

A Tale of Two Michaels
The first enantioselective total synthesis of (−)-GB17 is reported. Construction of this unique naphthoquinolizinone skeleton was achieved by two stereoselective intramolecular Michael additions. The first of these Michael additions is controlled by the use of a chiral organocatalyst, while the second cyclization is under substrate control and proceeds with concomitant lactam formation.
Keywords: Galbulimima Alkaloids, Natural Product Synthesis, Michael Addition
The Galbulimima alkaloids are isolated from the bark of rain forest trees belonging to the Galbulimima genus that grow in regions of Northern Australia, Papua New Guinea and Indonesia.[1] Significant structural diversity exists within this alkaloid family,[2] but common to all members is the presence of a piperidine ring and a trans-decalin carbocylic core (Figure 1). Investigations into the biological activity of the Galbulimima alkaloids have focused in large part on himbacine (1): a potent muscarinic receptor antagonist that might serve as a lead for the development of therapeutic agents to treat Alzheimer’s disease.[3] Research at Schering–Plough centered on himbacine led to SCH 530348 (now Vorapaxar at Merck),[4] a promising thrombin receptor agonist whose clinical trial for treating acute coronary symdrome was halted in 2011 due to adverse side-effects.
Figure 1.
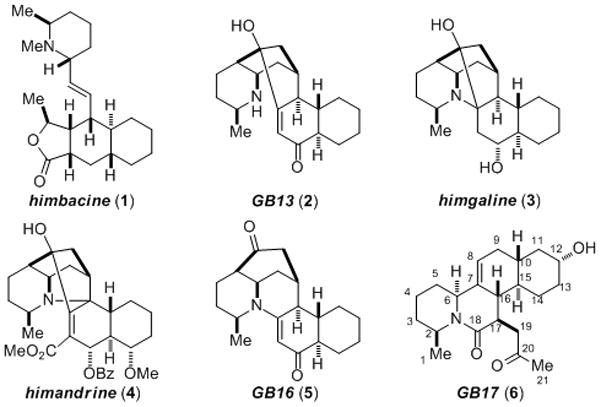
Selected Galbulimima Alkaloids
The combination of fascinating molecular structure and significant therapeutic potential has made the Galbulimima alkaloids attractive targets for total synthesis.[5] While early work focused on himbacine (1), most recent efforts have been directed at preparing the more complex members of the family. The first synthesis of GB13 (2) was reported by Mander and MacLachlan in 2003,[6a] with subsequent reports from the groups of Movassaghi, Chackalamannil, Evans, Sarpong and Ma.[6b–f] Himgaline (3) can be synthesized from GB13 (2), as shown by both Chackalamannil[6c] and Evans.[6e] Movassaghi and coworkers reported the only synthesis of himandrine (4) in 2009,[7] while the group of Ma reported the only synthesis of GB16 (5) that same year.[8] Our interest was drawn to GB17 (6), which was one of 19 new Galbulimima alkaloids whose isolation was reported by Taylor and coworkers in 1965.[1a] At that time only an elemental formula was reported – the structure of GB17 was only revealed in 2009 when Mander and coworkers reported an X-ray crystal structure.[2e] GB17 (6) possesses an unusual naphthoquinolizinone ring system that is unique amongst the family. We were drawn to the 1,4-dicarbonyl within 6 that we hoped might be formed by oxidative enolate coupling.[9] Herein we report the first enantioselective total synthesis of GB17 (6) through a concise and flexible strategy.
Our approach to GB17 (6) is outlined in retrosynthetic format in Scheme 1. We envisioned the C8 ketone 7 as a suitable precursor to 6 by way of carbonyl reduction and a late-stage oxidative enolate coupling to install the C17 2-propanone substituent. This allowed for the retrosynthetic clearing of several stereocenters and three of the compound’s four rings through application of two intramolecular Michael addition transforms. Thus, we arrived at aldehyde 10 as our first subtarget en route to GB17 (6).
Scheme 1.
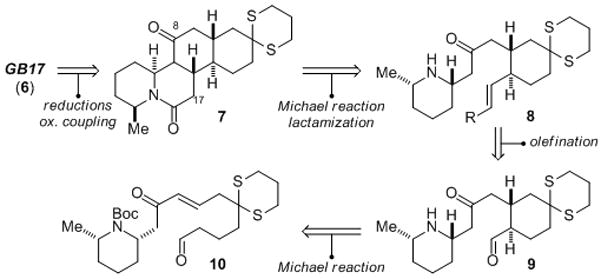
Retrosynthetic Analysis of (−)-GB17
The conversion of aldehyde 10 into the desired trans-cyclohexane 9 required the development of a stereoselective catalyst-controlled intramolecular Michael addition. We therefore conducted an initial model study utilizing readily prepared aldehyde 12 (Scheme 2).[10] Related catalytic intramolecular Michael additions had been reported for the analogous cyclopentanes by the groups of List and Hayashi.[11] A survey of several available organocatalysts revealed that the diphenylprolinol derivative D[12] provided the highest levels of both diastereo and enantiocontrol for the synthesis of cyclohexane 13 (20:1 dr, 99% ee). This reaction could be conducted on a 5 g scale using 5 mol% of D to provide essentially quantitative yield of the carbocycle 13, which could be converted into the potentially useful decalin synthon 14.[13] Interestingly, very poor results were obtained during attempts to carry out the intramolecular Michael reaction without the dithiane present, an outcome we interpret as arising from a lack of a favorable Thorpe–Ingold effect that is present in dithiane 12. In contrast, the corresponding cyclopentane reactions reported by List and Hayashi[11] do not seem to require such an effect.
Scheme 2.
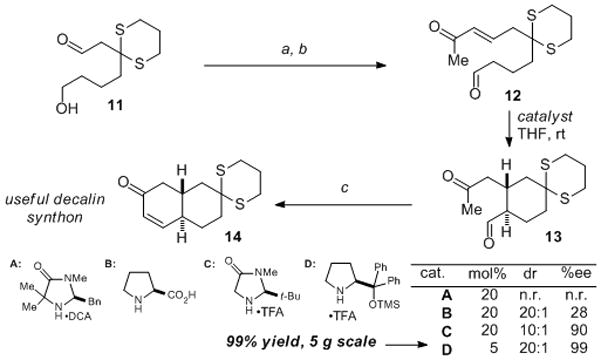
a) Dimethyl 2-oxopropylphosphonate (1.3 equiv), LiCl (2.3 equiv), iPr2EtN (1.8 equiv), 91%; b) (COCl)2 (1.6 equiv), Me2S(O) (4.2 equiv), Et3N (9.5 equiv), 89%; c) TsOH•H2O (10 mol%), MgSO4 (2.6 equiv), PhMe, 100 °C, 91%. Ts = p-toluenesulfonyl. D C A = dichloroacetic acid. TFA = trifluoroacetic acid.
With this valuable insight into our proposed catalyst-controlled cyclization, we began our GB17 (6) synthesis (Scheme 3). The enantioenriched phosphonate 16 was prepared from known cis-piperidine 15[14] in three steps. Condensation of 16 with the previously prepared aldehyde 11, under Masamune–Roush conditions[15] followed by oxidation, delivered the desired aldehyde 10 and set the stage for the first of our planned cyclization events. Exposure of aldehyde 10 to 5 mol% of organocatalyst ent-D under our previously developed conditions smoothly generated the desired trans-substituted cyclohexane in excellent yield (99%). A Wittig reaction of aldehyde 17 gave rise to the trans-enoate 18 in high yield (91%) and with >20:1 stereoselectivity. This sequence (i.e., 10 → 18) could be carried out in one-pot with no loss in yield or selectivity. At this juncture we set out to explore conditions to promote the second planned intramolecular Michael addition. Potassium tert-butoxide was found to be effective for this reaction, affording the desired decalin ring 19 in high yield as a 16:2:1:1 mixture of diastereomers. The major isomer could be readily separated (80% yield) and was converted into the tetracyclic lactam 20 by removal of the Boc-group and cyclization using Cs2CO3 in methanol at reflux. X-ray analysis of a single crystal of 20[13] revealed that the Michael addition had proceeded with the undesired sense of stereochemistry at C16 and that the C6 position of the piperidine had isomerized. 2D-NMR studies indicated that the piperidine inversion occurred under the conditions required to induce lactamization. This isomerization presumably occurred through an E1cB elimination/conjugate addition sequence of the β-disposed nitrogen, and while potentially problematic for our synthesis, more pressing was the issue of the C16 stereochemistry.
Scheme 3.
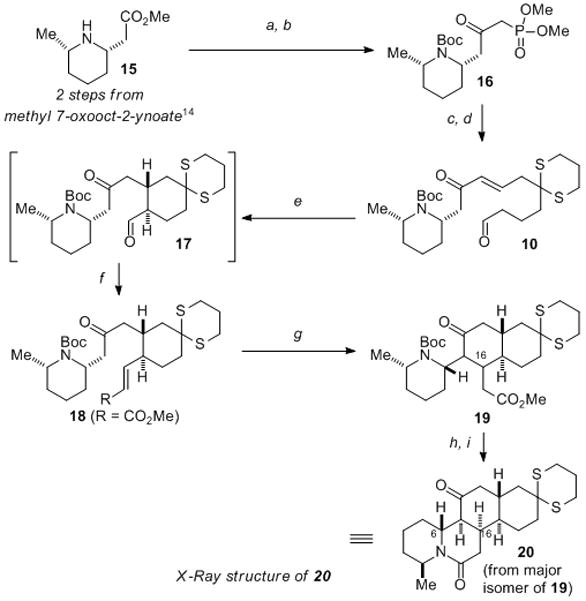
a) Dimethyl methylphosphonate (2.1 equiv), n-BuLi (2.1 equiv); b) i. (Boc)2O (3.0 equiv), Et3N (3.0 equiv), DMAP (9 mol%); ii. K2CO3 (1.0 equiv), MeOH, 75% from 15; c) iPr2EtN (2.0 equiv), LiCl (2.0 equiv), 11 (0.97 equiv), 99%; d) DMP (1.2 equiv), Py (7.0 equiv), 85%; e) cat. D (5 mol%), THF, 99%; f) Methyl (triphenylphosphoranylidene)acetate (1.3 equiv), THF, reflux, (91% from 10); g) KOt-Bu (15 mol%), MeOH, (80% of major isomer); h) CF3CO2H; i) Cs2CO3 (1.0 equiv), MeOH, reflux, 44% from 19. Boc = tert-butylcarbonyl, Py = pyridine, DMP = Dess–Martin periodinane.
We had anticipated that the correct C16 stereochemistry would be delivered from 18 by virtue of the enoate adopting a pseudo equatorial conformation I (Figure 2), but this leads to unfavorable interactions with the piperidine ring making the axial conformer II more favorable. Based upon the model proposed in Figure 2, we hypothesized that the corresponding cis-enoate might provide the desired C16 stereochemistry via conformer III; the corresponding axial conformer IV would now be significantly higher in energy due to severe allylic strain. It also appeared that the carbamate-protecting group enforced a 1,3-diaxial relationship between the substituents of the piperidine ring, giving rise to unfavorable steric interactions in the transition state leading to the desired isomer of tetracycle 19. It appeared to us, therefore, that a substrate possessing a cis-enoate and free piperidine nitrogen would significantly favor the desired stereochemical outcome.
Figure 2.

Models for the Intramolecular Michael Addition.
Our modified approach commenced with a Still–Gennari olefination[16] of aldehyde 17 to afford enoate 21 in 80% isolated yield (Scheme 3). Attempted cyclization of 21 using potassium tert-butoxide gave rise to a mixture of stereoisomeric products, with the major product corresponding to the same major isomer obtained from the cyclization of the trans-enoate 18. This result confirmed our suspicions regarding the impact of the Boc-group, and it was therefore removed by treatment with TFA. While exposure of this free piperidine to potassium tert-butoxide led to significant decomposition and no desired cyclization products, the use of sodium methoxide in methanol produced a remarkable transformation. Under these conditions, the desired Michael addition proceeded smoothly, and the intermediate ester 22 underwent lactamization in situ to produce the desired tetracyclic framework of GB17 (i.e., 23). A combined yield of 70% of the desired C16 isomers, differing only at C7, was obtained under these conditions. X-ray crystallographic analysis established the structure of 23[13] thereby confirming the stereochemistry of the Michael addition unambiguously (Scheme 3). It therefore appeared that the C16 stereocenter of 23 was set via conformer III (Figure 2) as we had hypothesized, but also that this stereochemistry allowed lactamization of 22 to proceed without isomerization of the C6 piperidine stereocenter.[17]
Our endgame sequence called for the conversion of the C8 ketone within 23 and 7-epi-23 into the requisite Δ7,8 alkene. This transformation was readily achieved by generation of the corresponding enol triflate of 23 via the more thermodynamically stable enolate, followed by palladium-catalyzed reduction (80% yield of 24, two steps).[18] Conversion of epi-23 into the corresponding enol triflate was problematic. Fortunately, we found that epi-23 could be quantitatively equilibrated upon reexposure to NaOMe to a 3:2 mixture of 23 and epi-23, allowing for easy recycling of material. From alkene 24, installation of the equatorially disposed C12 hydroxyl group proceeded first by cleavage of the thioketal,[19] then subsequent reduction of the thus formed ketone along the axial vector using sodium borohydride. Protection of the hydroxyl group as its TBS ether 25 was carried out as a prelude to installation of the propanone substituent (61% from 24). Disappointingly, our attempts to utilize oxidative enolate coupling to install the propanone unit were met with difficulties. Under a variety of conditions[20] we did not observe formation of the desired adduct, and so made recourse to an alternative strategy. We found alkylation of lactam 24 using NaHMDS and methallyl bromide provided the desired product 26 in 75% yield, along with 13% of the undesired axial epimer. Cleavage of the thioketal (66%) followed by reduction using sodium borohydride installed the requisite equatorial C12 hydroxyl group. Lastly, oxidative cleavage of the methallyl group afforded synthetic GB17 (6) that was spectroscopically identical to a natural sample (1H-NMR, 13C-NMR, HRMS).
Our stereoselective synthesis of the tetracyclic alkaloid GB17 was executed through a route utilizing two intramolecular Michael additions to forge key stereochemical elements. The development of the diphenylprolinol-catalyzed enantioselective intramolecular Michael addition was crucial to the success of the synthesis, and will likely find further applications to other natural product targets. The second intramolecular Michael addition provided an example of the profound effect enoate geometry can have during a substrate-controlled conjugate addition.[21] Future research on GB17 will focus on exploring its biological activity, which at this time remains unknown.
Supplementary Material
Scheme 4.
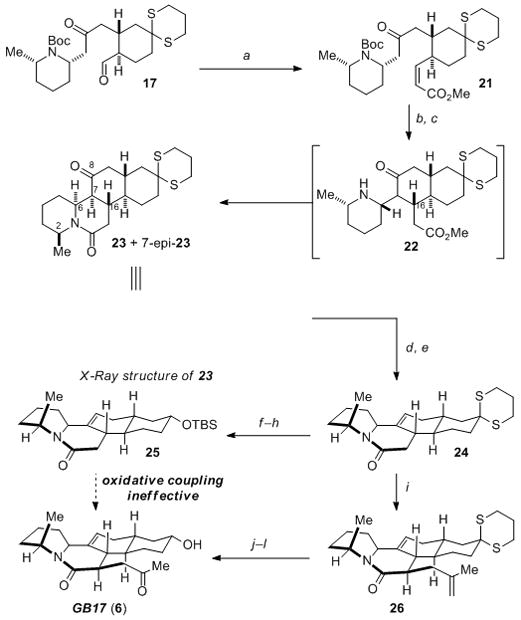
a) LiCl (2.0 equiv), iPr2EtN (2.0 equiv), methyl 2-(bis(2,2,2-trifluoroethoxy)phosphoryl)ethanoate (1.2 equiv), 80%; b) CF3CO2H; c) NaOMe (6.0 equiv), MeOH, 70% 23 + epi-23 from 21; d) NaHMDS (1.0 equiv), Tf2NPh (1.1 equiv), 86%; e) Pd(OAc)2 (7 mol%), Ph3P (14 mol%), HCO2H (2.0 equiv), Bu3N (3.0 equiv), 99%; f) Selectfluor (2.5 equiv), 69%; g) NaBH4 (1.5 equiv); h) TBSCl (8.0 equiv), imidazole (16 equiv), DMF, 89% over 2 steps; i) NaHMDS (4.5 equiv), 3-bromo-2-methylprop-1-ene (1.2 equiv), 75% 26 + 13% epi-26; j) f) Selectfluor (2.5 equiv), 66%; k) NaBH4 (4.5 equiv), 99%; l) OsO4 (1.4 mol%), NMO (1.0 equiv); NaIO4 (2.0 equiv), H2O, 50%. HMDS = hexamethyldisilazide, Tf = trifluoromethanesulfonyl, Selectfluor = 1-Chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate), NMO = N-methylmorpholine N-oxide.
Footnotes
This work was supported in part by Northwestern University (NU), Amgen, and the National Institutes of Health (1R01GM085322). We thank Professor Lewis N. Mander (The Australian National University) for providing an authentic sample of (−)-GB17.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Binns SV, Dunstan PJ, Guise GB, Holder GM, Hollis AF, McCredie RS, Pinhey JT, Prager RH, Rasmussen M, Ritchie E, Taylor WC. Aust J Chem. 1965;15:569–573. [Google Scholar]; b) Ritchie E, Taylor WC. In: The Alkaloids. Manske RHF, Holmes HI, editors. Vol. 9. Academic Press; New York: 1967. pp. 529–543. [Google Scholar]; c) Ritchie E, Taylor WC. In: The Alkaloids. Manske RHF, Holmes HI, editors. Vol. 13. Academic Press; New York: 1971. pp. 227–271. [Google Scholar]
- 2.Structural determination of some GB alkaloids. Himbacine: Pinhey JT, Ritchie E, Taylor WC. Aust J Chem. 1961;14:106–134.GB13: Mander LN, Prager RH, Rasmussen M, Ritchie E, Taylor WC. Aust J Chem. 1967;20:1473–1491.Himgaline: Mander LN, Prager RH, Rasmussen M, Ritchie E, Taylor WC. Aust J Chem. 1967;20:1705–1718.Himandrine: Guise GB, Mander LN, Prager RH, Rasmussen M, Ritchie E, Taylor WC. Aust J Chem. 1967;20:1026–1035.GB16 and GB17: Mander LN, Willis AC, Herlt AJ, Taylor WC. Tetrahedron Lett. 2009;50:7089–7092.
- 3.a) Darroch SA, Taylor WC, Choo LK, Mitchelson F. Eur J Pharmacol. 1990;782:131–136. doi: 10.1016/0014-2999(90)90501-v. [DOI] [PubMed] [Google Scholar]; b) Miller JH, Aagaard PJ, Gibson VA, McKinney MJ. Pharmacol Exp Ther. 1992;263:663–667. [PubMed] [Google Scholar]
- 4.Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS, Boykow G, Hsieh Y, Palamanda J, Agans-Fantuzzi J, Kurowski S, Graziano M, Chintala M. J Med Chem. 2008;51:3061–3064. doi: 10.1021/jm800180e. [DOI] [PubMed] [Google Scholar]
- 5.For a review of synthetic work, see: Rinner U, Lentsch C, Aichinger C. Synthesis. 2010;22:3763–3784.Bhattacharyya D. Tetrahedron. 2011;67:5525–5542.
- 6.a) Mander LN, McLachlan MM. J Am Chem Soc. 2003;125:2400–2401. doi: 10.1021/ja029725o. [DOI] [PubMed] [Google Scholar]; b) Movassaghi M, Hunt DK, Tjandra M. J Am Chem Soc. 2006;128:8126–8127. doi: 10.1021/ja0626180. [DOI] [PubMed] [Google Scholar]; c) Shah U, Chackalamannil S, Ganguly AK, Chelliah M, Kolotuchin S, Buevich A, McPhail A. J Am Chem Soc. 2006;128:12654–12655. doi: 10.1021/ja065198n. [DOI] [PubMed] [Google Scholar]; d) Evans DA, Adams DJ. J Am Chem Soc. 2007;129:1048–1049. doi: 10.1021/ja0684996. [DOI] [PubMed] [Google Scholar]; e) Larson KK, Sarpong R. J Am Chem Soc. 2009;131:13244–13254. doi: 10.1021/ja9063487. [DOI] [PubMed] [Google Scholar]; f) Zi W, Yu S, Ma D. Angew Chem Int Ed. 2010;49:5887–5890. doi: 10.1002/anie.201002299. [DOI] [PubMed] [Google Scholar]
- 7.Movassaghi M, Tjandra M, Qi J. J Am Chem Soc. 2009;131:9648–9650. doi: 10.1021/ja903790y.For an approach towards himandrine, see also: O’Connor PD, Mander LN, McLachlan MM. Org Lett. 2004;6:703–706. doi: 10.1021/ol036308n.O’Connor PD, Del Signore G, McLachlan MMW, Willis AC, Mander LN. Aust J Chem. 2010;63:1477–1491.
- 8.Zi W, Yu S, Ma D. Chem Asian J. 2011;6:573–579. doi: 10.1002/asia.201000556.See also reference 6f.
- 9.For recent examples of oxidative enolate coupling in natural product synthesis see, see: Konkol LC, Guo F, Sarjeant AA, Thomson RJ. Angew Chem, Int Ed. 2011;50:9931–9934. doi: 10.1002/anie.201104726.Herzon SB, Lu L, Woo CM, Gholap SL. J Am Chem Soc. 2011;133:7260–7263. doi: 10.1021/ja200034b.Martin CL, Overman LE, Rohde JM. J Am Chem Soc. 2010;132:4894–4906. doi: 10.1021/ja100178u.Clift MD, Thomson RJ. J Am Chem Soc. 2009;131:14579–14583. doi: 10.1021/ja906122g.Baran PS, Hafensteiner BD, Ambhaikar NB, Guerrero CA, Gallagher JD. J Am Chem Soc. 2006;128:8678–8693. doi: 10.1021/ja061660s.Baran PS, Guerrero CA, Ambhaikar NB, Hafensteiner BD. Angew Chem, Int Ed. 2005;44:606–609. doi: 10.1002/anie.200461864.
- 10.Prepared in three steps from dithiane. See Supporting Information for details.
- 11.a) Hechavarria Fonseca MT, List B. Angew Chem Int Ed. 2004;43:3958–3960. doi: 10.1002/anie.200460578. [DOI] [PubMed] [Google Scholar]; b) Hayashi Y, Gotoh H, Tamura T, Yamaguchi H, Masui R, Shoji M. J Am Chem Soc. 2005;127:16028–16029. doi: 10.1021/ja055740s. [DOI] [PubMed] [Google Scholar]
- 12.a) Hayashi Y, Gotoh H, Hayashi T, Shoji M. Angew Chem, Int Ed. 2005;44:4212–4215. doi: 10.1002/anie.200500599. [DOI] [PubMed] [Google Scholar]; b) Marigo M, Wabnitz TC, Fielenbach D, Jørgensen KA. Angew Chem, Int Ed. 2005;44:794–797. doi: 10.1002/anie.200462101. [DOI] [PubMed] [Google Scholar]
- 13.CCDC 854644–854646 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Supporting information for this manuscript contains details on the absolute configuration of 14.
- 14.Calvet-Vitale S, Vanucci-Bacqué C, Fargeau-Bellassoued MC, Lhommet G. Tetrahedron. 2005;61:7774–7782.For an alternative approach, see: Munchhof MJ, Meyers AI. J Am Chem Soc. 1995;117:5399–5400.
- 15.Blanchette MA, Choy W, Davis JT, Essenfeld AP, Masamune S, Roush WR, Sakai T. Tetrahedron Lett. 1984;25:2183–2186. [Google Scholar]
- 16.Still WC, Gennari C. Tetrahedron Lett. 1983;24:4405–4408. [Google Scholar]
- 17.When these same conditions were employed on the trans-enoate 18 (i.e., TFA followed by NaOMe), the same one-pot double cyclization ensued to deliver an approximate 1:1 ratio of the desired C16 stereoisomer 23 to the undesired isomer 20 in a combined yield of 50%. Interestingly, it appears that piperidine inversion is a prerequisite for lactamization of the incorrect C16 epimer.
- 18.Peterson GA, Kunng E, McCallum JS, Wulff WD. Tetrahedron Lett. 1987;28:1381–1384. [Google Scholar]
- 19.Liu J, Wong CH. Tetrahedron Lett. 2002;43:4037–4039. [Google Scholar]
- 20.The conditions of both Saegusa and Baran did not provide any desired product from lactam 25 in our hands. For representative conditions, see: Ito Y, Konoike T, Saegusa T. J Am Chem Soc. 1975;97:2912–2914.Ito Y, Konoike T, Harada T, Saegusa T. J Am Chem Soc. 1977;99:1487–1493.Baran PS, DeMartino MP. Angew Chem Int Ed. 2006;45:7083–7086. doi: 10.1002/anie.200603024.DeMartino MP, Chen K, Baran PS. J Am Chem Soc. 2008;130:11546–11560. doi: 10.1021/ja804159y.
- 21.For a review on intramolecular Michael additions, see: Little RD, Masjedizadeh MR, Wallquist O, McLoughlin JI. In: Organic Reactions. Paquett LA, editor. Vol. 47. John Wiley & Sons, Inc; New York: 1995. pp. 315–350.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.