=7.6Hz, 2H), 7.151 (dd, J=7.6, 7.2Hz, 1H), 7.355 (m, 6H), 7.462 (d, J=2.4Hz, 1H), 7.794 (s, 1H), 11.776 (s, 1H). HPLC TR 2.999 min; m/z+ 339.95 [M+H]+; m/z− 337.85 [M-H]−; (EM 339.07).
5-chloro-N-(3-(difluoromethoxy)phenyl)-2-hydroxybenzamide (3z)
Using the method described for compound 3n, 5-chlorosalicylic acid (0.86 g, 4.98 mmol) and 3-difluoromethoxyaniline (0.0.79 g, 4.98 mmol) reacted in refluxing xylenes (15 mL) in the presence of PCl3 (0.17 mL, 1.99 mmol). The crude product was recrystallized from EtOAc. 1H NMR (500 MHz, DMSO-d6) δ 11.82 – 11.49 (s, 1H), 10.63 – 10.27 (s, 1H), 7.92 – 7.87 (d, J = 2.7 Hz, 1H), 7.69 – 7.65 (t, J = 2.2 Hz, 1H), 7.56 – 7.51 (ddd, J = 8.2, 2.0, 1.0 Hz, 1H), 7.49 – 7.45 (dd, J = 8.8, 2.7 Hz, 1H), 7.44 – 7.39 (t, J = 8.2 Hz, 1H), 7.39 – 7.06 (t, J = 74.0 Hz, 1H), 7.04 – 6.99 (d, J = 8.8 Hz, 1H), 6.98 – 6.93 (dd, J = 8.1, 2.3 Hz, 1H). NMR HPLC TR 2.665 min; m/z+ 313.85 [M+H]+; m/z− 311.80 [M-H]−; (EM 313.03).
N-(3-chlorophenyl)-2-hydroxy-3,5-diiodobenzamide (3aa)
A mixture of 5-chlorosalicylic acid (0.573 g, 1.47 mmol) and 2-hydroxy-3,5-diiodobenzoic acid (0.154 mL, 1.47 mmol) in xylenes (15 mL) was brought to reflux whereupon all solids dissolved. A solution of PCl3 (0.051 mL, 0.588 mmol) in xylenes (5mL) was introduced in a dropwise fashion. After 1 hr, the reaction mixture was allowed to cool to 40° C and was decanted into rapidly stirring hexanes (40 mL) at rt. White crystals separated, and after 1 hr the product was collected by filtration, washed well with hexanes, and air-dried. 1H NMR (500 MHz, DMSO-d6) δ 12.87 – 12.83 (s, 1H), 10.72 – 10.68 (s, 1H), 8.38 – 8.34 (d, J = 2.0 Hz, 1H), 8.27 – 8.22 (d, J = 1.9 Hz, 1H), 7.86 – 7.81 (t, J = 2.1 Hz, 1H), 7.67 – 7.61 (ddd, J = 8.2, 2.1, 1.0 Hz, 1H), 7.47 – 7.40 (t, J = 8.1 Hz, 1H), 7.29 – 7.23 (ddd, J = 8.1, 2.1, 0.9 Hz, 1H). HPLC TR 3.351 min; m/z+ 499.70 [M+H]+; m/z− 497.65 [M-H] −; (EM 498.83).
N-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-2-hydroxybenzamide (3ab)
A mixture of 5-chlorosalicylic acid (1.73 g, 10.0 mmol) and 3,4-methylenedioxyaniline (1.51 g, 10.0 mmol) in xylenes (20 mL) was brought to reflux whereupon all solids dissolved. A solution of PCl3 (0.35 mL, 4.0 mmol) in xylenes (5mL) was introduced in a dropwise fashion. After 1 hr, the reaction solution was decanted from a dark residue which had accumulated in the reaction vessel. After cooling, decolorizing carbon (0.25 g) was introduced, and the mixture was heated at 80° C for 30 min. Decolorizing carbon was removed by filtration through a filter aid. The solution was washed with water, dried over MgSO4, and concentrated. Pure product was obtained by column chromatography (silica gel, 35 EtOAc: 65 hexanes. 1H NMR (500 MHz, DMSO-d6) δ 11.99 – 11.90 (s, 1H), 10.30 – 10.22 (s, 1H), 8.01 – 7.94 (d, J = 2.7 Hz, 1H), 7.50 – 7.43 (dd, J = 8.8, 2.7 Hz, 1H), 7.34 – 7.29 (d, J = 2.5 Hz, 1H), 7.14 – 7.08 (dd, J = 8.7, 2.5 Hz, 1H), 7.04 – 6.98 (d, J = 8.8 Hz, 1H), 6.89 – 6.82 (d, J = 8.7 Hz, 1H), 4.29 – 4.21 (qd, J = 3.7, 2.0 Hz, 4H). HPLC TR 2.482 min; m/z+ 305.90 [M+H]+; m/z− 303.80 [M-H]−; (EM 305.05).
Preparation of N-(3-ethoxyphenyl)-5-fluoro-2-hydroxybenzamide (3ac)
5-fluorosalicylic acid (0.312 g, 2.0 mmol), 3-ethoxyaniline (0.274 g, 2.0 mmol), PCl3 (0.174 mL, 2.0 mmol) and xylenes (15 mL) were placed in a sealed tube and heated to 150° C for 2 hr. Upon cooling to room temperature, the reaction solution was diluted with EtOAc and washed successively with saturated NaHCO3, water, and brine, then dried (MgSO4). Solvents were evaporated, and the residue was purified by preparative TLC (silica gel, 35 EtOAc: 65 hexanes). Significant decomposition was observed during the chromatography. Pure product (0.027 g, 4.9% yield) was obtained. 1H NMR (500 MHz, DMSO-d6) δ 11.59 (s, 1H), 10.36 (s, 1H), 7.74 (dd, J = 9.7, 3.2 Hz, 1H), 7.39 (t, J = 2.1 Hz, 1H), 7.28 (m, 3H), 7.00 (dd, J = 9.0, 4.6 Hz, 1H), 6.71 (ddd, J = 7.8, 2.5, 1.4 Hz, 1H), 4.03 (q, J = 6.9 Hz, 2H), 1.34 (t, J = 7.0 Hz, 3H). HPLC TR 2.532 min; m/z+ 275.95 [M+H]+; m/z− 549.00 [2M-H]−, 273.80 [M-H]−; (EM 275.10).
N-(4-amino-2-chlorophenyl)-5-chloro-2-hydroxybenzamide hydrochloride (5)
A slurry of niclosamide (4, 1.435 g, 4.38 mmol) in 1MeOH: 1 HOAc (20 mL) was treated with portions of Zn dust (3.0 g total) over 3 hr at rt. A mild exotherm was observed. As the reaction progressed, precipitates caused a thickening of the reaction mixture. MeOH was added from time to time to facilitate magnetic stirring. The reaction mixture was filtered through a plug of Celite and concentrated to an off-white residue. This was partitioned between EtOAc and saturated NaHCO3 (copious precipitate appeared in the aqueous phase). Water was added to the mixture to facilitate phase separation. The EtOAc phase was washed with brine, dried (MgSO4), and concentrated to a tan solid. This was recrystallized from EtOAc/hexanes to provide tan crystals (≥ 99% pure by LC-MS). The aniline product was dissolved in EtOAc by warming a suspension (0.2M) to 50°C, and HCl in dioxane (4M, 2 equivalents) was added in a dropwise manner. The resulting slurry was diluted with 1 volume of EtOAc, cooled to room temperature, and stirred for an additional 10 min. The product was collected by filtration and washed well with EtOAc. 1H NMR (500 MHz, DMSO-d6) δ 12.37 (s, 1H), 10.81 (t, J = 2.0 Hz, 1H), 8.26 (m, 1H), 7.99 (d, J = 2.8 Hz, 1H), 7.51 (dd, J = 8.8, 2.8 Hz, 1H), 7.38 (m, 1H), 7.16 (m, 2H). HPLC TR 1.125 min; m/z+ 339.85 [M+ CH3CN+2]+, 337.90 [M+ CH3CN+H]+, 298.85 [M+2]+ 296.85 [M+ H]+; m/z− 296.80 [M+2-H]− 294.80 [M-H]−; (EM 296.01).
4-chloro-2-((2-chloro-4-nitrophenyl)carbamoyl)phenyl benzoate (7a)
Benzoyl chloride (0.49 mL, 4.27 mmol) was added dropwise to a suspension of niclosamide (1.27 g, 3.88 mmol) in a solution of 4-dimethylaminopyridine (DMAP, 30 mg) in pyridine (15 mL) at rt. The suspension was warmed to 80°C whereupon all solids dissolved. Reaction continued at this temperature for 2 hr. The cooled reaction mixture was diluted with EtOAc (100 mL) and was washed successively with 1N HCl until the aqueous wash was acidic (about pH 1) to litmus. The EtOAc phase was washed with brine, dried (MgSO4), and concentrated to an off-white solid. The crude product was recrystallized from EtOAc/hexanes. 1H NMR (500 MHz, DMSO-d6) δ 10.56 (s, 1H), 8.31 (m, 1H), 8.25 (m, 1H), 8.08 (m, 2H), 7.90 (m, 2H), 7.25 (m, 1H), 7.54 (m, 3 H).). HPLC TR 2.990 min; m/z+ 430.95 [M+ H]+; m/z− 428.80 [M-H]−; (EM 430.01).
(S)-1-(4-chloro-2-((2-chloro-4-nitrophenyl)carbamoyl)phenyl) 2-methyl pyrrolidine-1,2-dicarboxylate (7b)
A: (S)-methyl 1-(chlorocarbonyl)pyrrolidine-2-carboxylate. L-proline methyl ester hydrochloride (2.62 g, 15.82 mmol) was partitioned between saturated NaHCO3 (50 mL) and CH2Cl2 (50 mL). The CH2Cl2 phase was dried (MgSO4) and concentrated to an oil. This was dissolved in THF (20 mL) and DIEA (3.03 mL, 17.40 mmol) was added. The resulting solution was added dropwise to a solution of phosgene in toluene (12.5%, 24.0 mL, 20.88 mmol) previously cooled to 0°C. The reaction mixture was allowed to warm to room temperature over one hr, then was filtered and concentrated to provide an oil. This was used in the next step without purification. B: (S)-1-(4-chloro-2-((2-chloro-4-nitrophenyl)carbamoyl)phenyl) 2-methyl pyrrolidine-1,2-dicarboxylate. A solution of (S)-methyl 1-(chlorocarbonyl)pyrrolidine-2-carboxylate (from Step A, 0.397 g, 2.07 mmol) and DMAP (10 mg) in CHCl3 (15 mL)was added to a slurry of niclosamide (645 mg, 1.973 mmol) in pyridine (15 mL). This was warmed to 80°C (all solids dissolved) and maintained 1 hr. The cooled solution was diluted with EtOAc (100 mL). The resulting mixture was washed successively with 1N HCl until the aqueous wash was acidic (about pH 1) to litmus. The EtOAc solution was then washed with brine, dried (MgSO4), and concentrated to an off-white solid. The crude product was recrystallized from EtOAc/hexanes. The 1H NMR spectrum is complex and reveals the existence of rotational isomers, presumably due to restricted rotation about the pyrolidine-1-carbamoyl bond. Analysis of HPLC and MS data reveals a single compound in ≥95% purity. 1HNMR (300 MHz, DMSO-d6): δ 1.823 – 2.003 (m, 3H, pyrrolidine C4 methylene and pyrrolidine C3 syn-H), 2.213 (m, 1H, pyrrolidine C3 anti-H), 3.450 (m, 1H, pyrrolidine C5), 3.599 (m, 1H, pyrrolidine C5), 3.600 (s, 1.5 H, methyl ester), 3.621 (s, 1.5 H, methyl ester), 4.252 (m, 0.5 H, pyrrolidine methine), 4.541 (m, 0.5H, pyrrolidine methine), 7.108 (d, J = 8.5 Hz, 0.33 H, H3), 7.216 (d, J = 8.5 Hz, 0.33 H, H3), 7.361 (d, J = 9 Hz, 0.33H, H3), 7.545 (m, 0.33H, H4), 7.665 (m, 0.66H, H4), 7.763 (m, 0.66H, H6), 7.974 (m, 0.33H, H6), 8.133 (m, 0.66H, H6′), 8.287 (m, 1H, H5′), 8.302 (m, 0.33H, H3′), 8.539 (m, 0.33H, H3′), 8.446 (m, 0.33H, H3′), 8.822 (d, J = 9.5 Hz, 0.33H, H6′), 10.313 (s, 0.33H, NH), 10.385 (s, 0.33H, NH), 11.367 (br s, 0.17H, NH), 12.519 (br s, 0.17H, NH). HPLC TR 2.743 min; m/z+ 482.00 [M+ H]+; m/z− 479.90 [M-H]−; (EM 481.04). Note: The optical purity of this material was not determined.
4-chloro-2-((2-chloro-4-nitrophenyl)carbamoyl)phenyl morpholine-4-carboxylate (7c)
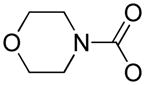
4-Morpholine carbonyl chloride (0.353 mL, 3.08 mmol) was added dropwise to a suspension of niclosamide 4 (0.503 g, 1.54 mmol) in pyridine (8.0 mL) containing DMAP (10 mg). This was raised to reflux (all solids dissolved) and maintained 3 hr. The hot solution was admitted via pipette dropwise into rapidly stirring distilled water (100 mL) whereupon a fine white solid precipitated. This mixture was stirred at room temperature for 2 hr. The crude product was collected by filtration, washed well with water (100 mL) and 1N HCl (200 mL), and then dried under a stream of room air for 2 hr. The product was then dried in a vacuum oven (28″ Hg, 50°C) for 60 hr. The off-white product was recrystallized from EtOAc to give white crystals. 1H NMR (500 MHz, CDCl3) δ 8.60 (m, 2H), 8.45 (m, 1H), 8.33 (m, 1H), 7.96 (m, 1H), 7.62 (m, 1H), 7.24 (m, 1H), 3.78–3.87 (m, 8H). HPLC TR 2.569 min; m/z+ 440.00 [M+ H]+; m/z− 437.85 [M-H]−; (EM 439.03).
4-chloro-2-((2-chloro-4-nitrophenyl)carbamoyl)phenyl 4-methylpiperazine-1-carboxylate (7d)
4-Methyl-1-piperazinecarbonyl chloride (0.603 g, 3.02 mmol) was added to a suspension of niclosamide 4 (0.495 g, 1.51 mmol)in pyridine (8.0 mL) containing DMAP (10 mg). The mixture was raised to reflux for 1 hr. The hot solution was introduced by pipette to rapidly stirring water at rt. After 1 hr, the solids were collected by filtration and added to 50 mL rapidly stirring 1N HCl. This was stirred for 30 min, then the crude product was collected by filtration and dried in a vacuum oven (28″ Hg, 50°C) for 18 hr, then recrystallized from EtOH. A small portion of the product was converted to the free base by partitioning between EtOAc and saturated NaHCO3. TLC (SiO2, 5 MeOH: 95 CHCl3) demonstrated a single component, Rf = 0.37. 1H NMR (400 MHz, CDCl3) δ 8.91 (s, 1H), 8.81 (d, J = 9.2 Hz), 8.34 (d, J = 2.4 Hz), 8.22 (dd, J = 9.2, 2.4 Hz), 7.87 (d, J = 2.8 Hz), 7.51 (dd, J = 2.8, 8.4 Hz), 7.13 (d, J = 8.4 Hz), 3.68 (m, 2H), 3.56 (m, 2H), 2.39 (m, 4H), 2.28 (s, 3H). HPLC TR 1.664; m/z+ 452.95 [M+ H]+; m/z− 450.90 [M-H]−; (EM 452.07).
N-(2-chloro-4-nitrophenyl)-5-fluoro-2-methoxybenzamide (10a)
A solution of 5-fluoro-2-methoxybenzoic acid (0.340 g, 2.0 mmol), 2-chloro-4-nitroaniline (0.173 g, 1.0 mmol), and O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU, 0.76 g, 2.0 mmol) in MeCN (4.0 mL) was treated with DIEA (0.10 mL) and the resulting solution was stirred at rt for 16 hr. EtOAc (10 mL) was added, and the organic phase was washed with 50% 1N HCl in saturated NaCl, then brine, dried over MgSO4, and concentrated to afford a yellow solid. This was recrystallized from EtOAc: hexanes. 1H NMR (500 MHz, DMSO-d6) δ 13.78 (s, 1H), 12.88 (s, 1H), 8.76 (dd, J = 4.4, 1.4 Hz, 1H), 8.53 (dd, J = 8.4, 1.4 Hz, 1H), 7.51 (dd, J = 8.4, 4.4 Hz, 1H), 7.37 (m, 2H), 7.13 (dd, J = 9.1, 4.3 Hz, 1H), 3.79 (s, 3H). HPLC TR 2.176 min; m/z+ 288.90 [M - Cl]+; (EM 324.03).
5-fluoro-N-(2-fluoro-4-nitrophenyl)-2-methoxybenzamide (10b)
A solution of 5-fluoro-2-methoxybenzoic acid (0.510 g, 3.0 mmol), 2-fluoro-4-nitroaniline (0.156 g, 1.0 mmol), and O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU, 1.140 g, 3.0 mmol) in MeCN (6.0 mL) was treated with DIEA (0.150 mL) and the resulting solution was stirred at room temperature for 2 hr. The reaction mixture was added to rapidly stirring water, and the resulting precipitate was washed well with water, then dissolved in EtOAc, dried over MgSO4, and concentrated to a yellow solid. The crude product was recrystallized from EtOAc/hexanes. 1H NMR (500 MHz, DMSO-d6) δ 13.78 (s, 1H), 12.89 (s, 1H), 8.76 (dd, J = 4.4, 1.4 Hz, 1H), 8.53 (dd, J = 8.4, 1.4 Hz, 1H), 7.51 (dd, J = 8.4, 4.4 Hz, 1H), 7.40 (dd, J = 8.8, 3.3 Hz, 1H), 7.35 (ddd, J = 9.1, 8.1, 3.3 Hz, 1H), 3.79 (s, 3H). HPLC TR 2.190 min; m/z+ 288.90 [M-F]+; m/z− 292.85 [M-CH3]−; (EM 308.06).
tert-butyl 2-((chlorocarbonyl)(methyl)amino)acetate (12)
A solution of sarcosine tert-butyl ester hydrochloride 11 (4.214 g, 23.20 mmol) in CH2Cl2 (40 mL) was shaken with saturated NaHCO3 in a separatory funnel. The organic phase was dried over MgSO4 and concentrated to a clear oil (2.298 g, 68% yield). A solution of phosgene in toluene (20%, 10.9 mL, 23.75 mmol) was cooled to −25°C, and a solution of sarcosine tert-butyl ester (2.298 g, 15.78 mmol) and DIEA (5.5 mL, 31.66 mmol) in CH2Cl2 (10 mL) was introduced in a dropwise fashion. The solution was allowed to warm to room temperature over 1 hr, and was then washed with 1N HCl (50 mL) and EtOAc sufficient to form two layers was added. The organic phase was washed with water, then brine, and dried over MgSO4. This solution was used without further manipulation.
2-(((4-chloro-2-((2-chloro-4-nitrophenyl)carbamoyl)phenoxy)carbonyl) (methyl)amino)acetic acid (13a)
A. tert-Butyl 2-(((4-chloro-2-((2-chloro-4-nitrophenyl)carbamoyl)phenoxy)carbonyl)(methyl)amino)acetate. A solution of 12 (15.78 mmol) in EtOAc (ca. 0.5M) was introduced dropwise to a refluxing solution of niclosamide (2.56 g, 7.92 mmol) in pyridine (80 mL containing DMAP (50 mg). After 30 min, 100 mL solvent was distilled from the reaction mixture. The remaining reaction mixture was cooled to room temperature, diluted with EtOAc (50 mL), and washed successively with 1N HCl until the aqueous wash was acidic (about pH 1) to litmus. The organic phase was washed with brine, dried (MgSO4), and concentrated to an off-white solid. This was used without further purification. B. 2-(((4-chloro-2-((2-chloro-4-nitrophenyl) carbamoyl) phenoxy) carbonyl) (methyl)amino) acetic acid. The solid from Step A (542 mg, 1.09 mmol) was dissolved in CH2Cl2 (15 mL) and CF3COOH (15 mL) was added. After 16 hr the reaction solution was concentrated to an oily residue. This was dissolved in CHCl3 (25 mL) and concentrated. The CHCl3 chase was repeated to leave a sinterable foam. This was layered with 35 EtOAc: 65 hexanes (50 mL), and warmed to 45°C under rapid stirring for 30 min. Hexane (30 mL) was added, and the stirring mixture was allowed to cool to room temperature over 20 min. The product was collected by filtration. The 1H NMR spectrum was complex and revealed the existence of rotational isomers, presumably due to restricted rotation about the sarcosine-1-carbamoyl bond. Analysis of HPLC and MS data reveals a single compound in ≥95% purity. TR 2.382 min; m/z+ 441.85 [M+ H]+; m/z− 324.75 [M-sarcosine-H]−; (EM 441.01).
4-chloro-2-((2-chloro-4-nitrophenyl)carbamoyl)phenyl (2-hydrazinyl-2-oxoethyl)(methyl) carbamate hydrochloride (13b)
A. Tert-butyl 2-(2-(((4-chloro-2-((2-chloro-4-nitrophenyl) carbamoyl)phenoxy)carbonyl)(methyl)amino)acetyl)hydrazinecarboxylate. Tert-butyl carbazate (417 mg, 3.16 mmol) was added to a solution of 13a (1.27 g, 2.87 mmol) in THF (8.7 mL) with stirring at rt. A solution of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (660 mg, 3.44 mmol) in CH2Cl2 (17.4 mL) was introduced dropwise at rt. The homogeneous solution was stirred at room temperature for 14 hr, then concentrated. The residue was partitioned between EtOAc and 1 N HCl. The EtOAc phase was washed with water, then with brine, dried (MgSO4), and concentrated to an off-white solid 1.20 g (75.1%). This was used without further manipulation. B. 4-Chloro-2-((2-chloro-4-nitrophenyl)carbamoyl)phenyl (2-hydrazinyl-2-oxoethyl)(methyl)carbamate hydrochloride. The product from step A (2.52 g, 5.70 mmol) was dissolved in a solution of HCl in dioxane (4.0 M, 10 mL) and the solution was stirred at room temperature for 30 min, then concentrated to an off-white solid which was washed well with EtOAc, then hexanes, and dried under a stream of air to provide a white solid (2.50 g, 89%). 1H NMR spectrum was complex and revealed the existence of two rotational isomers, presumably due to restricted rotation about the sarcosine-1-carbamoyl bond. 1H NMR (500 MHz, DMSO-d6) δ 11.09 (s, 1H), 10.45 (s, 1H), 10.40 (s, 1H), 8.41 (dd, J = 2.3, 1.5 Hz, 1H), 8.31 – 8.25 (m, 1H), 8.10 (m, 1H), 7.80 (dd, J = 6.2, 2.6 Hz, 1H), 7.71 – 7.65 (m, 1H), 7.36 (d, J = 8.7 Hz, 1H), 7.26 (d, J = 8.7 Hz, 1H), 4.21 (s, 1H), 4.05 (s, 1H), 3.07 (s, 1.5H), 2.92 (s, 1.5H). HPLC TR 2.03 min; m/z+ 455.95 [M+ H]+; m/z− 453.95 [M-H]−; (EM 308.06).
2-(((2-((2,4-bis(trifluoromethyl)phenyl)carbamoyl)-4 chlorophenoxy)carbonyl) (methyl) amino) acetic acid (14 a)
A. tert-Butyl 2-(((2-((2,4-bis(trifluoromethyl)phenyl)carbamoyl)-4-chlorophenoxy)carbonyl)(methyl)amino)acetate. N-(3,5-bis(triflouromethyl)phenyl-5-chloro-2-hydroxybenzamide (3j, 6.47 g, 16.86 mmol) was added to a solution of 12 (4.20 g, 20.22 mmol) in pyridine (35 mL) and the mixture was warmed to 80° C for three hours. The reaction solution was cooled to room temperature, diluted with EtOAc (200 mL) and washed four times with 1N HCl (final wash ph about 1 to litmus), once with brine, and dried (MgSO4). Concentration afforded an off-white solid which was triturated with hexanes to provide the product (7.01 g, 75%) which was used without further purification. B.2-(((2-((2,4-bis(trifluoromethyl)phenyl)carbamoyl)-4-chlorophenoxy)carbonyl)(methyl)amino) acetic acid. The product from step A (5.43 g, 9.70 mmol) was dissolved in CH2Cl2 (15 mL) and CF3COOH (7.5 mL) was added. The reaction mixture was stirred at room temperature for 14 hr, then the reaction mixture was concentrated and the residue was chased twice with CHCl3 (25 mL). The 1H NMR spectrum was complex and revealed the existence of rotational isomers, presumably due to restricted rotation about the sarcosine-1-carbamoyl bond. Analysis of HPLC (TR = 2.65 min) and MS data (m/z+ 499.05, [M+H]+, m/z− 381.80, [M-C4H6NO]−) reveals a single compound in ≥95% purity.
2-((2,4-bis(trifluoromethyl)phenyl)carbamoyl)-4-chlorophenyl(2-hydrazinyl-2-oxoethyl) (methyl)carbamate hydrochloride (14b)
A. tert-Butyl 2-(2-(((2-((2,4-bis (trifluoromethyl) phenyl)carbamoyl)-4-chlorophenoxy) carbonyl) (methyl)amino) acetyl) hydrazine carboxylate. A solution of 2-(((2-((2,4-bis(trifluoromethyl)phenyl)carbamoyl)-4-chlorophenoxy)carbonyl) (methyl)amino)acetic acid (14a, 540 mg,1.08 mmol) in CH2Cl2 (5.0 mL0 was treated with tert-butyl carbazate (172 mg, 1.30 mmol) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (249 mg, 1.30 mmol). The solution was stirred at room temperature for 4 hr, then concentrated, and the residue was dissolved in EtOAc (15 mL), washed with 1N HCl, then with brine, dried over MgSO4, and concentrated to a scinterable foam. The 1H NMR spectrum was complex and revealed the existence of rotational isomers, presumably due to restricted rotation about the sarcosine-1-carbamoyl bond. Analysis of HPLC (TR = 2.81 min) and MS data (m/z+ 634.90 [M+Na]+, m/z− 610.85, [M-H]−) reveals a single compound in ≥95% purity. B. 2-((2,4-bis(trifluoromethyl)phenyl)carbamoyl)-4-chlorophenyl (2-hydrazinyl-2-oxoethyl)(methyl) carbamate hydrochloride (14b, 2.263 g, 3.69 mmol) was dissolved in a solution of HCl in dioxane (15 mL). After stirring 2 hr at room temperature, the reaction mixture was concentrated and the residue was chased twice with 15 mL portions of CHCl3, providing a scinterable foam (1.90 g, 94%). 1H NMR (500 MHz, DMSO-d6) δ 11.03 (s, 1H), 10.95 (s, 1H), 9.15 (s, 1H), 9.08 (s, 1H), 8.36 (s, 4H), 7.84 (s, 2H), 7.80 (dd, J = 5.2, 2.7 Hz, 2H), 7.70 – 7.64 (m, 1H), 7.38 (d, J = 8.7 Hz, 1H), 7.26 (d, J = 8.7 Hz, 1H), 4.17 (q, J = 18.2, 13.8 Hz, 3H), 3.97 (s, 2H), 3.79 (s, 2H), 3.00 (s, 3H), 2.83 (s, 3H). HPLC TR 2.38 min; m/z+ 512.90 [M+ H]+; m/z− 510.80 [M-H]−; (EM 512.07).
Biology
In Vitro Evaluation of Inhibition of T. gondii tachyzoites
Test compounds were dissolved in DMSO to make a 10mM solution, and subsequently diluted with IMDM-C to the concentrations used in bioassay. In the in vitro experiments DMSO concentration was not greater than 0.1% unless otherwise specified. RH-YFP parasites, which stably express Yellow Florescent Protein, were used. Tachyzoites were extracted from HFF cells by double passage through a 25-gauge12 needle, centrifuged for 15 minutes at 1500 RPM, and resuspended in IMDM-C. Confluent monolayers of HFF cells were infected with parasites in 96-well plates (Falcon 96 Optilux Flat-bottom) with 3,500 parasites in 100 uL per well. One hour after inoculation, test compounds and control media were added for a final volume of 200uL per well. Parasite proliferation was assessed using [3H]-Uracil incorporation13 or YFP Fluorescence assay.
T. gondii Parasite and cell culture
Human Foreskin Fibroblast (HFF) cells were maintained in confluent monolayers in Iscoves’s Modified Dulbecco’s Medium supplemented with 10% fetal bovine serum, 1% GlutaMAX and 1% penicillin-streptomycin-fungizone (IMDM-C). T. gondii tachyzoites were cultivated in HFF monolayers12,13. Parasites and cells were maintained at 33°C or 37°C and 5% CO2. The strains of parasite used in this study include RH, RH-YFP, Me49 strain14, andTgGoatUS4 isolate15.
[3H]-Uracil and [3H]-Thymidine incorporation assays
25 μL of 0.1mCi/mL [3H]-Uracil or [3H]-Thymidine was added to each well 24 hours before reading plates. At harvesting, contents of the wells were transferred onto a 96-well UniFilter GF/C filter plates using a Filtermate 196 cell harvester (Packard). [3H]-Uracil and [3H]-Thymidine incorporation was measured using a Microplate Scintillation Luminescence Counter (Packard) 12,13.
YFP Fluorescence assay
72 hours after initiation of the in vitro challenge assay, parasite proliferation was assessed by reading fluorescence of YFP parasites with a Synergy H4 Hybrid Reader (BioTek) and Gen5 1.10 software, using a bottom optics positions, excitation wavelength of 514nm, and emission wavelength of 540nm12.
In vitro toxicity assay
HFF cells were grown to ~30% confluence in 96-well plates. Inhibitory compounds and control media were added to wells in concentrations equal to those being tested in challenge assays. After 72 hours, [3H]-Thymidine incorporation assay was conducted to assess cell growth12,13. Alternatively, toxicity was assessed using WST-1 cell proliferation reagent (Roche). Confluent HFF cells were treated with inhibitory and control compounds. On the final day of experiment, 10uL of WST-1 reagent was added to each well. Plates were incubated for 1 hour in the dark at 37°C, and absorbance was measured using Synergy H4 Hybrid Reader (BioTek) fluorometer at 420nM12.
Anti-Plasmodial SYBR Green I - Based Fluorescence (MSF) Assay
D6 (CDC/Sierra Leone) and TM91-C235 (WRAIR, Thailand) laboratory strains of P. falciparum were used for each drug sensitivity assessment. The parasite strains were maintained continuously in long-term cultures as previously described in Johnson et al11, and P. falciparum strains in late-ring or early-trophozoite stages were cultured in predosed 384-well microtiter drug assay plates in 38 μl culture volume per well at a starting parasitemia of 0.3% and a hematocrit of 2%. Pre-dosed, sterile, 384 well black optical bottom microtiter drug plates for use in the MSF assay were produced using a Tecan EVO Freedom Liquid Handling System (Tecan US, Durham, NC). Dose response plates were produced at a final concentration ranging from 0.5 – 10000 ng/ml in quadruplicate (twelve two-fold serial dilutions of each test compound or chloroquine control in DMSO. Each run was validated by a batch control plate with chloroquine (Sigma-Aldrich Co., Catalog #C6628) at a final concentration of 2000 ng/ml. The cultures were incubated for 72 hours at 37°C, 5% CO2, 5% O2 and 90% N2. Lysis buffer (38 μl per well), consisting of 20mM Tris HCl, 5mM EDTA, 1.6% Triton X, 0.016% saponin, and SYBR green I dye at a 20x concentration (Invitrogen, Catalog #S-7567) was then added to the assay plates utilizing the Tecan EVO Freedom system for a final SYBR Green concentration of 10x. Plates were incubated in the dark at room temperature in the dark for 24 hours. Compound activity was assessed by examining for the relative fluorescence units (RFU) per well using the Tecan Genios Plus (Tecan US, Inc., Durham, NC). GraphPad Prism (GraphPad Software Inc., San Diego, CA) using the nonlinear regression (sigmoidal dose-response/variable slope) equation was used to determine IC50 values.
Determination of Static or Cidal Effects. RH-YFP tachyzoites were treated with each compound at 1 μM under one of four conditions: a) parasites were treated for four days, then compound was removed; b) parasites were treated for ten days, then compound was removed; c) compound was refreshed at four days and removed at ten; or d) compound was maintained for the duration of the experiment. The four and ten day time points were taken to reveal the impact of extended exposure of the parasites to the test substance. Compounds were refreshed at four days to examine whether compound degradation could contribute to an observed static effect. Parasite growth was assessed at 11, 17, and 25 days.
In vivo Toxicity and Oocyst assays
HLA B07 transgenic mice were produced at Pharmexa-Epimmune (San Diego, CA, USA) and bred at the University of Chicago. All studies were conducted with Institutional Animal Care and Use Committee at the USDA, the University of Chicago, and the University of Strathclyde.
Infection of mice with T. gondii oocysts
Oocysts were obtained by feeding infected tissues of Swiss Webster mice to cats, sporulated in 2% sulfuric acid on a shaker for one week, and stored at 4°C until used (Dubey 2010). Oocysts were counted in a disposable hemocytometer and diluted 10-fold from 10−1 to 10−7 to reach an end point of ≅ 1 oocyst. All ten-fold dilutions were made in 50 ml tubes with 2% sulfuric acid (5 ml aliquot + 45 ml sulfuric acid), and dilutions were stored at 4° C, to avoid variability in inocula preparations. For inoculation of mice, oocysts from the designated dilution were neutralized with 3.3% sodium hydroxide with neutral red as indicator (approximately the same volume as the inoculum). The resultant mixture was inoculated orally into 5 mice for each dilution (unless indicated otherwise) via a gastric needle with a blunt bulb (22 gauge, 50 mm long, Cadence Science catalogue no. 7920), without washing to avoid variability of the inocula during washing procedure. All orally inoculated mice were housed in autoclavable rodent cages with biohazard signs to incinerate bedding and food for 10 days to avoid spread of T. gondii because some oocysts pass unencysted in mouse feces16.
Bioassay of T. gondii in mice
Mice were observed daily for the duration of the experiment. All mice were examined for T. gondii infection. Impression smears of tissues (usually mesenteric lymph nodes or lungs) were examined microscopically for tachyzoites. Survivors were bled six to eight weeks later and 1:25 dilution of their sera were tested for T. gondii antibodies using the (MAT) as described17. The last infective dilution was considered to have 1 viable organism. The inoculated mice were considered infected with T. gondii when tachyzoites or tissue cysts were found in tissues. Seroconversion at 6 weeks was considered as indication of the presence of live parasites in the inocula. However, brains of all mice that survived 6 weeks were examined for tissue cysts, irrespective of serological results. With the strains of T. gondii used here, tissue cysts are found in all seropositive mice.
Evaluation of efficacy of 14a in a mouse model of T.gondii oocyst infection
Five groups of B7 female mice weighing approximately 25g were used. Group 1 received only 14a (100 mg/kg, 2.5 mg/mL, 1.0 mL) by oral gavage. Group 2 received only 14a (25 mg/kg, 0.5 mg/mL, 1.0 mL) by oral gavage. Group 3 received 14a (100 mg/kg, 2.5 mg/mL, 1.0 mL) and 100 ME49 oocysts by oral gavage. Group 4 received 14a (25 mg/kg, 0.5 mg/mL, 1.0 mL) and 100 ME49 oocysts by oral gavage. Group 5 received only 100 ME49 oocysts by oral gavage.
Evaluation of efficacy of 14b in a mouse model of T.gondii oocyst infection
Five groups of B7 female mice weighing approximately 25g were used. Group 1 received only 14b (100 mg/kg, 2.5 mg/mL, 1.0 mL) by oral gavage. Group 2 received only 14b (25 mg/kg, 0.5 mg/mL, 1.0 mL) by oral gavage. Group 3 received 14b (100 mg/kg, 2.5 mg/mL, 1.0 mL) and 100 TgGoatUS4 oocysts by oral gavage. Group 4 received 14a (25 mg/kg, 0.5 mg/mL, 1.0 mL) and 100 TgGoatUS4 oocysts by oral gavage. Group 5 received only 100 TgGoatUS4 oocysts by oral gavage.
Insertional Mutagenesis Experiments
THdhxgTRP tachyzoites were transfected with pLK47 vector plasmid12. Parasites were extracted from HFF cells and resuspended in 1mL cytomix electroporation buffer solution (120mM KCl, 150uM CaCl2, 5mM K2HPO4, 5mM KH2PO4, 25mM HEPES, 2mM EDTA and 5mM MgCl2 in sterile H2O). Equivalent amounts of plasmid DNA and cytomix solution containing 10 × 107 parasites were combined for a total volume of 400uL and electroporated using BioRad electroporator at 1.5k, 25fF and 100 Ω. This resulted in a random insertion of the plasmid in the parasite genome and random disruption of a gene. Successfully transfected parasites were selected by month-long treatment with mycophenolic acid (25ug/mL) and xanthine (50ug/mL). After one month, the mixed mutant population was exposed to 3i, 3j, 7a or 14b to select for those mutants whose gene disruption conferred resistance to the drug. No parasite growth was noted after six weeks of exposure to mycophenolic acid, xanthine and any of the four test compounds.
Acknowledgments
We thank Dr. Laura Knoll (University of Wisconsin) for providing the plasmid pLK47 construct used generate insertional mutants, Kristen Wroblewski for assistance in statistical analyses of in vivo studies, Daniel Lee for assistance with preparation of the manuscript, Dr. Kaipeen Yang (Snowdon, Inc.) for expert technical assistance in the synthesis of test compounds, and Boris Streipen, (University of Georgia) for providing RH-YFP parasites. This work was supported by NIAID-AT-SBIR 1R43AI078763-01A1, and by Institute for Genomics, Genetics, and Systems Biology, University of Chicago, and by gifts from the Mann and Cornwell, Taub, Rooney-Alden, Engel, Pritzker, Harris, Zucker, and Mussilami families. SPM is supported by an MRC Career Development fellowship. We thank K. Wroblewski for assistance with statistical analyses.
Abbreviations
- CDC
Centers for Disease Control and Prevention
- DIEA
diisopropylethylamine
- DMAP
dimethylaminopyridine
- DMSO
dimethylsulfoxide
- ESI
electrospray interface
- EtOAc
ethyl acetate
- FDA
Food and Drug Administration
- FIBS
fibroblasts
- HATU
and O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HFF
human foreskin fibroblasts
- IC
inhibitory concentration
- IC50
half-maximal inhibitory concentration
- IC90
concentration necessary to achieve 90% inhibition
- IMDM-C
Iscove’s Modified Dulbecco’s Media
- LC-MS
high performance liquid chromatography-mass spectral analysis
- m/z
mass to charge ratio
- MAT
modified agglutination test
- MeCN
acetonitrile
- MIC
minimum inhibitory concentration
- MSF
Malaria SYBR Green I - Based Fluorescence Assay
- OD
optical density
- P. falciparum
Plasmodium falciparum
- P/S
pyrimethamine and sulfadiazine in combination
- RH-YFP
tachyzoites expressing YFP
- SYBR
SYBR Green Dye I
- T. gondii
Toxoplasmosis gondii
- Tr
chromatographic retention time
- WST-1
water-soluble tetrazolium salt 1
- YFP
yellow fluorescent protein
Footnotes
Disclaimers. The project described was supported by Award Number R43AI078763 from the National Institute of Allergy and Infectious Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. The opinions of researchers at Walter Reed Army Institute of Research are their own and do not reflect the views of the US Army or the Department of Defense.
There are no conflicts of interest.
References
- 1.McLeod R, Boyer K, Karrison T, Kasza K, Swisher C, Roizen N, Jalbrzikowski J, Remington J, Heydemann P, Noble AG, Mets M, Holfels E, Withers S’, Latkany P, Meier P. Outcome of treatment for congenital toxoplasmosis, 1981–2004: the National Collaborative Chicago-Based, Congenital Toxoplasmosis Study. Clin Infect Dis. 2006;42:1383–1394. doi: 10.1086/501360. [DOI] [PubMed] [Google Scholar]
- 2.Roizen N, Kasza K, Karrison T, Mets M, Noble AG, Boyer K, Swisher C, Meier P, Remington J, Jalbrzikowski J, McLeod R, Kipp M, Rabiah P, Chamot D, Estes R, Cezar S, Mack D, Pfiffner L, Stein M, Danis B, Patel D, Hopkins J, Holfels E, Stein L, Withers S, Cameron A, Perkins J, Heydemann P. Impact of visual impairment on measures of cognitive function for children with congenital toxoplasmosis: implications for compensatory intervention strategies. Pediatrics. 2006;118:e379–390. doi: 10.1542/peds.2005-1530. [DOI] [PubMed] [Google Scholar]
- 3.McLeod R, Khan AR, Noble GA, Latkany P, Jalbrzikowski J, Boyer K. Severe sulfadiazine hypersensitivity in a child with reactivated congenital toxoplasmic chorioretinitis. Pediatr Infect Dis J. 2006;25:270–272. doi: 10.1097/01.inf.0000202070.59190.9a. [DOI] [PubMed] [Google Scholar]
- 4.NDA #018669 (Bayer)
- 5.Weinbach EC, Garbus J. Mechanism of action of reagents that uncouple oxidative phosphorylation. Nature. 1969;221:1016–1018. doi: 10.1038/2211016a0. [DOI] [PubMed] [Google Scholar]
- 6.Niclocide package insert (Miles—US), Rev 8/88, Rec 4/89.
- 7.Chang Y, Wu Y, Hsieh H, Chen C, Chen C. Journal of Food Drug Analysis. 2006;14:329–333. [Google Scholar]
- 8.McEvoy GK, editor. AHFS Drug information 88. Bethesda, MD: American Society of Hospital Pharmacists; 1988. pp. 40–41. [Google Scholar]
- 9.a) 3079297 US Patent. ; b) Waisser K, Hladuvkova J, Kunes J, Kubicova L, Klimesova V, Karajannis P, Kaustova J. Synthesis and antimycobacterial activity of salicylanilides substituted in position 5. Chemical Papers. 2001;55(2):121–129. [Google Scholar]
- 10.Plouffe D, Brinker A, McNamara C, Henson K, Kato N, Kuhen K, Nagle A, Adrian F, Matzen JT, Anderson P, Nam TG, Gray NS, Chatterjee A, Janes J, Yan SF, Trager R, Caldwell JS, Schultz PG, Zhou Y, Winzeler EA. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc Natl Acad Sci U S A. 2008;105:9059–9064. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson JD, Dennull RA, Gerena L, Lopez-Sanchez M, Roncal NE, Waters NC. Assessment and Continued Validation of the Malaria SYBR Green I-Based Fluorescence Assay for Use in Malaria Drug Screening. Antimicrob Agents Chemother. 2007;51:1926–1933. doi: 10.1128/AAC.01607-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fomovska A, Huang Q, El Bissati K, Mui EJ, Witola WH, Cheng G, Zhou, Sommerville C, Roberts CW, Bettis S, Prigge ST, Afanador GA, Hickman MR, Lee PJ, Leed SE, Auschwitz JM, Pieroni M, Stec J, Muench SP, Rive DW, Kozikowski AP, Mcleod R. Novel N-Benzoyl-2-hydroxybenzamide Disrupts Unique Parasite Secretory Pathway. Antimicrob Agents Chemother. 2012;56:2666–2682. doi: 10.1128/AAC.06450-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mui EJ, Jacobus D, Milhous WK, Schiehser G, Hsu H, Roberts CW, Kirisits MJ, McLeod R. Triazine inhibits Toxoplasma gondii tachyzoites in vitro and in vivo. Antimicrob Agents Chemother. 2005;49:3463–3467. doi: 10.1128/AAC.49.8.3463-3467.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lunde MN, Jacobs L. Antigenic differences between endozoites and cystozoites of Toxoplasma gondii. Journal of Parasitology. 1983;69:806–808. [PubMed] [Google Scholar]
- 15.Dubey JP, Rajendran C, Ferreira LR, Martins J, Kwok OCH, Hill DE, Villena I, Zhou H, Su C, Jones JL. High prevalence and genotypes of Toxoplasma gondii isolated from goats from a retail meat store destined for human consumption in the USA. International Journal for Parasitology. 2011;41:827–833. doi: 10.1016/j.ijpara.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 16.Dubey JP, Frenkel JK. Experimental Toxoplasma infection in mice with strains, producing oocysts. Journal of Parasitology. 1973;59:505–512. [PubMed] [Google Scholar]
- 17.Dubey JP, Desmonts G. Serological responses of equids fed Toxoplasma gondii oocysts. Equine Veterinary Journal. 1987;19:337–339. doi: 10.1111/j.2042-3306.1987.tb01426.x. [DOI] [PubMed] [Google Scholar]