

Abstract
Although arachidonic acid cascade has been shown to be involved in sepsis, little is known about the role of prostaglandin D2 and its newly found receptor, chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2), on the septic response. Severe sepsis is associated with the failure of neutrophil migration. To investigate whether CRTH2 influences neutrophil recruitment and the lethality during sepsis, sepsis was induced by cecal ligation and puncture (CLP) surgery in mice. CRTH2 knockout (−/−) mice were highly resistant to CLP-induced sepsis, which was associated with lower bacterial load and lower production of TNF-α, IL-6, and CCL3. IL-10, an anti-inflammatory cytokine, was higher in CRTH2−/− mice, blunting CLP-induced lethality in CRTH2−/− mice. Neutrophil accumulation in the peritoneum was more pronounced after CLP in CRTH2−/− mice, which was associated with higher CXCR2 level in circulating neutrophils. Furthermore, sepsis caused a decrease in the level of acetylation of histone H3, an activation mark, at the CXCR2 promoter in WT neutrophils, suggesting that CXCR2 expression levels are epigenetically regulated. Finally, both pharmacological depletion of neutrophils and inhibition of CXCR2 abrogated the survival benefit in CRTH2−/− mice. These results demonstrate that genetic ablation of CRTH2 improved impaired neutrophil migration and survival during severe sepsis, which was mechanistically associated with epigenetic mediated CXCR2 expression. Thus, CRTH2 is a potential therapeutic target for polymicrobial sepsis.
Introduction
Neutrophils are key cells in the innate immune response during sepsis, releasing cytokines, antimicrobial proteins, and phagocytizing invading microbes. Neutrophil migration into the infectious site has been shown to be markedly impaired during severe sepsis, which is associated with downregulation of the chemokine receptor CXCR2, resulting in the failure of pathogen clearance and worsening severity (1–3). Therefore, investigating neutrophil function during sepsis is especially important for further understanding the pathophysiology of sepsis.
Arachidonic acid, a major lipid mediator, is released from membrane phospholipids by phospholipase A2 (PLA2). Then prostaglandin (PG) G2 is synthesized from arachidonic acid by cyclooxygenase (COX), followed by the production of thromboxane A2 and prostaglandins (4). PGD2, a major prostanoid produced mainly by mast cells, has been implicated in the pathogenesis of allergic diseases (5). PGD2 is also released from Th2 cells, dendritic cells, and macrophage-like cell lines in response to various inflammatory stimuli (6–8). The released PGD2 can bind to two G protein-coupled receptors: the D prostanoid receptor (DP1) and the chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2). CRTH2 has been shown to be expressed on various human immune cells including Th2 cells, a subpopulation of CD8+ T cells, basophils, eosinophils, but not on human Th1 cells (9–12). Meanwhile mouse CRTH2 is expressed on Th1 cells, Th2 cells, eosinophils, monocytes, and neutrophils (13, 14). CRTH2 plays major roles on activation and chemotaxis of Th2 cells, eosinophils, and basophils (6). In addition, CRTH2 contributes to double-strand RNA-induced enhancement of airway inflammation (15). Interestingly, PGD2 and one of its metabolites, 15-deoxy-12,14-PGJ2 (15d-PGJ2), are both ligands for CRTH2. 15d-PGJ2 is also the ligand for peroxisome proliferator-activated receptor gamma (PPAR-γ) (16).
Products of the arachidonic acid cascade have been shown to be involved in sepsis (17). For example, the secretory form of PLA2 is induced in septic mice and its inhibition improved survival (18). PGE2 (19), PGI2 (20), and thromboxane A2 (21) also have been shown to play important roles in sepsis. However, the role of PGD2 and its receptor CRTH2 in sepsis is largely unknown.
Here, we investigated the role of CRTH2 on the pathogenesis of polymicrobial sepsis using the cecal ligation and puncture (CLP) mouse model. We observed that CRTH2−/− mice were highly resistant to sepsis, which was associated with lower bacterial load, lower production of proinflammatory cytokines, and enhanced neutrophil accumulation via an epigenetic based mechanism that maintained the expression of CXCR2.
Materials and Methods
Mice
Female WT BALB/c mice (7–10 weeks old) were purchased from Charles River Laboratories Japan, Inc (Yokohama, Japan). CRTH2 knockout (−/−) mice (22) on BALB/c background were bred in the Keio University Animal Research Facility. Mice were housed under specific pathogen-free conditions, and all animal experiments were approved by the Animal Use Committee at Keio University School of Medicine.
In vivo experimental sepsis model induced by cecal ligation and puncture
CLP surgery was performed as previously described (23, 24). Briefly, after ligation, the cecum was punctured five or seven times with a 21-gauge needle for survival studies, and five times for other studies. For survival studies, CLP mice were monitored for indicated periods after CLP surgery. In some experiments, mice were anesthetized, bled, and euthanized at specific time points after CLP surgery. Peritoneal lavage fluid also was collected with 3 ml of sterile PBS and a portion was used for bacterial culture. The collected lavage fluid was centrifuged at 500 g for 6 min at 4°C. The supernatant was stored at −80°C for ELISA and the pellet containing peritoneal exudate cells was lysed with hypotonic buffer to remove RBCs and the total cell number was counted with a hematocytometer. Differential cell counts were analyzed by Diff-Quick staining method. The whole blood obtained by cardiac puncture was centrifuged at 8000 rpm for 8 min at 4°C. The supernatant was collected and stored at -80°C until use for ELISA.
For in vivo neutralization of IL-10 or IL-12, WT mice intraperitoneally received either 250 µg of rabbit anti-mouse monoclonal antibody to IL-10 or to IL-12 (R–D systems, Minneapolis, MN) or rabbit IgG control (R–D systems). After 1 h, mice were subjected to CLP surgery and the survival was monitored for 96 h. For pharmacological neutrophil depletion studies, mice received 250 µg of antibodies to mouse Gr-1 (clone RB6-8C5; R&D systems) or mouse Ly-6G (clone 1A8; Biolegend, San Diego, CA) 24 h before CLP surgery. For CXCR2 inhibition studies, mice received intraperitoneally, 10 mg/kg of SB225002 (Cayman Chemical, Ann Arbor, MI), a CXCR2 antagonist. After 1 h, mice were subjected to CLP surgery. The dose of all agents in these studies was determined according to previous reports (25–27). In addition, we confirmed the antibody-mediated IL-10 depletion using ELISA. The degree of inhibition of IL-10 level was more than 78% (data not shown).
Enzyme-linked immunosorbent assay
The level of PGD2 and 15d-PGJ2 was measured using a PGD2-MOX Express EIA Kit (Cayman Chemical) and a 15d-PGJ2 ELISA Kit (Assay designs, Ann Arbor, MI), respectively. The expression of CXCL1 (KC) and CXCL2 (MIP-2α) was measured by ELISA (R–D systems). The detection limit of PGD2, 15d-PGJ2, CXCL1, and CXCL2 was 16 pg/ml, 36.8 pg/ml, 2.0 pg/ml, and 1.5 pg/ml, respectively.
Quantitative real time PCR
Total RNA was isolated using the RNeasy Mini kit (Qiagen, Valencia, CA). Reverse transcription was performed as previously described (24). Quantitative real-time PCR (qRT-PCR) analysis was performed using the 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA). Premixed PCR primers and Taqman probes for Gpr44, Ptgdr, Pla2g2a, Pla2g4a, Ptgs1, Ptgs2, Ptgds, Hpgds, and Pparg were purchased from Applied Biosystems. Gapdh (Applied Biosystems) was used for loading control.
Bacterial load
Peritoneal lavage fluids and peripheral blood were serially diluted with sterile saline. Equal volumes of each dilution was applied to trypticase soy agar (TSA) plates with 5% sheep blood (BD™ Trypticase™ Soy Agar II with 5% Sheep Blood: BD Diagnostics, Sparks, MD) and were incubated overnight at 37°C. After overnight incubation, the number of bacteria colonies was counted and was expressed as colony-forming units (CFU) per indicated volume of peritoneal lavage fluids.
Multiplex cytokine assay
Murine TNF-α, IL-1β, IL-6, CCL3, IL-12 (p70), and IL-10 were measured using a multiplex assay according to the manufacturer’s instruction (Luminex 200; Luminex, Austin, TX). Data were analyzed using the Luminex Manager software (Luminex). The detection limit was 1.2 pg/ml.
Purification of peripheral polymorphonuclear leukocytes (neutrophils)
Peripheral blood leukocytes were isolated from whole blood by RBC sedimentation with 2% Dextran 500 (Sigma-Aldrich, St. Louis, MO). Peripheral neutrophils for qRT-PCR and Chromatin immunoprecipitation (ChIP) assays were isolated by positive selection using anti-Ly-6G microbead kit according to manufacturer’s instruction (Miltenyi Biotec, Bergisch Gladbach, Germany). The percentage of neutrophils was routinely more than 95%.
Peripheral polymorphonuclear leukocytes (PMNs) for chemotaxis assays were isolated from peripheral blood leukocytes by negative selection using immunomagnetic beads according to the previous report with minor modification (28). Peripheral leukocytes were incubated with rat anti-mouse primary antibodies specific to CD4, CD5, CD45R/B220, ICAM-1, Ter119 (Biolegend), and F4/80 (AbD Serotec, Raleigh, NC) for 15 min at 4°C. Next, the cells were washed and goat anti-rat IgG microbeads (Miltenyi Biotec) were added and incubated for 15 min at 4°C. The cell suspension was then applied onto a MS column (Miltenyi Biotec). The PMNs passed through the column by negative selection, while T cells, B cells, NK T cells, NK cells, RBC, and monocytes were captured in the column. The percentage of neutrophils was routinely more than 97%.
Flow cytometry Analysis
Whole blood was collected by cardiac puncture. After RBC lysis with AKC buffer (0.15 M NH4Cl, 1.0 mM KHCO3, 0.1 mM Na2EDTA; pH 7.4), peripheral leukocytes were incubated with anti-CD16/CD32 monoclonal antibody (BD PharMingen, Franklin Lakes, NJ) followed by staining with PE-conjugated anti-CXCR2 antibody (R&D Systems) and FITC-conjugated anti-Gr-1 antibody (BD PharMingen). Then the cells were washed twice, and fixed with 4% paraformaldehyde. For intracellular staining with GRK2, the fixed cells were permeabilized with perm/wash buffer (BD PharMingen) and were stained with rabbit anti-mouse GRK2 antibody (Abcam, Cambridge, MA), followed by staining with anti-rabbit IgG Alexa Fluor 488 (Invitrogen). The cells were analyzed by flow cytometry with Epics XL-MCL (Beckman Coulter, Brea, CA). The data was analyzed using FlowJo 7.6.1 software (Tree Star, Inc, Ashland, OR).
Chemotaxis assay
Chemotaxis assay was performed as previously described with minor modifications (29). Recombinant murine CXCL2 (30 ng/ml, R𠄽 systems) was incubated in 300µl of RPMI medium supplemented with 0.5% FBS, and was added to the lower compartment of a chemotaxis apparatus, separated from the top chamber by a 5µm pore size filter (ChemoTx; Neuro Probe Inc., Gaithersburg, MD). Peripheral PMNs (5 × 104 cells / 50 µl) isolated from WT and CRTH2−/− mice 2 h after CLP surgery were placed in the upper compartment. After 1 hour of incubation at 37 °C, the medium on the upper surface of the filter was removed, and replaced by 0.5 mM EDTA in PBS, followed by another hour of incubation. After 1 h, the medium on the upper surface was removed, the plate was centrifuged at 400 g at 4°C for 20 min, the filter was gently removed, and the supernatant aspirated. The neutrophil peroxidase activity in the lower chamber was determined by incubating the plate for 10 min with substrate solution (0.5 mM o-phenylenediamine, 10 mM H2O2, and 0.1% Triton X-100 in Tris buffer, pH 8.0), followed by the addition of 4 M H2SO4 to stop the reaction, and the absorbance was measured at 490 nm.
Chromatin immunoprecipitation assay
A ChIP assay was carried out using a EZ-Magna ChIP™ A Chromatin Immunoprecipitation Kit (Millipore, Temecula, CA) according to manufacturer’s instruction. Peripheral neutrophils (1 × 106 cells) pooled from three mice 2 h after sham or CLP surgery were used. ChIP antibodies for Acetyl H3 (06-599) and H3K27me3 (07-449) were purchased from Millipore. Antibody for H3K4me3 (ab8580) was purchased from Abcam. Rabbit IgG (Millipore) was used as a control. ChIP primers for mouse CXCR2 promoters were designed by using primer express 3.0 (Applied Biosystems) and were as follows: forward, 5'- AGGCATAGGCTGGGAAGGAA; reverse, 5'-TCATGCCTTCTGATGGCTTCT.
Statistical analysis
Data are expressed as mean ± SEM, and analyzed for statistical significance using Student’s t test or ANOVA, followed by Tukey’s test for multiple comparisons. For survival studies, a log-rank test was used. P values less than 0.05 were considered statistically significant.
Results
Expression of PGD2 and arachidonic acid cascade enzymes during CLP-induced sepsis
In the initial experiments, we found that PGD2 and its metabolite 15d-PGJ2, both ligands for CRTH2 (13), were significantly increased in peritoneal lavage fluid at 20 h after CLP surgery (Fig. 1A, 1B). Interestingly, the levels were comparable between WT and CRTH2−/− mice (Supplemental Fig. 1). As mRNA of CRTH2 (Gpr44) and DP1 (Ptgdr), receptors for PGD2, has been detected in various immune cells (13, 14), we next examined the gene expression of CRTH2 and DP1 in peritoneal exudate cells recovered from the infectious focus: peritoneal cavity. Both CRTH2 and DP1 mRNA were detected in peritoneal exudate cells (Fig. 1C, 1D). There was no significant difference in CRTH2 expression between sham and CLP mice (Fig. 1C), while DP1 mRNA expression 4 h after surgery was lower in CLP mice than in sham-operated mice (Fig. 1D).
FIGURE 1.
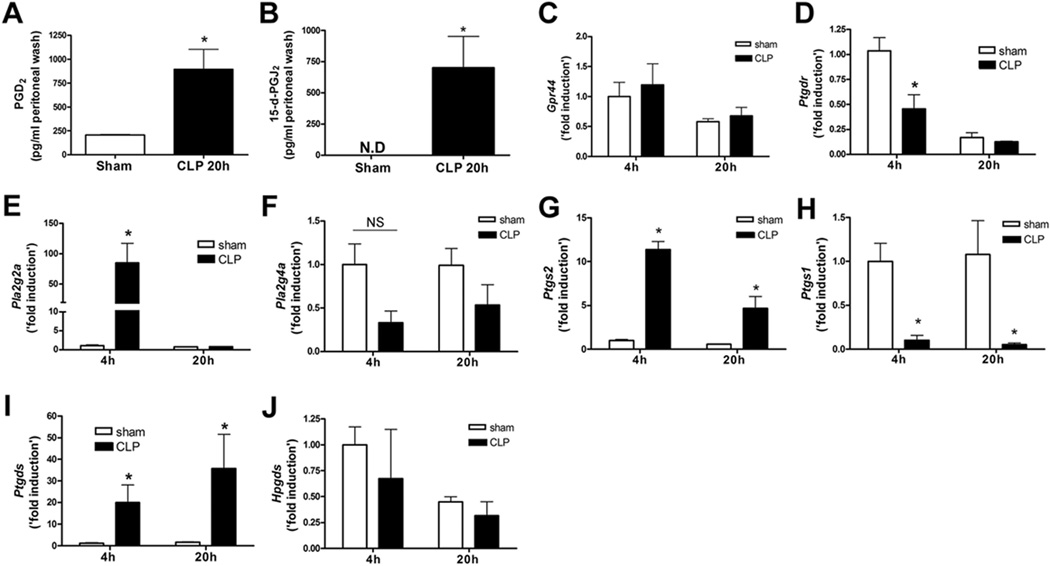
Upregulation of PGD2 and arachidonic acid cascade enzymes during CLP-induced sepsis. Female BALB/c mice were subjected to sham or five-puncture CLP surgery. A and B, The peritoneal lavage fluid was collected at 20 h after the surgery. The level of PGD2 (A) and 15-deoxy-delta-12,14-PGJ2 (15d-PGJ2) (B) in the cell-free peritoneal lavage fluid was measured by ELISA. C and D, The peritoneal exudate cells were collected 4 and 20 h after the surgery. The mRNA expression of CRTH2 (Gpr44) (C) and DP1 (Ptgdr) (D) in the peritoneal exudate cells was measured by quantitative real time PCR (qRT-PCR). The gene expression of secretory phospholipase A2 (PLA2) group IIA (Pla2g2a) (E), cytosolic PLA2 group 4A (Pla2g4a) (F), COX-2 (Ptgs2) (G), COX-1 (Ptgs1) (H), L-PGDS (Ptgds) (I), and H-PGDS (Hpgds) (J) in the peritoneal exudate cells was measured by qRT-PCR. Data are expressed as mean ± SEM. n = 5–6 in each group. All data of qRT-PCR shown are ‘fold induction’ relative to 4 h after sham operation. *, p < 0.05, compared with sham operated group. NS, not significant. These results are representative of two or three different experiments.
Since we identified an increase in the levels of the CRTH2 ligands, PGD2 and 15d-PGJ2, we next examined whether the upstream enzymes involved in arachidonic acid metabolism were also changed in CLP-induced sepsis. PLA2 releases arachidonic acid from membrane phospholipids. The gene expression of secretory PLA2 group IIA (Pla2g2a) but not cytosolic PLA2 group 4A (Pla2g4a) was significantly increased in peritoneal exudate cells at 4 h after CLP surgery (Fig. 1E, 1F). We next examined the expression of COX-1 (Ptgs1) and COX-2 (Ptgs2), enzymes that metabolize PGG2 from arachidonic acid. COX-2, an inducible isoform of COX, was highly upregulated in the peritoneal exudate cells after CLP surgery (Fig. 1G), whereas COX-1, a constitutive isoform of COX, was dramatically suppressed (Fig. 1H). In addition, lipocalin-type PGD2 synthase (LPGDS) (Ptgds) but not hematopoietic PGD2 synthase (HPGDS) (Hpgds), both of which are PGD2 synthetases, was also upregulated after CLP surgery (Fig. 1I, 1J). Together, these results confirmed that enzymes of the arachidonic acid cascade are involved in CLP-induced sepsis.
CRTH2−/− mice were resistant to CLP-induced lethality
To investigate the role of CRTH2 on CLP-induced lethality, WT and CRTH2−/− (Gpr44−/−) mice were subjected to five- or seven-puncture CLP surgery and the survival was monitored for 96 h. CRTH2−/− mice were highly resistant to both five and seven-puncture CLP-induced lethality (Fig. 2A, 2B). In CRTH2−/− mice, all of the mice that survived to 96 h were still alive at day 10 (data not shown). By 96 h post CLP, all the WT mice were dead.
FIGURE 2.
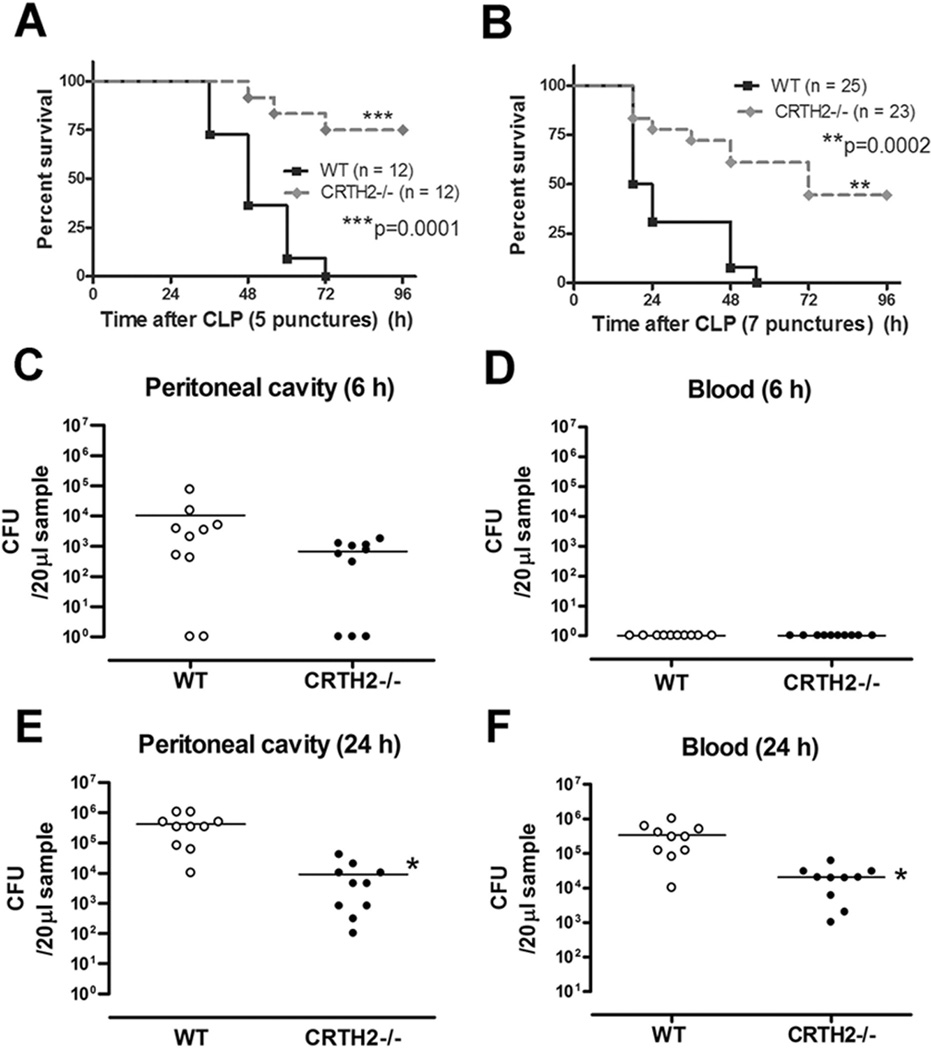
Improved survival and enhanced bacterial clearance in lethal sepsis induced by CLP in CRTH2−/− mice. A, WT (n = 12) and CRTH2−/− (n = 12) mice were subjected to five-puncture CLP surgery and survival was monitored for 96 h. B, WT (n = 25) and CRTH2−/− (n = 23) mice were subjected to seven-puncture CLP surgery and survival was monitored for 96 h. The data of (B) are the composite of three independent experiments. ***, p = 0.0001; **, p = 0.0002, compared with WT group. C-F, Peritoneal lavage fluid and blood was harvested at 6 and 24 h after five-puncture CLP surgery. Peritoneal lavage fluid and blood were serially diluted and plated on the Trypticase Soy Agar (TSA) plates. Bacteria levels at 6 h after CLP surgery in peritoneal lavage fluid (C) and blood (D), and at 24 h in peritoneal lavage fluid (E) and blood (F) were expressed as colony forming units (CFU) per 20 µl. These data are the composite of two independent experiments. The horizontal bar indicates the mean for each group. n = 10–12 in each group. *, p < 0.05, compared with WT mice.
We next examined whether resistance to CLP-induced sepsis in CRTH2−/− mice was associated with enhanced bacterial clearance. There was no difference between WT and CRTH2−/− mice in regards to bacterial count of peritoneal lavage fluid and blood at 6 h after CLP (Fig. 2C, 2D). However, the bacterial load in both the peritoneal cavity and blood at 24 h after CLP was lower in CRTH2−/− mice, as compared with WT mice (Fig. 2E, 2F), indicating that CRTH2 deficiency results in enhanced bacterial clearance as a function of time.
To examine if the resistance to sepsis in CRTH2−/− mice is associated with nitrosative stress in the infectious site, metabolites of NO (nitrite and nitrate) in peritoneal fluid in WT and CRTH2−/− mice were measured by the Griess method. No differences in the levels of nitrite and nitrate in peritoneal fluid after CLP surgery were seen when CRTH2−/− mice were compared to WT mice (Supplemental Fig. 2).
Cytokine levels were altered and IL-10 induction was protective in CRTH2−/− mice during CLP-induced sepsis
Severe sepsis is associated with excessive production of proinflammatory cytokines called the “cytokine storm” (30). To investigate if the survival benefit of sepsis-induced lethality in CRTH2−/− mice was correlated with modulated level of cytokines, we examined the cytokine expression profile in peritoneal lavage fluid and serum. Twenty hours after CLP surgery, the level of proinflammatory cytokines TNF-α, IL-1β, IL-6, and CCL3 was significantly lower in CRTH2−/− mice than in WT mice (Fig. 3A). Serum levels of TNF-α, IL-6, and CCL3 were also significantly lower 20 h post CLP surgery in CRTH2−/− mice compared to WT mice (Fig. 3B). These results suggest that the suppression of excessive proinflammatory cytokine induction in the infectious site (peritoneal lavage fluid) and systemic circulation (serum) from CRTH2−/− mice contributes to the resistance to CLP-induced sepsis.
FIGURE 3.
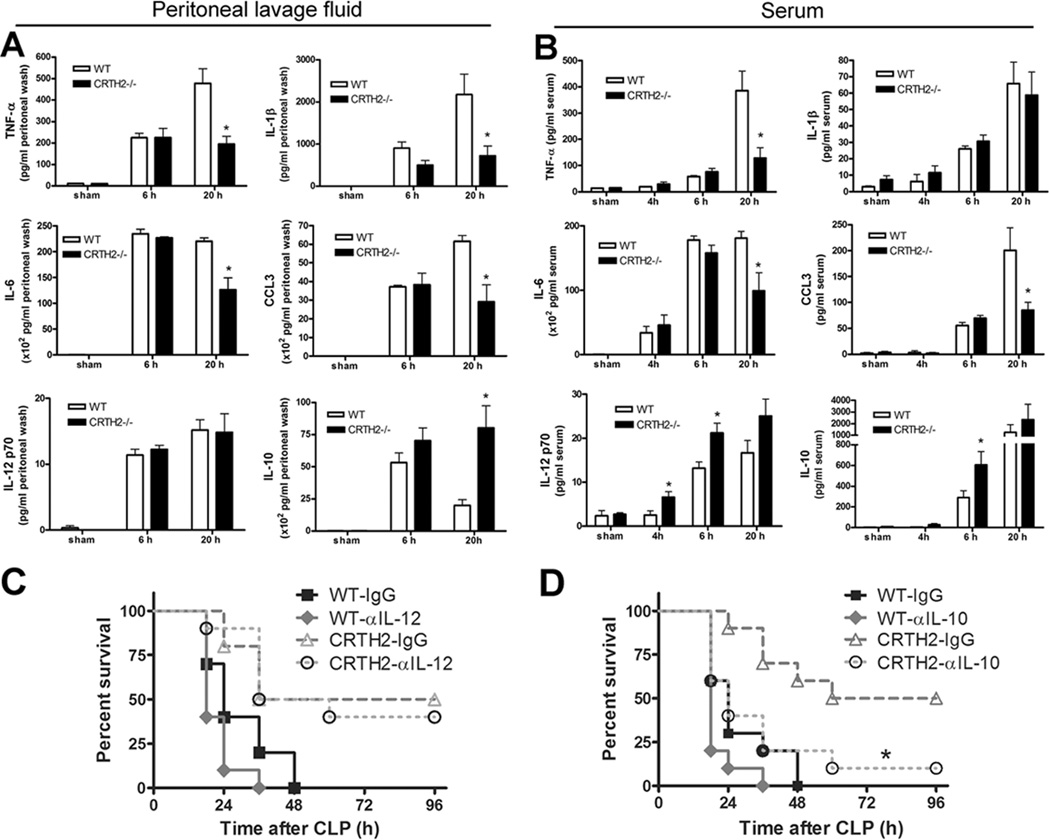
Altered cytokine levels and protective roles of IL-10 in CRTH2−/− mice after CLP surgery. A, At 6 and 20 h after five-puncture CLP surgery, peritoneal lavage fluid was collected and the level of TNF-α, IL-1β, IL-6, CCL3, IL-12p70, and IL-10 was measured by multiplexed immunobead-based assay. B, Serum was also collected at indicated time points and the level of TNF-α, IL-1β, IL-6, CCL3, IL-12p70, and IL-10 was measured by multiplexed immunobead-based assay. Data are expressed as mean ± SEM pg per ml of peritoneal lavage fluid or serum. n = 5–6 in each group. *, p < 0.05, compared with WT group. These results are representative of two different experiments. C, WT and CRTH2−/− mice received either control IgG (250 µg/mouse) or anti-IL-12 antibody (250 µg/mouse) 1 h before seven-puncture CLP surgery. The survival was monitored for 96 h. These data are the composite of two independent experiments. n = 9–10 mice in each group. D, WT and CRTH2−/− mice received either control IgG (250 µg/mouse) or anti-IL-10 antibody (250 µg/mouse) 1 h before seven-puncture CLP surgery. The survival was monitored for 96 h. These data are the composite of two independent experiments. n = 10 in each group. *, p < 0.05, compared with CRTH2−/− mice group which received control IgG.
As IL-12 and IL-10 play protective roles during sepsis (31–33), we examined levels of these in serum and peritoneal lavage fluid. IL-12p70 levels at 4 and 6 h in serum, but not in peritoneal lavage fluid, were higher in CRTH2−/− mice than in WT mice (Fig. 3A, 3B). However, inhibition of IL-12 did not worsen the mortality in CRTH2−/− mice, suggesting that enhanced IL-12 is not essential for the survival improvement in CRTH2−/− mice (Fig. 3C). We next examined IL-10 and found that the levels were higher both in peritoneal lavage fluid and serum recovered from CRTH2−/− mice (Fig. 3A, 3B). Relatedly, the survival benefit in CRTH2−/− mice was abrogated by pharmacological inhibition of IL-10, suggesting that increased IL-10 production, at least in part, contributes to the protection against sepsis in CRTH2−/− mice (Fig. 3D).
CXCR2 is critical for enhanced accumulation of neutrophils into the peritoneal cavity in CRTH2−/− mice during CLP-induced sepsis
We demonstrated that IL-10 plays a protective role against sepsis in CRTH2−/− mice. Since peritoneal neutrophils is a major producer of IL-10 during sepsis (34) and neutrophil migration into the site of inflammation is shown to be impaired during sepsis, resulting in an increase in mortality (1, 35), we examined the neutrophil influx into the infectious focus: the peritoneal cavity. CRTH2−/− mice exhibited a significantly higher accumulation of neutrophils but not of mononuclear cells into peritoneal cavity compared to WT mice, resulting in the higher total numbers of leukocyte influx (Fig. 4A). These results demonstrate that the enhanced neutrophil influx to the infection focus during sepsis in CRTH2−/− mice, contribute to the improvement of their survival.
FIGURE 4.
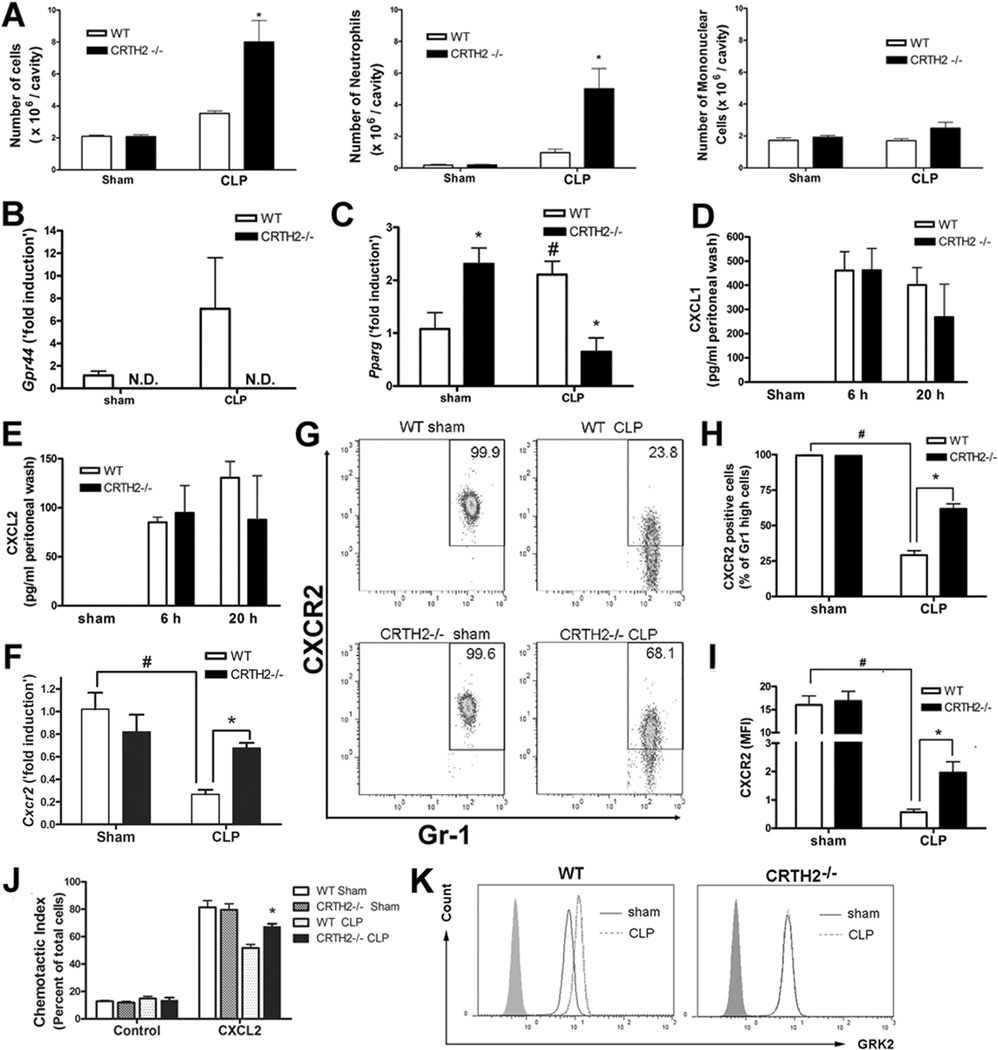
Essential roles of CXCR2 on enhanced accumulation of neutrophils during CLP-induced sepsis in CRTH2−/− mice. A, At 24 h after five-puncture CLP surgery, peritoneal exudate cells were collected by peritoneal lavage with 3 ml PBS. The number of peritoneal cells is expressed per peritoneal cavity. B and C, Peripheral neutrophils were purified from peripheral blood 4 h after sham or five-puncture CLP surgery as described in the methods section. The mRNA expression of CRTH2 (Gpr44) (B) and PPAR-gamma (Pparg) (C) in peripheral neutrophils were measured by qRT-PCR. N.D., not detected. D and E, The level of CXCL1 (D) and CXCL2 (E) in peritoneal lavage fluid at 24 h after sham or five-puncture CLP surgery was measured by ELISA. Data expressed as mean ± SEM. n = 5–6 in each group. *, p < 0.05, compared with WT group. These results are representative of two different experiments. F-H, Peripheral neutrophils were purified from peripheral blood 2 h after five-puncture CLP surgery as described in Materials and Methods. CXCR2 mRNA levels in peripheral neutrophils were measured by qRT-PCR (F). Data are expressed as mean ± SEM. n = 4 in each group. The level of CXCR2 in peripheral neutrophils was measured by flow cytometry (G–I). Representative flow cytometry data in Gr1 high positive gated cells (neutrophils) is shown (G). The percentage of CXCR2 positive cells in Gr1 high positive cells (H) and the mean fluorescence intensity (MFI) of CXCR2 is shown (I). Data of (H) and (I) expressed as mean ± SEM. n = 5–6 in each group. #, p < 0.05, compared with sham-operated group. *, p < 0.05, compared with WT group. J, Peripheral PMNs were purified from peripheral blood 2 h after CLP surgery. Chemotaxis assay was performed by using CXCL2 (30 ng/ml), as the ligand, or not (control cells) in a Boyden chamber. Data are expressed as percentage of migrated cells in total cells. n = 4 in each group. *, p < 0.05, compared with CLP-operated WT mice. K, Neutrophils were purified from peripheral blood 2 h after five-puncture CLP surgery. The histogram data of GRK2 positive cells in Gr1 high positive cells are shown. The data shown are representative of three independent experiments.
We further investigated what contributes to higher neutrophil influx in CRTH2−/− mice. We confirmed that CRTH2 mRNA was detected and tended to be upregulated in peripheral neutrophils 2 h after CLP surgery (Fig. 4B). Because PPAR-γ, a ligand activated transcription factor belonging to the nuclear hormone receptor family, is upregulated in peripheral blood neutrophils during severe sepsis causing impaired neutrophil migration (36), we next examined if CRTH2 deficiency causes inhibition of PPAR-γ in circulating neutrophils. We confirmed that PPAR-γ mRNA (Pparg) was significantly increased in WT neutrophils after CLP surgery compared to sham surgery. Also, PPAR-γ mRNA expression was significantly lower in neutrophils recovered from CLP CRTH2−/− mice, compared to those from CLP WT mice, while its expression level was significantly higher in CRTH2−/− peripheral neutrophils after sham surgery (Fig. 4C). These results suggest that suppressed PPAR-γ expression in CRTH2−/− peripheral neutrophils is correlated with enhanced neutrophil influx into the peritoneal cavity during sepsis.
As CXCL1 and CXCL2 are chemoattractant chemokines that enhance neutrophil recruitment, we examined the levels of CXCL1 and CXCL2. The levels of CXCL1 and CXCL2 in peritoneal lavage fluid were not significantly different between WT and CRTH2−/− mice (Fig. 4D, 4E). These results suggest that neither CXCL1 nor CXCL2 induction contributes to the enhanced neutrophil influx into the peritoneal cavity in CRTH2−/− mice.
CXCR2, a receptor for ELR-positive CXC chemokines including CXCL1 and CXCL2, plays a pivotal role in the recruitment of neutrophils (37) and its level in circulating neutrophils was downregulated during sepsis, resulting in impaired neutrophil migration into the infectious sites (38). Therefore, we examined if enhanced neutrophil recruitment in CRTH2−/− mice is associated with higher CXCR2 levels in circulating neutrophils. The mRNA and protein levels of CXCR2 were decreased after CLP surgery in both WT and CRTH2−/− neutrophils, when compared with neutrophils from sham treated mice. CXCR2 levels were significantly higher in CRTH2−/− neutrophils, when compared with WT neutrophils from CLP mice (Fig. 4F–I). Also, CXCL2 dependent chemotaxis of peripheral PMNs isolated at 2 h after CLP surgery was higher in CRTH2−/− mice than that in WT mice (Fig. 4J), and was associated with higher levels of CXCR2 in CRTH2−/− neutrophils. As CXCR2 levels have been shown to be negatively regulated by G protein-coupled receptor kinase (GRK) 2 during sepsis (27), we next measured the GRK2 level in circulating neutrophils. GRK2 level was increased in WT neutrophils, while the level was not changed in CRTH2−/− neutrophils (Fig. 4K). These results suggest that higher CXCR2 levels are, at least in part, responsible for the enhanced neutrophil recruitment in CRTH2−/− mice.
The level of CXCR2 in circulating neutrophils was epigenetically supported during sepsis
To investigate the mechanism for higher CXCR2 levels in CRTH2−/− neutrophils during sepsis, we hypothesized that CXCR2 levels may be epigenetically regulated and thus, examined the histone modifications at the CXCR2 promoter in circulating neutrophils by ChIP assay. The levels of trimethylation of histone H3 at lysine 4 (H3K4me3), a mark for transcription activation, and trimethylation of histone H3 at lysine 27 (H3K27me3), a mark for transcription repression, at the CXCR2 promoter were not significantly changed after CLP surgery (Fig. 5A, 5B). In contrast, acetylation of histone H3 (Acetyl H3), a mark for transcription activation, was significantly lower in WT neutrophils recovered from CLP mice, when compared with sham-operated mice. Also, Acetyl H3 levels were significantly higher in CRTH2−/− neutrophils when compared with WT neutrophils after CLP surgery (Fig. 5C). These results suggest that CXCR2 reduction during sepsis is epigenetically supported by a decrease in Acetyl H3 and the observed higher levels of Acetyl H3 correlated with higher CXCR2 levels in CRTH2−/− neutrophils as compared with WT neutrophils after CLP surgery.
FIGURE 5.
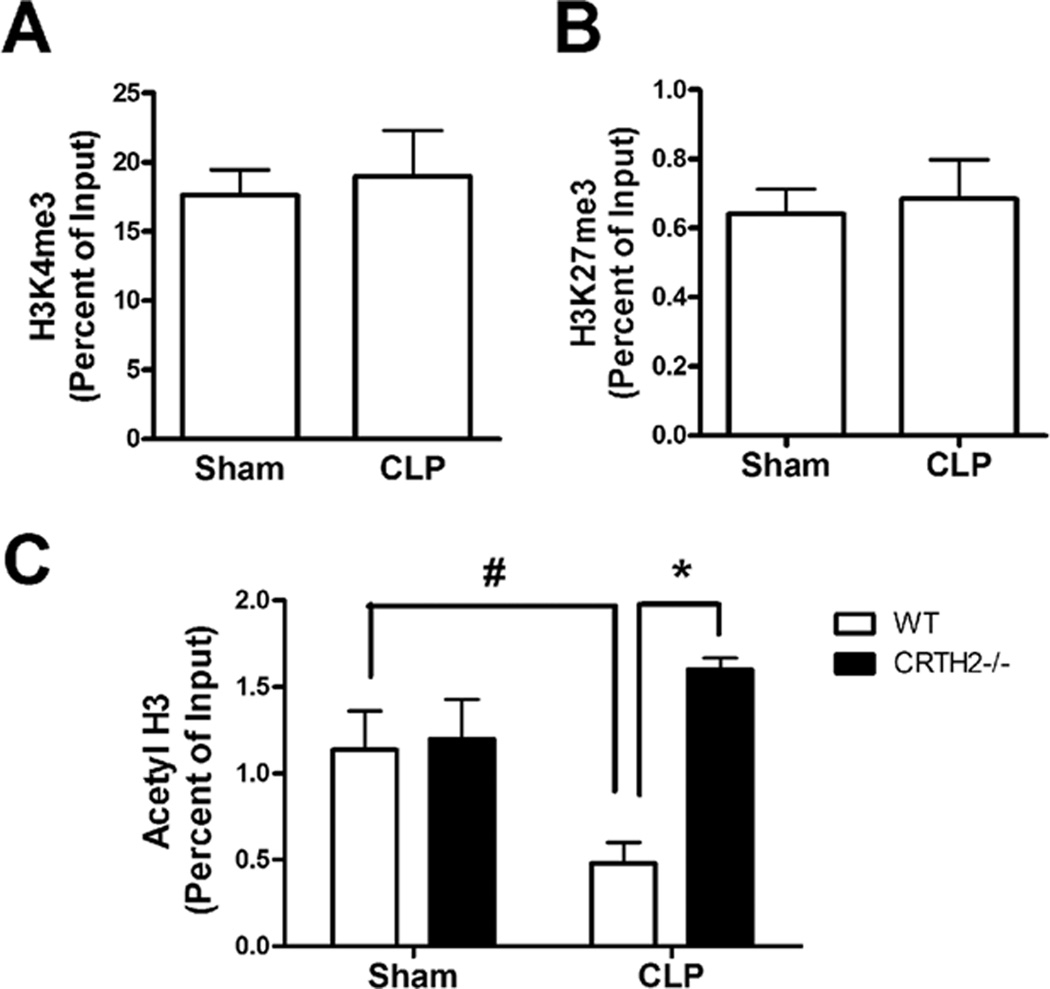
Epigenetic regulation of CXCR2 gene expression in peripheral neutrophils during sepsis. WT and CRTH2−/− peripheral blood were collected at 2 h after sham or five-puncture CLP surgery. The blood from three mice was pooled. Peripheral neutrophils were purified from the pooled blood as described and were used as an individual sample. A and B, ChIP assay was performed to determine the status of H3K4 tri-methylation (H3K4me3) (A) and H3K27 tri-methylation (H3K27me3) (B) on the promoter region of CXCR2 in neutrophils from WT and CRTH2−/− mice. C, The acetylation of histone H3 (Acetyl H3) on the promoter region of CXCR2 was also measured in these neutrophils by ChIP assay. n = 3 in each group. #, p < 0.05, compared with sham-operated group. *, p < 0.05, compared with WT mice. These results are representative of three different experiments.
CXCR2-expressing neutrophils were protective against sepsis in CRTH2−/− mice
To investigate whether enhanced neutrophil recruitment and higher CXCR2 levels in circulating neutrophils from CRTH2−/− mice contribute to their resistance to CLP-induced lethality, we depleted neutrophils using anti-Gr-1 or anti-Ly-6G antibodies, and inhibited CXCR2 using CXCR2 antagonist SB225002. Depletion of neutrophils by administration of antibody for Gr-1 dampened the survival benefit in CRTH2−/− mice (Fig. 6A). As Gr-1 antibody recognizes not only Ly-6G (neutrophils) but also Ly-6C expressing monocytes, T cells, and dendritic cells, we next used Ly-6G antibody for more specific neutrophil depletion. Neutrophil depletion by Ly-6G antibody also worsened the survival in CRTH2−/− mice (Fig. 6B) which was associated with higher bacterial load (Fig. 6C) and suppressed IL-10 induction in neutrophil-depleted CRTH2−/− mice (Fig. 6D), suggesting that enhanced IL-10-expressing neutrophil recruitment in CRTH2−/− mice is important for enhanced bacterial clearance and improved survival. Moreover, pharmacological inhibition of CXCR2 resulted in the abrogation of the prolonged survival in CRTH2−/− mice after CLP-induced sepsis (Fig. 6E). These results indicate that CXCR2-mediated increased neutrophil influx in CRTH2−/− mice contributes to protection against sepsis.
FIGURE 6.
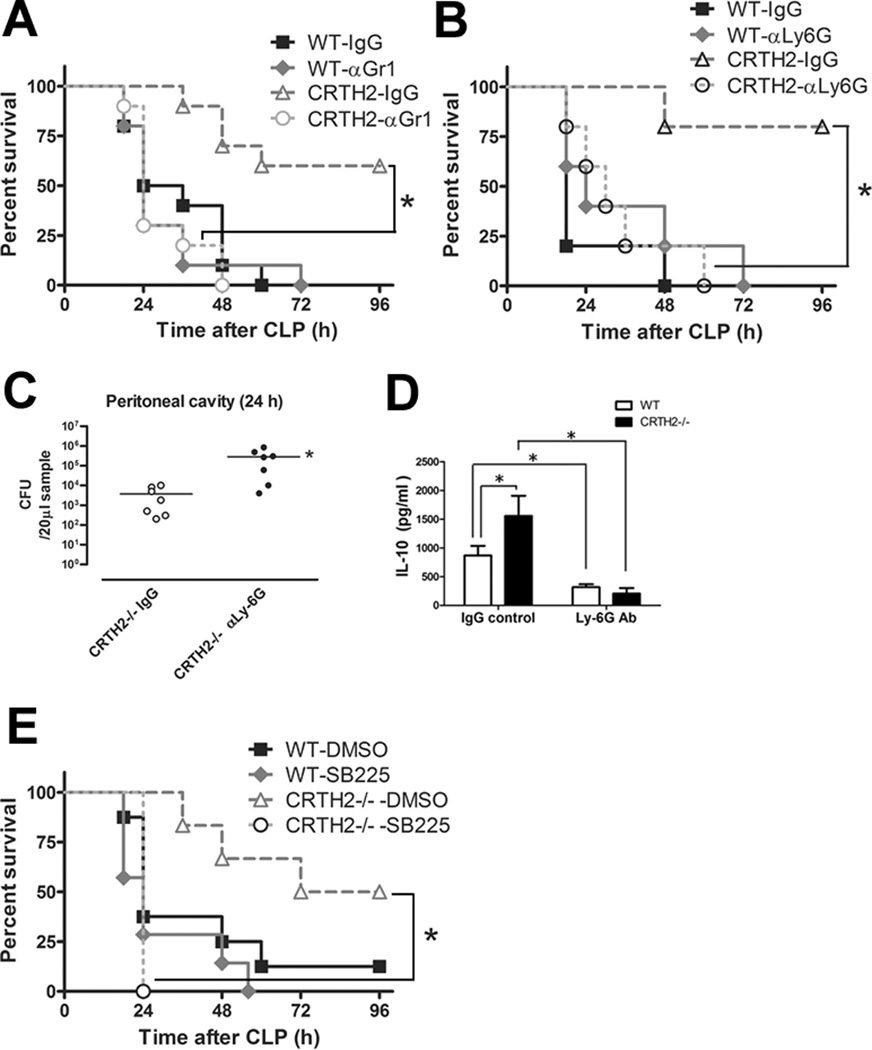
The roles of neutrophils and CXCR2 expression on the protection against CLP-induced sepsis in CRTH2−/− mice. A, WT and CRTH2−/− mice received either control IgG (250 µg/mouse) or anti-Gr-1 antibody (250 µg/mouse) 24 h before seven-puncture CLP surgery. The survival was monitored for 96 h. The data are the composite of two independent experiments. n = 10 in each group. *, p < 0.05. B-D, WT and CRTH2−/− mice received either control IgG (250 µg/mouse) or anti-Ly-6G antibody (250 µg/mouse) 24 h before seven-puncture CLP surgery. The survival was monitored for 96 h. n = 5 in each group. *, p < 0.05 (B). Peritoneal lavage fluids recovered at 24 h after CLP surgery from CRTH2−/− mice which were received either control IgG or anti-Ly-6G antibody were serially diluted and plated on the TSA plates. Bacteria levels were expressed as colony forming units (CFU) per 20 µl. The horizontal bar indicates the mean for each group. n = 7 in each group. *, p < 0.05, compared with control IgG-treated group (C). The levels of IL-10 in peritoneal lavage fluids recovered 24 h after CLP surgery were measured by ELISA. n = 5 in each group. *, p < 0.05 (D). E, WT and CRTH2−/− mice received either control DMSO or CXCR antagonist SB225002 (10 mg/kg body weight mouse) 1 h before seven-puncture CLP surgery. The survival was monitored for 96 h. The data are composite of two independent experiments. n = 10 in each group. *, p < 0.05.
To investigate the role of CRTH2 in PMN function, phagocytic activity was measured in both WT and CRTH2−/− thioglycollate-elicited PMNs, the major class of PMNs used in in vitro assays including phagocytosis (39). We found that the phagocytic activity of these PMNs was higher in CRTH2−/− mice (Supplemental Fig. 3).
We further tried to investigate the molecular mechanisms linking RTH2-deficiency to altered CXCR2 expression. LPS/TLR4/MyD88-mediated signaling pathway, a major signaling pathway during sepsis, was suppressed in CRTH2−/− bone marrow neutrophils, which may be associated with impaired LPS-induced GRK2 induction and higher CXCR2 expression in CRTH2−/− neutrophils (Supplemental Fig. 4).
Discussion
In this study, we demonstrated several salient findings that demonstrate the importance of the PGD2 receptor CRTH2 in sepsis. First, PGD2 was induced and CRTH2 was detected during CLP-induced sepsis. Second, CRTH2 deficiency significantly improved many of the biological parameters used to monitor experimental polymicrobial sepsis. Third, the protection found in CRTH2−/− mice was associated with improved bacterial clearance, suppressed proinflammatory cytokine induction, and enhanced anti-inflammatory cytokine IL-10 induction. Fourth, neutrophil recruitment into the infectious focus was increased in CRTH2−/− mice, which was associated with higher levels of CXCR2 expression by circulating neutrophils. Fifth, CXCR2 expression was epigenetically supported by acetylation of histone H3 in the promoter of this gene. Finally, CXCR2-expressing neutrophils are essential for protection against CLP-induced lethality in CRTH2−/− mice.
In the present study, PGD2 was highly expressed during CLP-induced sepsis, but the cellular sources of PGD2 during experimental sepsis remain unknown. We postulate that since dendritic cells and macrophages play key roles during sepsis (40), and because mast cells have been shown to produce PGD2 (6, 7), these cell types may be cellular sources of PGD2 production during sepsis.
PGD2 metabolite 15d-PGJ2, a ligand for both CRTH2 and PPAR-γ (16), was identified in the peritoneal cavity after CLP surgery. A previous study showed that intraperitoneal administration of exogenous 15d-PGJ2 contributed to reducing mortality in an endotoxin shock model (41). Therefore we hypothesized that the 15d-PGJ2-mediated PPARγ pathway may be a compensatory pathway that is enhanced in response to the abrogation of PGD2- and 15d-PGJ2-mediated CRTH2 pathway in CRTH2−/− mice. The end result was that a deficient CRTH2 pathway contributed to protection and prolonged survival during the septic response. However, the level of 15d-PGJ2 in the peritoneal lavage was not different between WT and CRTH2−/− mice, while PPAR-γ levels were lower, in CRTH2−/− neutrophils compared to WT neutrophils after CLP surgery. Next, we showed that COX-2 was highly upregulated while COX-1 was significantly downregulated in peritoneal exudate cells. The reason for the suppression of COX-1 levels is unclear, however, this may be simply because of the change in the major cell population of peritoneal exudate cells: from mononuclear cells to neutrophils. Another possible reason is the reduction of Sp1, one of the most important transcription factors for COX-1. A previous study showed that COX-1 was decreased in the lung after intratracheal administration of LPS, a principle mediator of sepsis caused by gram-negative bacteria (42), and the decrease was due to a reduction of Sp1 (43).
Proinflammatory cytokine levels in both peritoneal lavage fluid and serum recovered from CLP-treated CRTH2−/− mice were reduced, when compared with WT mice. As the MyD88 level was suppressed in LPS-stimulated CRTH2−/− neutrophils, we speculate that altered cytokine production is, at least in part, associated with suppressed TLR4/MyD88/NF-κB-mediated signaling pathway in CRTH2−/− cells including neutrophils. Interestingly, IL-10, an anti-inflammatory cytokine, was higher in CRTH2−/− mice and its neutralization resulted in an abrogation of the protection against CLP-induced lethality in CRTH2−/− mice. These results are consistent with a previous study showing that endogenous IL-10 protects mice against sepsis (33). The reason why increased IL-10 in CRTH2−/− mice is protective is unclear, but this may be associated with suppression of TNF-α production by IL-10 in CRTH2−/− mice. In fact, a previous study reported that IL-10 contributes to resistance to LPS-induced lethality by suppressing TNF-α production (44).
Neutrophils have been shown to be the first cells to migrate to foci of microbial infection and a major IL-10 expressing cell in the peritoneal cavity during sepsis (34). We have confirmed that peritoneal neutrophils from CLP mice produce IL-10 (data not shown). It was reported that CRTH2 mRNA is expressed in neutrophils obtained from naïve mice (14). We confirmed that CRTH2 mRNA was expressed in peripheral neutrophils in our models and showed that neutrophil influx into the peritoneal cavity was augmented in CRTH2−/− mice after CLP surgery. A previous study reported that IL-10−/− mice showed increased neutrophil recruitment to the peritoneal cavity during sepsis (45), suggesting that our results showing enhanced IL-10 levels in CRTH2−/− mice does not cause increased neutrophil recruitment. Therefore, we tried to investigate new mechanism that causes enhanced accumulation of neutrophils. As impaired neutrophil chemotaxis during sepsis was reported to be mediated by activation of PPARγ (36), we examined PPARγ level in peripheral neutrophils to determine if this activity was mechanistically important. We found that PPARγ mRNA level after CLP surgery was lower in CRTH2−/− neutrophils as compared with WT neutrophils, suggesting that lower PPARγ level may correlate with enhanced neutrophil chemotactic activity and recruitment to the peritoneal cavity during sepsis in the CRTH2−/− mice.
In this study, we showed that CXCR2 was unaffected by sham surgery but was decreased after CLP surgery and the decrease was greater in peripheral neutrophils from WT mice than from CRTH2−/− mice. Moreover, the study of pharmacological CXCR2 inhibition demonstrated that CXCR2-mediated neutrophil recruitment is a key event for protection against CLP-induced lethality in CRTH2−/− mice. These results were consistent with a previous report showing that neutrophil recruitment was impaired during sepsis, and was correlated with suppressed CXCR2 levels by peripheral neutrophils (46). We further demonstrated a novel mechanism for the regulation of CXCR2 gene regulation during sepsis: epigenetic dependent histone modification. Previous studies have shown that post–septic immune cells including dendritic cells, helper T cells, and regulatory T cells were involved in epigenetic gene regulation, which contributed to immune paralysis after sepsis (47). In this study, we showed that gene expression in neutrophils was also epigenetically regulated; CXCR2 reduction in WT neutrophils after CLP surgery was associated with decreased levels of Acetyl H3, a mark for transcription activation (48), at the CXCR2 promoter. Furthermore, higher Acetyl H3 levels were associated with higher CXCR2 levels in CRTH2−/− neutrophils when compared to WT neutrophils during sepsis. However, the data demonstrating that CXCR2 levels during CLP in CRTH2−/− neutrophils were decreased compared to the levels after sham surgery seems to be inconsistent with the result showing the Acetyl H3 levels actually tended to increase in CRTH2−/− neutrophils after CLP surgery. Since gene expression depends on a complex combination of both genetic and epigenetic factors, we hypothesize that Acetyl H3 marks in the CXCR2 gene promoter in concert with other yet unknown factors mechanistically explain fluctuations in CXCR2 expression.
The reason why transcription activation markers of Acetyl H3 and H3K4me3 were inconsistent (i.e. Acetyl H3 but not H3K4me3 was increased) is unclear. However, since histone acetylation and methylation are correlated with gene promoters and can cooperatively regulate gene transcription with other epigenetic modifications, a specific epigenetic modification may predominantly regulate gene transcription over other modifications in specific gene promoter region (49, 50).
Another possible mechanism contributing to higher CXCR2 levels in CRTH2−/− neutrophils is inactivation of G protein-coupled receptor kinase-2 (GRK2). GRK2 is a protein kinase that downregulates chemokine receptors such as CXCR2 (51). A previous study demonstrated that GRK2 contributed to decreasing CXCR2 levels in a polymicrobial sepsis model (46). The higher CXCR2 level in CRTH2−/− neutrophils may be due in part, to impaired GRK2 induction in CRTH2−/− neutrophils, contributing to the protection against sepsis. In fact, GRK2 level was lower in neutrophils from CRTH2−/− CLP mice and LPS-stimulated CRTH2−/− neutrophils as compared with WT neutrophils. The molecular mechanism linking CRTH2-deficiency to higher CXCR2 expression in CRTH2−/− neutrophils is unclear. However, impaired-LPS/TLR4/MyD88-mediated signaling by CRTH2-deficiency may contribute to lower LPS-induced GRK2 expression, and thus higher CXCR2 expression, leading to enhanced neutrophil recruitment during sepsis.
In addition to the role of CXCR2, there may be other factors which contribute to enhanced neutrophil recruitment in CRTH2−/− mice during sepsis. There are several steps in neutrophil recruitment: mobilization, rolling, adhesion, and transmigration (chemotaxis). All these phases of neutrophil recruitment are reported to be modulated during sepsis (35). Therefore, enhanced neutrophil influx in CRTH2−/− mice may be not only due to a CXCR2-mediated increase in neutrophil transmigration, but also altered steps of mobilization, rolling, and adhesion. In addition, activated dendritic cells have been shown to promote neutrophil chemotaxis (52), and may contribute to enhanced neutrophil recruitment in CRTH2−/− mice regardless of neutrophil expression of CRTH2. Future studies will try to further unravel the complex interactions leading to the protective survival effects in septic CRTH2−/− mice.
In summary, we have defined a role for CRTH2 in innate immunity during CLP-induced sepsis. Genetic ablation of CRTH2 resulted in protection against CLP-induced sepsis. This protection was associated with increased bacterial clearance, suppressed proinflammatory cytokine production, and enhanced anti-inflammatory cytokine IL-10 production. Moreover, higher CXCR2 levels in peripheral neutrophils contributed to this protection by enhancing neutrophil recruitment into the infectious locus. Interestingly, the CXCR2 level was epigenetically regulated by histone modification, specifically Acetyl H3, also contributing to enhanced neutrophil recruitment. Our results shed new light on the role of CRTH2 during sepsis and contribute to our understanding of the pathophysiology of sepsis. Additionally, CRTH2 and the factors regulating it have therapeutic potential as targets for polymicrobial sepsis and septic shock in the clinical setting.
Supplementary Material
Acknowledgments
We thank Dr. Makoto Arita, University of Tokyo, Mrs. Miyuki Yamamoto and the Core Instrumentation Facility, Keio University School of Medicine, Dr. Katsuhide Okunishi, University of Michigan, for technical assistance, and Dr. Judith Connett, University of Michigan for critical reading of the manuscript.
This work was supported in part by Grant-in-Aid for Young Scientists (B) from the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT) (to M.I.), Strategic Research Foundation Grant-aided Project for Private Universities from MEXT (to M.I.), grant from Ohyama Health Foundation (to M.I.), grant from Takeda Science Foundation (to M.I.), grant from Daiwa Securities Health Foundation (to M.I.), Keio Gijuku Fukuzawa Memorial Fund for the Advancement of Education and Research (to M.I.), and National Institutes of Health grant HL31237 (to S.L.K.)
Abbreviations used in this article
- PLA2
phospholipase A2
- PG
prostaglandin
- COX
cyclooxygenase
- DP1
D prostanoid receptor 1
- CRTH2
chemoattractant receptor-homologous molecule expressed on Th2 cells
- 15d-PGJ2
15-deoxy-12,14-PGJ2
- PPAR-γ
peroxisome proliferator-activated receptor gamma
- CLP
cecal ligation and puncture
- qRT-PCR
quantitative real time PCR
- TSA
trypticase soy agar
- CFU
colony forming units
- ChIP
Chromatin immunoprecipitation
- PMN
polymorphonuclear leukocyte
- LPGDS
lipocalin-type PGD2 synthase
- HPGDS
hematopoietic PGD2 synthase
- GRK2
G protein-coupled receptor kinase-2
- H3K4me3
trimethylation of histone H3 at lysine 4
- H3K27me3
trimethylation of histone H3 at lysine 27
- Acetyl H3
acetylation of histone H3
References
- 1.Pinheiro da Silva F, Soriano FG. Neutrophils recruitment during sepsis: Critical points and crossroads. Front Biosci. 2009;14:4464–4476. doi: 10.2741/3542. [DOI] [PubMed] [Google Scholar]
- 2.Alves-Filho JC, de Freitas A, Spiller F, Souto FO, Cunha FQ. The role of neutrophils in severe sepsis. Shock. 2008;30(Suppl 1):3–9. doi: 10.1097/SHK.0b013e3181818466. [DOI] [PubMed] [Google Scholar]
- 3.Johnston RA, Mizgerd JP, Shore SA. CXCR2 is essential for maximal neutrophil recruitment and methacholine responsiveness after ozone exposure. Am J Physiol Lung Cell Mol Physiol. 2005;288:L61–L67. doi: 10.1152/ajplung.00101.2004. [DOI] [PubMed] [Google Scholar]
- 4.Granstrom E. The arachidonic acid cascade. The prostaglandins, thromboxanes and leukotrienes. Inflammation. 1984;8(Suppl):S15–S25. doi: 10.1007/BF00915709. [DOI] [PubMed] [Google Scholar]
- 5.Saito S, Tsuda H, Michimata T. Prostaglandin D2 and reproduction. Am J Reprod Immunol. 2002;47:295–302. doi: 10.1034/j.1600-0897.2002.01113.x. [DOI] [PubMed] [Google Scholar]
- 6.Pettipher R. The roles of the prostaglandin D(2) receptors DP(1) and CRTH2 in promoting allergic responses. Br J Pharmacol. 2008;153(Suppl 1):S191–S199. doi: 10.1038/sj.bjp.0707488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mrabet-Dahbi S, Metz M, Dudeck A, Zuberbier T, Maurer M. Murine mast cells secrete a unique profile of cytokines and prostaglandins in response to distinct TLR2 ligands. Exp Dermatol. 2009;18:437–444. doi: 10.1111/j.1600-0625.2009.00878.x. [DOI] [PubMed] [Google Scholar]
- 8.Tajima T, Murata T, Aritake K, Urade Y, Hirai H, Nakamura M, Ozaki H, Hori M. Lipopolysaccharide induces macrophage migration via prostaglandin D(2) and prostaglandin E(2) J Pharmacol Exp Ther. 2008;326:493–501. doi: 10.1124/jpet.108.137992. [DOI] [PubMed] [Google Scholar]
- 9.Nagata K, Tanaka K, Ogawa K, Kemmotsu K, Imai T, Yoshie O, Abe H, Tada K, Nakamura M, Sugamura K, Takano S. Selective expression of a novel surface molecule by human Th2 cells in vivo. J Immunol. 1999;162:1278–1286. [PubMed] [Google Scholar]
- 10.Cosmi L, Annunziato F, Galli MIG, Maggi RME, Nagata K, Romagnani S. CRTH2 is the most reliable marker for the detection of circulating human type 2 Th and type 2 T cytotoxic cells in health and disease. Eur J Immunol. 2000;30:2972–2979. doi: 10.1002/1521-4141(200010)30:10<2972::AID-IMMU2972>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 11.Nagata K, Hirai H, Tanaka K, Ogawa K, Aso T, Sugamura K, Nakamura M, Takano S. CRTH2, an orphan receptor of T-helper-2-cells, is expressed on basophils and eosinophils and responds to mast cell-derived factoRs) FEBS Lett. 1999;459:195–199. doi: 10.1016/s0014-5793(99)01251-x. [DOI] [PubMed] [Google Scholar]
- 12.Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, Ichimasa M, Sugamura K, Nakamura M, Takano S, Nagata K. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. 2001;193:255–261. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim N, Luster AD. Regulation of immune cells by eicosanoid receptors. ScientificWorldJournal. 2007;7:1307–1328. doi: 10.1100/tsw.2007.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeshita K, Yamasaki T, Nagao K, Sugimoto H, Shichijo M, Gantner F, Bacon KB. CRTH2 is a prominent effector in contact hypersensitivity-induced neutrophil inflammation. Int Immunol. 2004;16:947–959. doi: 10.1093/intimm/dxh096. [DOI] [PubMed] [Google Scholar]
- 15.Shiraishi Y, Asano K, Niimi K, Fukunaga K, Wakaki M, Kagyo J, Takihara T, Ueda S, Nakajima T, Oguma T, Suzuki Y, Shiomi T, Sayama K, Kagawa S, Ikeda E, Hirai H, Nagata K, Nakamura M, Miyasho T, Ishizaka A. Cyclooxygenase-2/prostaglandin D2/CRTH2 pathway mediates double-stranded RNA-induced enhancement of allergic airway inflammation. J Immunol. 2008;180:541–549. doi: 10.4049/jimmunol.180.1.541. [DOI] [PubMed] [Google Scholar]
- 16.Monneret G, Li H, Vasilescu J, Rokach J, Powell WS. 15-Deoxy-delta 12,14-prostaglandins D2 and J2 are potent activators of human eosinophils. J Immunol. 2002;168:3563–3569. doi: 10.4049/jimmunol.168.7.3563. [DOI] [PubMed] [Google Scholar]
- 17.Cook JA. Eicosanoids. Crit Care Med. 2005;33:S488–S491. doi: 10.1097/01.ccm.0000196028.19746.42. [DOI] [PubMed] [Google Scholar]
- 18.Anderson BO, Moore EE, Banerjee A. Phospholipase A2 regulates critical inflammatory mediators of multiple organ failure. J Surg Res. 1994;56:199–205. doi: 10.1006/jsre.1994.1032. [DOI] [PubMed] [Google Scholar]
- 19.Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X, Jelinek I, Star RA, Mezey E. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15:42–49. doi: 10.1038/nm.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zardi EM, Zardi DM, Dobrina A, Afeltra A. Prostacyclin in sepsis: a systematic review. Prostaglandins Other Lipid Mediat. 2007;83:1–24. doi: 10.1016/j.prostaglandins.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 21.Suchner U, Katz DP, Furst P, Beck K, Felbinger TW, Thiel M, Senftleben U, Goetz AE, Peter K. Impact of sepsis, lung injury, and the role of lipid infusion on circulating prostacyclin and thromboxane A(2) Intensive Care Med. 2002;28:122–129. doi: 10.1007/s00134-001-1192-3. [DOI] [PubMed] [Google Scholar]
- 22.Satoh T, Moroi R, Aritake K, Urade Y, Kanai Y, Sumi K, Yokozeki H, Hirai H, Nagata K, Hara T, Utsuyama M, Hirokawa K, Sugamura K, Nishioka K, Nakamura M. Prostaglandin D2 plays an essential role in chronic allergic inflammation of the skin via CRTH2 receptor. J Immunol. 2006;177:2621–2629. doi: 10.4049/jimmunol.177.4.2621. [DOI] [PubMed] [Google Scholar]
- 23.Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW, 3rd, Bland KI, Chaudry IH. Cecal ligation and puncture. Shock. 2005;24(Suppl 1):52–57. doi: 10.1097/01.shk.0000191414.94461.7e. [DOI] [PubMed] [Google Scholar]
- 24.Ishii M, Hogaboam CM, Joshi A, Ito T, Fong DJ, Kunkel SL. CC chemokine receptor 4 modulates Toll-like receptor 9-mediated innate immunity and signaling. Eur J Immunol. 2008;38:2290–2302. doi: 10.1002/eji.200838360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lyons A, Goebel A, Mannick JA, Lederer JA. Protective effects of early interleukin 10 antagonism on injury-induced immune dysfunction. Arch Surg. 1999;134:1317–1323. doi: 10.1001/archsurg.134.12.1317. discussion 1324. [DOI] [PubMed] [Google Scholar]
- 26.Nicoletti F, Di Marco R, Zaccone P, Magro G, Di Mauro M, Grasso S, Meroni PL. Endogenous interleukin-12 only plays a key pathogenetic role in non-obese diabetic mouse diabetes during the very early stages of the disease. Immunology. 1999;97:367–370. doi: 10.1046/j.1365-2567.1999.00836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alves-Filho JC, Sonego F, Souto FO, Freitas A, Verri WA, Jr., Auxiliadora-Martins M, Basile-Filho A, McKenzie AN, Xu D, Cunha FQ, Liew FY. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med. 2010;16:708–712. doi: 10.1038/nm.2156. [DOI] [PubMed] [Google Scholar]
- 28.Cotter MJ, Norman KE, Hellewell PG, Ridger VC. A novel method for isolation of neutrophils from murine blood using negative immunomagnetic separation. Am J Pathol. 2001;159:473–481. doi: 10.1016/S0002-9440(10)61719-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki Y, Asano K, Niimi K, Miyata J, Shiraishi Y, Fukunaga K, Shiomi T, Nakajima T, Oguma T, Sayama K, Ishizaka A. TP receptor-mediated release of eosinophil chemotactic activity from human bronchial smooth muscle cells. Eur J Pharmacol. 2008;600:133–139. doi: 10.1016/j.ejphar.2008.09.044. [DOI] [PubMed] [Google Scholar]
- 30.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 31.Steinhauser ML, Hogaboam CM, Lukacs NW, Strieter RM, Kunkel SL. Multiple roles for IL-12 in a model of acute septic peritonitis. J Immunol. 1999;162:5437–5443. [PubMed] [Google Scholar]
- 32.Moreno SE, Alves-Filho JC, Alfaya TM, da Silva JS, Ferreira SH, Liew FY. IL-12, but not IL-18, is critical to neutrophil activation and resistance to polymicrobial sepsis induced by cecal ligation and puncture. J Immunol. 2006;177:3218–3224. doi: 10.4049/jimmunol.177.5.3218. [DOI] [PubMed] [Google Scholar]
- 33.van der Poll T, Marchant A, Buurman WA, Berman L, Keogh CV, Lazarus DD, Nguyen L, Goldman M, Moldawer LL, Lowry SF. Endogenous IL-10 protects mice from death during septic peritonitis. J Immunol. 1995;155:5397–5401. [PubMed] [Google Scholar]
- 34.Kasten KR, Muenzer JT, Caldwell CC. Neutrophils are significant producers of IL-10 during sepsis. Biochem Biophys Res Commun. 2010;393:28–31. doi: 10.1016/j.bbrc.2010.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reddy RC, Standiford TJ. Effects of sepsis on neutrophil chemotaxis. Curr Opin Hematol. 2010;17:18–24. doi: 10.1097/MOH.0b013e32833338f3. [DOI] [PubMed] [Google Scholar]
- 36.Reddy RC, Narala VR, Keshamouni VG, Milam JE, Newstead MW, Standiford TJ. Sepsis-induced inhibition of neutrophil chemotaxis is mediated by activation of peroxisome proliferator-activated receptor-{gamma} Blood. 2008;112:4250–4258. doi: 10.1182/blood-2007-12-128967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olson TS, Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol. 2002;283:R7–R28. doi: 10.1152/ajpregu.00738.2001. [DOI] [PubMed] [Google Scholar]
- 38.Cummings CJ, Martin TR, Frevert CW, Quan JM, Wong VA, Mongovin SM, Hagen TR, Steinberg KP, Goodman RB. Expression and function of the chemokine receptors CXCR1 and CXCR2 in sepsis. J Immunol. 1999;162:2341–2346. [PubMed] [Google Scholar]
- 39.Baron EJ, Proctor RA. Elicitation of peritoneal polymorphonuclear neutrophils from mice. J Immunol Methods. 1982;49:305–313. doi: 10.1016/0022-1759(82)90130-2. [DOI] [PubMed] [Google Scholar]
- 40.Benjamim CF, Hogaboam CM, Kunkel SL. The chronic consequences of severe sepsis. J Leukoc Biol. 2004;75:408–412. doi: 10.1189/jlb.0503214. [DOI] [PubMed] [Google Scholar]
- 41.Kaplan JM, Cook JA, Hake PW, O'Connor M, Burroughs TJ, Zingarelli B. 15-Deoxy-delta(12,14)-prostaglandin J(2) (15D-PGJ(2)), a peroxisome proliferator activated receptor gamma ligand, reduces tissue leukosequestration and mortality in endotoxic shock. Shock. 2005;24:59–65. doi: 10.1097/01.shk.0000167108.88376.f2. [DOI] [PubMed] [Google Scholar]
- 42.Manthous CA, Hall JB, Samsel RW. Endotoxin in human disease. Part 1: Biochemistry, assay, and possible role in diverse disease states. Chest. 1993;104:1572–1581. doi: 10.1378/chest.104.5.1572. [DOI] [PubMed] [Google Scholar]
- 43.Ye X, Liu SF. Lipopolysaccharide down-regulates Sp1 binding activity by promoting Sp1 protein dephosphorylation and degradation. J Biol Chem. 2002;277:31863–31870. doi: 10.1074/jbc.M205544200. [DOI] [PubMed] [Google Scholar]
- 44.Marchant A, Bruyns C, Vandenabeele P, Ducarme M, Gerard C, Delvaux A, De Groote D, Abramowicz D, Velu T, Goldman M. Interleukin-10 controls interferon-gamma and tumor necrosis factor production during experimental endotoxemia. Eur J Immunol. 1994;24:1167–1171. doi: 10.1002/eji.1830240524. [DOI] [PubMed] [Google Scholar]
- 45.Sewnath ME, Olszyna DP, Birjmohun R, ten Kate FJ, Gouma DJ, van Der Poll T. IL-10-deficient mice demonstrate multiple organ failure and increased mortality during Escherichia coli peritonitis despite an accelerated bacterial clearance. J Immunol. 2001;166:6323–6331. doi: 10.4049/jimmunol.166.10.6323. [DOI] [PubMed] [Google Scholar]
- 46.Alves-Filho JC, Sonego F, Souto FO, Freitas A, Verri WA, Jr., Auxiliadora-Martins M, Basile-Filho A, McKenzie AN, Xu D, Cunha FQ, Liew FY. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med. 16:708–712. doi: 10.1038/nm.2156. [DOI] [PubMed] [Google Scholar]
- 47.Carson WF, Cavassani KA, Dou Y, Kunkel SL. Epigenetic regulation of immune cell functions during post-septic immunosuppression. Epigenetics. 2011;6:273–283. doi: 10.4161/epi.6.3.14017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 49.Chen C, Zhao M, Yin N, He B, Wang B, Yuan Y, Yu F, Hu J, Yin B, Lu Q. Abnormal histone acetylation and methylation levels in esophageal squamous cell carcinomas. Cancer Invest. 2011;29:548–556. doi: 10.3109/07357907.2011.597810. [DOI] [PubMed] [Google Scholar]
- 50.Zhao M, Liang G, Wu X, Wang S, Zhang P, Su Y, Yin H, Tan Y, Zhang J, Lu Q. Abnormal epigenetic modifications in peripheral blood mononuclear cells from patients with alopecia areata. Br J Dermatol. 2012;166:226–273. doi: 10.1111/j.1365-2133.2011.10646.x. [DOI] [PubMed] [Google Scholar]
- 51.Vroon A, Heijnen CJ, Kavelaars A. GRKs and arrestins: regulators of migration and inflammation. J Leukoc Biol. 2006;80:1214–1221. doi: 10.1189/jlb.0606373. [DOI] [PubMed] [Google Scholar]
- 52.Guo Z, Zhang M, Tang H, Cao X. Fas signal links innate and adaptive immunity by promoting dendritic-cell secretion of CC and CXC chemokines. Blood. 2005;106:2033–2041. doi: 10.1182/blood-2004-12-4831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.