

Abstract
5-hydroxymethylcytosine (5hmC) is a recently discovered base in the mammalian genome, produced upon oxidation of 5-methylcytosine (5mC) in a process catalyzed by TET proteins. The biological functions of 5hmC and further oxidation products of 5mC are under intense investigation, as they are likely intermediates in DNA demethylation pathways. Here we describe a novel protocol to profile 5hmC at a genome-wide scale. This approach is based on sodium bisulfite–mediated conversion of 5hmC to cytosine-5-methylenesulfonate (CMS); CMS-containing DNA fragments are then immunoprecipitated using a CMS-specific antiserum. The anti-CMS technique is highly specific with a low background, and is much less dependent on 5hmC density than anti-5hmC immunoprecipitation (IP). Moreover, it does not enrich for CA and CT repeats, as noted for 5hmC DNA IP using antibodies to 5hmC. The anti-CMS protocol takes 3 d to complete.
INTRODUCTION
In mammalian genomes, 5mC is important for biological processes such as imprinting1, silencing of genes2, transposons3 and chromosomal stability4. The recently discovered Fe(II) and 2-oxoglutarate–dependent dioxygenase ‘TET’ family of enzymes (TET1, TET2 and TET3 in humans) are capable of oxidizing the methyl group of 5mC to 5hmC5,6, a base that is now known to be present at significant levels in mammalian genomes5–9.
5hmC has several known biological properties that distinguish it from 5mC. During DNA replication, DNMT1 maintains symmetrical CpG methylation at sites across from 5mC but not 5hmC10, implying that Tet-mediated hydroxymethylation at CpGs could induce passive DNA demethylation. Methyl-CpG binding (MBD) domains, which are present on a number of proteins that induce repressive chromatin states (MeCP2, MBD1, MBD2), bind methylated CpGs but do not recognize 5hmCpGs efficiently11,12. Finally, Tet proteins can further oxidize 5hmC to 5-formylcytosine (5fC) and 5-carboxycytosines (5caC)9,13,14, which in turn can be removed by the DNA glycosylase TDG and potentially replaced with cytosine as part of a ‘DNA demethylation’ pathway14,15.
Tet1 and Tet2 are highly expressed in mouse embryonic stem (ES) cells (mESCs), and they regulate lineage specification upon ES cell differentiation16. Expression levels of Tet1 and Tet2, as well as the total amount of 5hmC in the genome, drop sharply upon differentiation of mouse ES cells16. TET2 is a tumor suppressor mutated in ~20% of human myeloid malignancies17 and in a smaller percentage of lymphoid malignancies18, and Tet2-deficient mice show expansion of the hematopoietic stem/progenitor cell compartment and develop cancers similar to human chronic myelomonocytic leukemias18–21. Furthermore, 5hmC expression levels are markedly lower in bone marrow samples from myeloid cancer patients with somatic TET2 mutations, compared with bone marrow from healthy controls22. 5hmC is also decreased in breast, prostate and colon cancer samples compared with normal cells, further suggesting a link between the amount of 5hmC and cancer23. Tet3 is essential for demethylation of the male pronucleus in fertilized mouse zygotes24,25, and Tet3-deficient mice show neonatal lethality25.
In light of the many biological roles of Tet proteins and 5hmC, it is important to be able to reliably map 5hmC in the genomic DNA of various cell types and to correlate its presence or absence with gene expression or other genomic events. Here we present an anti-5hmC mapping technique that relies on converting 5hmC to the modified base CMS, and then precipitating with the antibody against CMS. In the accompanying protocol in this issue, we introduce a combined enzymatic and chemical technique, called glucosylation, periodate oxidation and biotinylation (GLIB), for mapping 5hmC26. As discussed below, both methods are superior to antibodies against 5hmC in sequencing applications27.
Sodium bisulfite sequencing is commonly used to distinguish cytosine and 5mC. Cytosine reacts with sodium bisulfite and undergoes deamination to uracil, whereas reaction and deamination of 5mC is roughly 100 times slower28,29. Therefore, after subsequent PCR and sequencing, cytosine is read as ‘T’ and 5mC as ‘C’30. In contrast, 5hmC reacts efficiently with sodium bisulfite to yield a distinct adduct, CMS29. CMS is read as ‘C’ during sequencing (Fig. 1), and is therefore indistinguishable from 5mC under the standard bisulfite protocol12,31. However, we anticipated that the bulky, negatively charged CMS adduct would be highly immunogenic. The CMS-specific antiserum generated by our laboratory and used in this protocol is of high titer and is very specific for CMS, and it does not cross-react with unmodified, methylated or hydroxymethylated DNA (Fig. 2a)22. This antibody will be commercialized soon, and currently it can be obtained from our laboratory by request.
Figure 1.
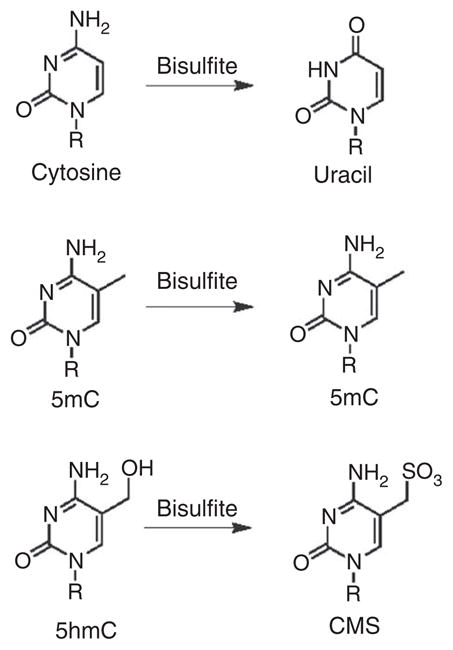
Effect of sodium bisulfite treatment on C, 5mC and 5hmC. Top, bisulfite-mediated deamination of cytosine to uracil (U) at high pH. Middle, 5mC remains as 5mC during bisulfite conversion because of the slow reaction rate. Bottom, bisulfite quickly converts 5-hydroxymethylcytosine to form CMS. This adduct does not readily undergo deamination.
Figure 2.

Testing antibody specificity and comparing the density dependence of anti-5hmC and anti-CMS methods. (a) Left, cytosine-, 5mC-, 5hmC- or CMS-containing oligonucleotides (201 bp) were 32P-end-labeled and precipitated with the CMS-specific antibody. The IP efficiency was measured by counting the radioactivity in the IP material. Right, unmethylated lambda and mESC genomic DNA were sheared to 200 bp and end-labeled with 32P. Genomic DNA was subsequently precipitated with the CMS-specific antibody. IP efficiency was measured as described above. Error bars show s.d. (n = 3). (b) Density dependence of 5hmC-specific and CMS-specific antisera demonstrated using the dot-blot method. Twofold dilutions of a 201-bp oligonucleotide with different densities of 5hmC were spotted on a nitrocellulose membrane. The data were plotted as dot-blot intensity normalized to the total amount of 5hmC in the spot (i.e., 1 pmol of an oligonucleotide in which all 81 cytosines are replaced with 5hmC contains 81 pmol 5hmC, equivalent to 5 pmol of an oligonucleotide generated with a 20:80 mixture of 5hmC:C, which contains ~16 5hmC on average). Top, anti-5hmC dotblot. Bottom, anti-CMS dotblot. (c) Precipitation of 5hmC-containing DNA fragments via anti-CMS IP and anti-5hmC IP. PCR amplicons (201 bp, 81 cytosines) that contained varying amounts of 5hmC were bisulfite-converted or left untreated, 32P-end-labeled and precipitated using the appropriate method. The data shown in this figure are reproduced with permission from refs. 22 and 27. pAb, polyclonal antibody.
Techniques for enrichment of a given target are said to be ‘density dependent’ if they are more efficient in precipitating molecules that contain a high (rather than a low) spatial concentration of the target in question. For example, available 5mC-specific antibodies are markedly more efficient in precipitating DNA fragments of a given size that contain ten 5mCs instead of one, and are therefore density dependent12,32. Density dependence has both advantages and disadvantages. A highly density-dependent method for 5mC precipitation will enrich methylated CpG islands to the exclusion of the rest of the genome. A method that efficiently precipitates a single 5mC will not distinguish between regions of dense and sparse methylation, which is a serious problem considering that 5mC is very widespread in vertebrate genomes33. It is not inherently obvious what the ‘right’ level of density dependence is for the precipitation of a given target. When considering 5hmC, however, a key consideration is the scarcity of the 5hmC mark relative to 5mC. In hematopoietic organs and cells, 5hmC is found at only 1–3% the amount of 5mC9,22, even though Tet2 is important for hematopoietic cell development21,22. We therefore considered it important that the 5hmC precipitation methods efficiently precipitate sparsely hydroxymethylated DNA.
To test the efficiency of both GLIB and anti-CMS IP methods in 5hmC recognition and precipitation, we generated PCR products containing varying ratios of cytosine and 5hmC to simulate DNA containing high or low concentrations of 5hmC. The polyclonal antibodies we raised against CMS are in fact more sensitive and specific than any 5hmC-specific antibodies we have generated or tested. They were more sensitive and less density dependent in dot-blotting applications22 (Fig. 2b). Precipitation efficiency of the different 5hmC pull-down techniques is described in Pastor et al.27 (Fig. 2c). Briefly, the antibody against CMS did have some density dependence and incomplete pull-down (20% precipitation of a singly hydroxymethylated oligo 70% pull-down of a heavily hydroxymethylated oligo) but also very low background (0.1% pull-down of methylated or unmethylated DNA)27 (Fig. 2c). Anti-CMS therefore precipitated a DNA fragment with one 5hmC ~200 times as efficiently as a fragment with no 5hmCs. The commercially available Active Motif 5hmC-specific antibody showed inferior performance (2–3% background pull-down, 30% pull-down of a single 5hmC, 80% pull-down of a heavily hydroxymethylated PCR product) and, in our hands, was sensitive to even slight changes in the precipitation protocol (W.A.P., Y.H. and A.R., unpublished observations).
Applied to genomic DNA, both the GLIB and anti-CMS methods show higher degrees of total pull-down when applied to samples with large amounts of 5hmC (HEK293 cells expressing TET1), less pull-down from an endogenous source containing an intermediate level of 5hmC (mESCs) and still less pull-down when applied to a DNA source almost entirely lacking 5hmC (HEK293 cells; Fig. 3).
Figure 3.

Precipitation of genomic DNA using the anti-CMS method. (a) For testing the efficiency of precipitation from genomic DNA, three sources with varying amounts of 5hmC were used. HEK293 cells contain little 5hmC, mouse ES cells contain intermediate levels and HEK293 cells expressing the TET1 catalytic domain (293 TET1) contain large amounts, as demonstrated by an anti-5hmC dot blot. (b) DNA from the HEK293, ES and HEK293 TET1 cells was sonicated, end-labeled and precipitated using the anti-CMS method. Note that the degree of pull-down in each sample correlates with the extent of hydroxymethylation observed in part a. Error bars show s.d. (n = 3). The data shown in this figure are reproduced with permission from ref. 27.
The GLIB and CMS methods were applied to map 5hmC in mESCs, and 5hmC-enriched regions of the genome (termed HERGs) were identified. Even though only ~5% of the genome was defined as 5hmC enriched, roughly 80% of GLIB and CMS HERGs overlapped and all biologically relevant trends were almost identical in the GLIB and CMS data sets. The primary difference was that the CMS HERGs were on average slightly smaller than GLIB HERGs (1,168 bases versus 1,422 bases), probably because smaller DNA fragments were used in the CMS as compared with the GLIB sequencing (200-bp average fragment size for CMS versus 300 bp for GLIB). In summary, the two techniques have strongly cross-validated each other27.
The performance of anti-5hmC mapping studies has been spottier. When data from multiple studies34–37 were analyzed independently, pull-down with the 5hmC-specific antibody yielded low signal over background compared with the anti-CMS and GLIB methods38. In addition, the antibody against 5hmC showed substantial enrichment for areas immediately adjacent to CA and CT repeats. Although it is possible that these reflect real deposits of 5hmC, CA and CT repeats do not contain substantial amounts of 5mC, and the peaks are probably an artifact arising from an ability of the antibody against 5hmC to precipitate regions rich in unmodified cytosine. The GLIB and anti-CMS methods do not produce enrichment at these repeats38.
Formyl and carboxycytosine are unlikely to be identified by the CMS technique: they would not be predicted to form the CMS adduct14,39, and moreover they are relatively rare bases in genomic DNA from ES cells and other cells/organisms9.
Recently, three methods have been developed for identification of 5hmC at a single-base resolution. The first method, developed by Song et al.40, involves the addition of large covalently attached adducts to 5hmC using ‘click’ chemistry. The adducts stall DNA polymerase during replication, and the delay can be measured, thereby identifying sites of hydroxymethylation. This method requires the use of a Pacific Biosciences STS instrument, which cannot yet be used to measure modification in mammals. The other methods rely on the critical observation that 5mC and 5hmC are read as ‘C’ in bisulfite sequencing, whereas cytosine, 5fC and 5caC are deaminated and read as ‘T’. The second method, developed by Booth et al.39, entails oxidation of 5hmC to 5fC using KRuO4. Sodium bisulfite sequencing is performed on the same sample before and after KRuO4 treatment: 5hmC is read as ‘C’ before treatment and ‘T’ after treatment, whereas other bases are unaffected, thus allowing quantification of 5hmC at a given site. The third method involves protection of 5hmC by glucosylation with β-glucosyltransferase (BGT), followed by oxidation of all 5mC to 5caC using a high concentration of recombinant Tet protein41. Upon subsequent bisulfite treatment, only 5hmC bases will resist deamination and will therefore be identifiable. Although these latter two methods are broadly applicable and can be used on most sequencing platforms, they require a very high read coverage to identify the rare 5hmC base, making them prohibitively expensive for most applications at present.
In our published analysis, we used bisulfite-treated input DNA as a control for anti-CMS IP. Two other negative controls are possible. For experiments in mouse systems, anti-CMS IP can be undertaken with Dnmt1−/−, Dnmt3a−/− and Dnmt3b−/− ES cells, which do not contain 5hmC35. Alternatively, although we have not yet tested this, anti-CMS IP could be undertaken with DNA that has been treated with recombinant BGT42. Glucosylation of 5hmC by BGT would be expected to block conversion to CMS and eliminate pull-down with the CMS-specific antibody.
The CMS method has drawbacks that must be understood before use. The CMS technique relies on bisulfite conversion of DNA. To achieve efficient bisulfite conversion, DNA must be well sheared and dissolved. In addition, special mapping algorithms must be used to map the lower-complexity bisulfite-treated genome43. Moreover, to eliminate PCR bias, it is also essential to sequence the bisulfite-treated input DNA (before IP), which is used for normalization of the reads from the CMS-IP DNA during bio-informatics analysis. A meta-analysis by Matarese et al.38 showed that input bisulfite-treated DNA showed an apparent enrichment at CGI promoters; this is probably because more PCR bias is introduced in a less-complex AT-rich genome (3 bases) compared with a normal genome (4 bases).
It is strongly recommended that new users perform quick validation with synthetic substrates to confirm efficient precipitation before undertaking expensive sequencing of genomes (Box 1).
Box 1. Optimization of CMS-IP conditions ● TIMING 2 d.
Prepare DNA oligonucleotides containing different amounts of 5hmC ● TIMING ~8 h
-
1
Generate a 201-bp DNA oligonucleotide using PCR (DNA sequence and primer information is listed in Fig. 5). 5hmC-containing oligonucleotides are generated by replacing dCTP with hmdCTP; oligonucleotides containing different ratios of 5hmC and C are made using different ratios of dCTP to hmdCTP (The dCTP to hmdCTP ratio is listed in Table 1). Note that other ~200-bp amplicons containing 5hmC can be used for this application as well.
-
2
Set up the PCR using the following recipe and program.
Reagent Volume (μl) Final Template, 5 ng μl−1 2 10 ng Primer mixture, 10 μM 2 0.2 μM dNTP, 10 mM 5 1 mM Reaction buffer, 10× 5 1× Taq polymerase (5 U μl−1) 1 0.1 U ddH2O Up to 50 Cycle Denature Anneal Extend Hold 1 95 °C, 2 min — — — 2–36 95 °C, 20 s 65 °C, 30 s 72 °C, 2 min — 37 — — 72 °C, 10 min — 38 — — — 4 °C, ∞ -
3
Run the PCR product on a 2% (wt/vol) agarose gel (120 V, ~20 min) and purify it with a Qiagen gel extraction kit, according to the manufacturer’s instructions.
-
4
Treat with sodium bisulfite according to the manufacturer’s instructions.
-
5
End-label bisulfite-treated DNA oligonucleotide with 32P using T4 PNK enzyme (NEB) in a 50-μl volume as tabulated below. Incubate at 37 °C for 1 h.
Reagent Volume (μl) Final DNA, 250 ng X 250 ng T4 PNK (10 U μl−1) 1 0.4 U T4 PNK buffer, 10× 2.5 1× ATP [γ-32P] 1 ddH2O Fill to 25 ! CAUTION ATP [γ-32P] is a radioactive material. Radiation should be handled carefully and only after training, according to rules set by the facility at which the experiments are conducted.
-
6
Remove unincorporated ATP [γ-32P] by processing through a G50 Illustra MicroSpin column (GE Healthcare), according to the manufacturer’s instructions.
■ PAUSE POINT DNA can be stored at − 20 °C for 2 weeks, but as γ-32P has a half-life of 2 weeks, sensitivity will drop as the DNA is stored.
Optimize CMS-IP conditions ● TIMING 5 h to 1 d
-
7
Denature 1 μl (typically 10 ng) of labeled DNA in 29 μl of TE buffer and 20 μl of NaOH/EDTA buffer and incubate at 95 °C for 12 min.
-
8
Immediately put the DNA mixture into an ice slurry for 10 min, and then add 50 μl of chilled 2 M ammonium acetate (pH 7.0).
-
9
Mix the denatured DNA mixture with TE buffer, 50 μl of 10× IP buffer and various amounts of CMS-specific antiserum to yield a total volume of 500 μl, and incubate at 4 °C for 2 h.
-
10
For each reaction, preincubate various amounts of protein G beads with PBS-BSA twice for 10 min, resuspend in an equal volume of 1× IP buffer and add to the DNA and antibody mixture and incubate at 4 °C for 2 h or overnight.
-
11
Spin the mixture briefly and put on a magnet for 3 min. Transfer 20% of the clear supernatant to a new tube and remove the rest of the clear supernatant.
-
12
Add 500 μl of 1× IP buffer to the beads and rotate at 4 °C for 5 min.
-
13
Repeat Steps 11 and 12 twice.
-
14
Transfer 20% of unbound supernatant and washes into scintillation vials and transfer the beads to another scintillation vial.
-
15
Count the radioactivity using a scintillation counter and calculate the IP efficiency based on the counts per minute (c.p.m.)from unbound and bound fractions (measured IP)/(measured IP + 5× measured pooled supernatant and washes).
▲ CRITICAL STEP Measured c.p.m. tend to drop as beads settle in the scintillation counter, and thus vials must be inverted repeatedly before counting in order to ensure accuracy.
-
16
Compare the IP efficiency with the theoretical IP efficiency in Table 1. Compare with the optimized values in ref. 27 and Figure 4.
Experimental design
Genomic DNA is isolated from cells or tissues and sheared using a Covaris Adaptive Focused Acoustics (AFA) instrument. The anti-CMS method is then implemented as described below. In the anti-CMS method, unmodified lambda DNA should be added to the genomic DNA before treatment with sodium bisulfite, as a control to determine C to T conversion efficiency; meanwhile, an input sample should be used to serve as a background for anti-CMS IP. More strictly, genomic DNA sequentially treated with BGT, sodium bisulfite and CMS-IP can be used as a pull-down background as well because glucosylation masks 5hmC.
As the CMS-specific antiserum we have used is a rabbit polyclonal antiserum, an optimization step is required for each new batch of the antibody to control for variations in the antiserum obtained from different animals. This optimization step is described in Box 1 (see also Table 1) and Figures 4 and 5.
TABLE 1.
Calculated 5hmC amount and IP efficiency per amplicon.
| No. of 5hmC molecules per amplicon | 81 | 20 | 4 | 2 | 1 | 0.5 | 0.25 | 0 |
|---|---|---|---|---|---|---|---|---|
| 5hmdCTP (%) | 100 | 24.69 | 4.94 | 2.47 | 1.23 | 0.62 | 0.31 | 0 |
| dCTP (%) | 0 | 75.31 | 95.06 | 97.53 | 98.77 | 99.38 | 99.69 | 100 |
| Theoretical IP efficiency | 100 | 100 | 84.4 | 61.9 | 38.7 | 22.0 | 11.8 | 0 |
Figure 4.

Optimization of the anti-CMS IP method. (a) A 201-bp DNA oligonucleotide containing an average of hmC bases per DNA molecule was end-labeled and immunoprecipitated with increasing amounts of CMS-specific antiserum and 40 μl of protein G beads. A volume of 1 μl of CMS-specific antiserum was chosen as the optimal condition. (b) A 201-bp 5hmC-containing DNA oligonucleotide was end-labeled and immunoprecipitated with 1 μl of CMS-specific antiserum and varying amounts of protein G beads. A volume of 25 μl of protein G beads was chosen as optimal. Error bars show s.d. (n = 3).
Figure 5.

Sequence of hmC-containing oligos for optimization of pull-down. See Box 1 for procedural details.
An overview of the working procedure is shown in Figure 6. At least two biological replicates are recommended to obtain statistically significant results.
Figure 6.

Flowchart of the anti-CMS method.
MATERIALS
REAGENTS
DNA sample (RNA-free and suggested starting amount is > 10 μg)
Sodium hydroxide (NaOH; Sigma-Aldrich, cat. no. 221465)
EDTA (Sigma-Aldrich, cat. no. E9884)
Ammonium acetate (Sigma-Aldrich, cat. no. A1542)
-
Phenol:chloroform:isoamylalcohol (25:24:1; Sigma-Aldrich, cat. no. P2069)
! CAUTION Phenol:chloroform:isoamyl alcohol (25:24:1) is irritating to the eyes, skin and respiratory system. Please handle it in a fume hood while wearing gloves.
-
Chloroform:isoamylalcohol (24:1; Sigma-Aldrich, cat. no. 25666)
! CAUTION Chloroform:isoamyl alcohol (25:24:1) is toxic and is irritating to the eyes, skin and respiratory system. Please handle it in a fume hood while wearing gloves.
Triton X-100 (Sigma-Aldrich, cat. no. T8787)
-
Sodium dodecyl sulfate (SDS; Sigma-Aldrich, cat. no. 436143)
! CAUTION In solid powder form, SDS is irritating to the eyes and the respiratory system. When handling dry SDS powder, wear gloves, goggles and a face mask.
Sodium chloride (NaCl; Sigma-Aldrich, cat. no. S3014)
GenElute-LPA (Sigma-Aldrich, cat. no. 56575)
Sodium phosphate, monobasic (Fisher Scientific, cat. no. S397-500)
Sodium phosphate, dibasic (Fisher Scientific, cat. no. S375-500)
Sodium acetate (Sigma-Aldrich, cat. no. S2889-250G)
Unmethylated lambda DNA (Promega, cat. no. D1521)
MethylCode bisulfite conversion kit (Invitrogen, cat. no. MECOV-50)
MinElute PCR cleanup kit (Qiagen, cat. no. 28004)
Paired-end DNA sample prep kit (Illumina, cat. no. PE-102-1001)
Methylated adapters (Illumina, cat. no. ME-100-0010)
UltraPure agarose (Invitrogen, cat. no. 16500500)
AmpurXP beads (Beckman, cat. no. A63880)
Dynabeads protein G (Invitrogen, cat. no. 100-04D)
PBS (10×; Meditech CellGro, cat. no. 46-013-CM)
Qubit dsDNA high-sensitivity (HS) assay kit (Invitrogen, cat. no. Q32851)
dNTPs (Promega, cat. no. U1511)
Illustra MicroSpin G-50 columns (GE Healthcare, cat. no. 27-5330-01)
Proteinase K (Roche, cat. no. 03115852001)
pfuTurbo C× Hotstart DNA polymerase (Agilent, cat. no. 600410)
Choice-Taq DNA polymerase (Denville Scientific, cat. no. CB4050-2)
T4 DNA polymerase (NEB, cat. no. M0203S)
Klenow fragment (3′→5′ exo–; NEB, cat. no. M0212M)
T4 Polynucleotide kinase (PNK; NEB, cat. no. M0201S)
Ethanol (Sigma-Aldrich, cat. no. E7023)
BSA (Sigma-Aldrich, cat. no. A7906)
Tris (Sigma-Aldrich, cat. no. 154563)
Agarose (Invitrogen, cat. no. 16500500)
EQUIPMENT
Adaptive Focused Acoustics (AFA) S2 (Covaris)
DynaMag-2 (Invitrogen, cat. no. 123-21D)
Gel running apparatus (Thermo Scientific)
NanoDrop 1000 (Thermo Scientific)
Thermocycler (Applied Biosystems, cat. no. 2720)
Nutator (Southwest Science, cat. no. SB3D2300)
Thermomixer (Eppendorf)
Desktop centrifuge (Eppendorf, cat. no. 5424)
Qubit 2.0 fluorometer (Invitrogen, cat. no. Q32866)
StepOnePlus real-time PCR system (Life Technologies)
Microcentrifuge (VWR, cat. no. C1413-VWR230)
Bioanalyzer (Agilent)
P20 pipette tips
REAGENT SETUP
▲ CRITICAL All of the reagents listed below can be stored indefinitely at room temperature (20 °C) but must be discarded if a visible precipitate is formed.
TE buffer TE contains 10 mM Tris (pH 8.0) and 1 mM EDTA.
TAE buffer TAE contains 40 mM Tris (pH 8.1), 20 mM acetate and 2 mM EDTA.
Sodium phosphate buffer, 1 M, pH 7.0 Sodium phosphate buffer contains 57.7 ml of 1 M Na2HPO4 and 42.3 ml of 1 M NaH2PO4.
IP buffer, 10× IP buffer contains 100 mM sodium phosphate (pH 7.0), 1.4 M NaCl and 0.5% (vol/vol) Triton X-100.
Proteinase K digestion buffer Proteinase K digestion buffer contains 50 mM Tris-HCl (pH 8.0), 10 mM EDTA and 0.5% (wt/vol) SDS.
PROCEDURE
DNA preparation and size selection ● TIMING 3 h, depending on the number of samples
-
1|
Pre-chill the Covaris AFA S2 instrument to 4 °C and degas for 30 min.
-
2|
Dilute 10 μg of genomic DNA into 100 μl of TE buffer and transfer it to a Covaris microtube (6 × 16 mm). For CMS-IP, spike in 50 ng of unmethylated lambda DNA to check bisulfite conversion efficiency.
▲ CRITICAL STEP The DNA input amount can be increased or decreased. The ratio of genomic DNA to unmethylated lambda DNA should be kept at 200:1.
▲ CRITICAL STEP If the amount of DNA is increased, shear no more than 10 μg of DNA at a time.
-
3|
Shear genomic DNA in a microtube using Covaris AFA, according to the manufacturer’s instructions. The target fragment size depends on the sequencing platform and application. For CMS-IP on the Illumina sequencing platform, the recommended target fragment size is 200 bp.
-
4|
Size selection can be performed in two ways: by using AmpurXP beads (option A) or by gel extraction (option B) (both methods are equally recommended).
-
AmpurXP beads (all steps are performed in 1.5-ml DNA LoBind microtubes) ● TIMING 2 h
Clean up sheared DNA using the Qiagen MinElute PCR cleanup kit according to the manufacturer’s instructions and elute in 20 μl of elution buffer.
-
Dilute DNA into 200 μl of nuclease-free water and mix with 100 μl of AmpurXP beads by vortexing the beads for 10 s (this ratio between DNA and beads is to select the fragments < 250 bp) and then pulse-spin.
▲ CRITICAL STEP Use no more than 5 μg of DNA for each reaction in order to prevent the overloading of beads, which leads to inaccurate size selection.
Incubate the mixture at room temperature for 10–20 min.
Place the tube in a DynaMag-2 magnetic rack for ~5–10 min until the solution clears.
Carefully transfer the supernatant into a new 1.5-ml tube without disturbing the pellet.
Incubate 300 μl of the supernatant from Step 4A(v) with 60 μl of AmpurXP beads by vortexing the mixture for 10 s (this ratio of DNA to beads is to select fragments > 100 bp) and then pulse-spin.
Repeat Steps 4A(iii) and 4A(iv).
Remove and discard the supernatant without disturbing the beads.
Wash the beads three times with 200 μl of 70% (vol/vol) ethanol without disturbing the beads.
Remove the 70% (vol/vol) ethanol carefully. Take the tubes containing the beads off the magnets and spin briefly in a minicentrifuge.
Place the tubes containing the beads back onto the magnets again for 1–2 min until all the beads migrate to the wall of the tube.
Remove residual ethanol carefully using P20 pipette tips and dry the beads at room temperature for < 3 min.
Remove the tubes containing the beads from the magnets, resuspend the beads in 30 μl of nuclease-free water, soak the beads for 2 min and then resuspend them by vigorously pipetting more than 20 times.
Incubate the beads at room temperature for 5 min, and then apply them to the magnets again for 3 min.
-
Carefully transfer the clear supernatant to another clean tube.
■ PAUSE POINT DNA can be stored at − 20 °C for months.
-
Gel extraction ● TIMING 2 h
Run the fragmented genomic DNA on a 2% (wt/vol) agarose gel (120 V, ~20 min) and excise DNA of the desired size ±50 bp.
-
Clean the excised DNA with a Qiagen MinElute gel extraction kit according to the manufacturer’s instructions and elute the DNA in 30 μl of nuclease-free water buffer.
■ PAUSE POINT DNA can be stored at − 20 °C for months.
-
CMS-IP ● TIMING ~3 d
▲ CRITICAL The PROCEDURE describes the preparation of DNA libraries using the Illumina paired-end library sample preparation kit with methylated adapters. However, CMS-IP for high-throughput sequencing also can be used on other similar sequencing platforms. For example, to perform CMS-IP on SOLiD platform, use the SOLiD library preparation kit and methylated adapter provided by Applied Biosystems, and then perform CMS-IP and sequencing as described in Steps 5–41. All the experiments are performed using DNA LoBind 1.5-ml microtubes.
-
5|
Set up a reaction to end-repair 5 μg of sheared and size-selected genomic DNA (from Step 4) as tabulated below. Incubate at 20 °C for 30 min according to the Illumina paired-end sample preparation guide.
Reagent Volume (μl) Final DNA sample 30 5 μg T4 DNA ligase buffer with 10 mM ATP, 10× 10 1× dNTP, 10 mM 4 0.4 mM T4 DNA polymerase (3 U μl−1) 5 0.15 U T4 Polynucleotide kinase (PNK; 10 U μl−1) 5 0.5 U ddH2O Up to 100 ? TROUBLESHOOTING
-
6|
Clean up the reaction using the Qiagen MinElute PCR cleanup kit according to the manufacturer’s instructions and elute twice in 16 μl of elution buffer.
-
7|
Set up a reaction to add ‘A’ overhangs to the DNA fragments using dATP and Klenow enzyme, as tabulated below. Incubate at 37 °C for 30 min.
Reagent Volume (μl) Final DNA sample 32 Klenow buffer, 10× 5 1× dATP, 1 mM 10 0.2 mM Klenow exo - (5 U μl−1) 3 0.33 U ddH2O Up to 50 -
8|
Clean up the reaction using the Qiagen MinElute PCR cleanup kit according to the manufacturer’s instructions and elute in 10 μl of elution buffer.
-
9|
Set up a reaction to ligate paired-end methylated adapters to DNA fragments as tabulated below. Incubate at 16 °C overnight in a thermocycler in PCR tubes.
Reagent Volume (μl) Final DNA sample 10 DNA ligase buffer, 10× 5 1× PE methylated adapter mix 10 T4 DNA ligase (400 U μl−1) 1 8 U ddH2O Up to 50 ▲ CRITICAL STEP The adapter amount listed above is used for an initial DNA amount of 5 μg; if the starting DNA material is less, reduce the adapter amount correspondingly.
-
10|
Remove excess adapters by using AmpureXP beads. Mix 50 μl of ligation mixture with 65 μl of AmpureXP beads and pipette up and down more than 20 times.
-
11|
Incubate the beads and ligation mixture for 10 min and place the tubes on magnets for 5 min until all the beads are attached to the magnets.
-
12|
Remove the clear supernatant and wash the beads with 70% (vol/vol) ethanol twice without disturbing the beads on the magnet.
▲ CRITICAL STEP Do not perturb the beads on the magnet; if you do, it may reduce the recovery efficiency.
-
13|
Remove the 70% (vol/vol) ethanol carefully. Take the tubes containing the beads off the magnets and spin briefly in a minicentrifuge.
-
14|
Put the tubes containing the beads back onto the magnets again for 1–2 min until all the beads migrate to the wall of the tube.
-
15|
Remove residual ethanol carefully using P20 pipette tips and dry the beads at room temperature for < 3 min.
-
16|
Remove the tubes containing the beads from the magnets, resuspend the beads in 20 μl of TE buffer, soak them for 2 min and resuspend by vigorously pipetting more than 20 times.
-
17|
Incubate the beads at room temperature for 5 min and then apply them to the magnets again for 3 min.
-
18|
Carefully transfer the clear supernatant to another clean tube.
-
19|
Measure DNA concentration by NanoDrop or Qubit.
■ PAUSE POINT DNA can be stored at − 20 °C for months.
-
20|
Perform bisulfite conversion using the MethylCode conversion kit according to the manufacturer’s instructions.
Use < 500 ng of DNA in each reaction; for example, use three separate reactions of 500 ng each to bisulfite-treat a total of 1.5 μg of DNA.
▲ CRITICAL STEP DNA input amount for each bisulfite conversion reaction should be < 500 ng in order to yield maximal conversion efficiency (> 99%).
? TROUBLESHOOTING
-
21|
Measure the concentration of the bisulfite-converted DNA using a NanoDrop instrument and the RNA measurement program.
-
22|
Use 1 μg of bisulfite-converted DNA to perform CMS-IP. Retain 10 ng of DNA as input control; dilute the remaining 990 ng of DNA in 30 μl of TE buffer and denature by adding 20 μl of NaOH/EDTA buffer and then incubating at 95 °C for 12 min in the thermocycler.
▲ CRITICAL STEP DNA input amount can be reduced to 500 ng if the starting material is limited.
-
23|
Chill 2 M ammonium acetate (pH 7.0) on ice for 10–20 min.
-
24|
Immediately put the denatured, heated DNA into an ice slurry for 10 min.
-
25|
Add 50 μl of chilled 2 M ammonium acetate and incubate for 10 min on ice.
-
26|
Add 349 μl of TE buffer, 50 μl of 10× IP buffer and 1 μl of CMS-specific antiserum and incubate at 4 °C for 2 h.
-
27|
For each reaction, preincubate 30 μl of protein G beads with PBS-BSA for 10 min, spin the beads down and resuspend in 30 μl of 1× IP buffer (repeat this procedure twice).
-
28|
Add the DNA and antibody mixture from Step 26 to the beads and incubate at 4 °C for 2 h or overnight.
▲ CRITICAL STEP Our original experiments were performed using protein G beads alone; however, as protein A and protein G bind to distinct antibody isotypes, a mixture of protein A and protein G magnetic beads is likely to increase IP efficiency.
▲ CRITICAL STEP The ratio of the amount of CMS-specific antiserum to protein G beads is extremely important. Higher serum to beads ratio yields lower IP efficiency, as excess antibody-DNA fragment complexes will not be able to bind to the beads and will be washed away. The conditions listed are optimized for serum produced in our laboratory. For optimizing IP conditions for other antisera, please see Box 1.
-
29|
Put the IP mixture on a magnet for 3 min and then remove the clear supernatant carefully.
-
30|
Add 600 μl of 1× IP buffer to the beads and rotate at 4 °C for 5 min.
-
31|
Put the IP mixture on the magnet and then remove the supernatant carefully.
-
32|
Repeat Steps 30 and 31 two more times.
-
33|
Resuspend the beads in 250 μl of proteinase K digestion buffer with 0.3 mg ml−1 of proteinase K and incubate at 55 °C for 3 h or overnight with vigorous shaking (1,000 r.p.m.) in a thermomixer.
-
34|
Briefly centrifuge the bead-containing tube using a microcentrifuge and place the tube on the magnets for 3 min.
-
35|
Transfer the clear supernatant to another clean 1.5-ml microtube.
-
36|
Remove the bead-containing tube from magnets and resuspend in 250 μl of proteinase K digestion buffer. Incubate at 55 °C for 15 min with vigorous shaking (1,000 r.p.m.) in a thermomixer.
-
37|
Repeat Steps 34 and 35 and combine the clear supernatant into one tube.
-
38|
Mix the supernatant with an equal volume of phenol:chloroform:isoamyl alcohol (25:24:1) by inverting the tube more than 20 times and then centrifuging for 10 min at 20,000g at room temperature.
! CAUTION Phenol:chloroform:isoamyl alcohol (25:24:1) is toxic. Please handle it in a fume hood.
-
39|
Transfer the aqueous phase to another clean tube and mix with an equal volume of chloroform:isoamyl alcohol (24:1) by inverting the tube more than 20 times and then centrifuging for 10 min at 20,000g at room temperature.
! CAUTION Chloroform:isoamyl alcohol (24:1) is toxic. Please handle it in a fume hood.
-
40|
Transfer the aqueous phase to another clean tube and add a one-tenth volume of 5 M NaCl, 2.5 volumes of 100% (vol/vol) ethanol and 1 μl of 250 mg ml−1 linear polyacrylamide and mix well. Next, incubate for 1 h at 4 °C with rotation or at − 80 °C overnight without rotation.
-
41|
Centrifuge the mixture at 4 °C for 30 min at 20,000g. Carefully remove the clear supernatant without disturbing the DNA pellet.
-
42|
Add 800 μl of 70% (vol/vol) ethanol and centrifuge at room temperature for 10 min at 20,000g.
-
43|
Carefully remove the supernatant without disturbing the DNA pellet.
-
44|
Repeat Steps 42 and 43 once more.
-
45|
Dry the DNA pellet at room temperature for < 5 min.
-
46|
Resuspend the DNA pellet in 10 μl of nuclease-free water.
■ PAUSE POINT DNA can be stored at − 20 °C for months.
-
47|
Use half the amount of DNA from the input and the IP for each PCR in PCR tubes. The PCR conditions are listed below.
Reagent Volume (μl) Final DNA template 5 Forward primer, 25 μM 1 0.2 μM Reverse primer, 25 μM 1 0.2 μM dNTP, 10 mM 2 0.2 mM pfuTurbo C× buffer, 10× 5 1× pfuTurbo C× polymerase (2.5 U μl−1) 1 0.05 U ddH2O Up to 50 Cycle Denature Anneal Extend Hold 1 95 °C, 2 min — — — 2 98 °C, 30 s 3–8 98 °C, 15 s 60 °C, 30 s 72 °C, 4 min — 9 — — 72 °C, 10 min — 10 — — — 4 °C, ∞ ▲ CRITICAL STEP The number of PCR cycles can be increased to ten if the amount of library DNA obtained is insufficient; however, more PCR cycles could cause more clonal reads to be removed in the analysis of high-throughput sequencing data.
-
48|
Clean up the PCR and remove the primer dimers by using AmpurXP beads (Steps 10–19).
▲ CRITICAL STEP Use AmpurXP beads twice if there are residual primer dimers left after one application with AmpurXP beads.
? TROUBLESHOOTING
-
49|
Quantify the input and CMS-IP libraries by Qubit, quantitative PCR or Bioanalyzer.
▲ CRITICAL STEP Accurate library concentration quantification is very important for obtaining good-quality next-generation sequencing data.
-
50|
Load the library on an Illumina HiSeq instrument for next-generation sequencing.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
TABLE 2.
Troubleshooting table.
| Step | Problem | Possible reason | Solutions |
|---|---|---|---|
| 5 | Inaccurate genomic concentration | Genomic DNA is not completely dissolved and is very viscous; | Shear the DNA with needle or sharp tips; alternatively, after AFA shearing, measure the DNA concentration again |
| RNA contamination | Treat DNA with an increased amount of RNase and for a longer time | ||
| 20 | Incomplete bisulfite conversion (< 99%) | Genomic DNA amount is > 500 ng during reaction | Dilute DNA and perform the reaction |
| Bisulfite solution is too old | Freshly prepare the bisulfite solution | ||
| Reaction conditions are not optimized | Prolong the reaction time and change the reaction temperature | ||
| Treat samples twice with bisulfite; overtreatment with bisulfite could impair library integrity and can result in less-useful reads from high-throughput sequencing | |||
| 48 | Adapter and primer dimer contamination | Incomplete removal of the adapter and primer dimer | Remove the adapter and primer dimer once or several times more with AmpurXP beads |
● TIMING
Steps 1–4, DNA preparation and size selection: ~3 h, depending on the number of samples
Steps 5 and 6, end-repairing the DNA fragments: 1 h
Steps 7 and 8, adding A overhangs: 1 h
Step 9, adding methylated adapter: overnight
Steps 10–19, removing the excess adapter: 1 h
Step 20, bisulfite conversion: 4 h
Steps 21–28, CMS-IP: overnight
Steps 29–46: elution and cleanup of the IP fragments: 1 d
Steps 47–49, amplification and quality-checking of the library: 3 h
Step 50, running libraries on the sequencing instrument; variable, depending on the sequencing platform Box 1, optimization of CMS-IP conditions: 2 d
ANTICIPATED RESULTS
Five micrograms of DNA are used for each IP. After bisulfite treatment, ~1 μg of DNA can be recovered. After CMS-IP and PCR amplification, ~20 ng of libraries in 20 μl of nuclease-free water will be obtained for next-generation sequencing. The pattern of enrichment for 5hmC will vary depending on the species and sources of the DNA. Between 10 and 50 million reads will probably be necessary to reach saturation and to define all 5hmC-enriched regions27.
Acknowledgments
Y.H. is supported by a postdoctoral fellowship from the Leukemia and Lymphoma Society. This study was supported by US National Institutes of Health grants AI44432 and HD065812, grant RM1-01729 from the California Institute for Regenerative Medicine and Translational Research grant 6187-12 from the Leukemia and Lymphoma Society (to A.R.).
Footnotes
AUTHOR CONTRIBUTIONS A.R. conceptualized and directed the project. Y.H. and W.A.P. developed the CMS-IP method. J.A.Z.-M. contributed to optimizing the CMS-IP method.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Li Y, Sasaki H. Genomic imprinting in mammals: its life cycle, molecular mechanisms and reprogramming. Cell Res. 2011;21:466–473. doi: 10.1038/cr.2011.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson-Grusby L, et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat Genet. 2001;27:31–39. doi: 10.1038/83730. [DOI] [PubMed] [Google Scholar]
- 3.Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet. 1998;20:116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- 4.Bourc’his D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431:96–99. doi: 10.1038/nature02886. [DOI] [PubMed] [Google Scholar]
- 5.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ito S, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munzel M, et al. Quantification of the sixth DNA base hydroxymethylcytosine in the brain. Angew Chem Int Ed Engl. 2010;49:5375–5377. doi: 10.1002/anie.201002033. [DOI] [PubMed] [Google Scholar]
- 9.Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–950. doi: 10.1158/0008-5472.CAN-06-3123. [DOI] [PubMed] [Google Scholar]
- 11.Valinluck V, et al. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2) Nucleic Acids Res. 2004;32:4100–4108. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin SG, Kadam S, Pfeifer GP. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res. 2010;38:e125. doi: 10.1093/nar/gkq223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfaffeneder T, et al. The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew Chem Int Ed Engl. 2011;50:7008–7012. doi: 10.1002/anie.201103899. [DOI] [PubMed] [Google Scholar]
- 14.He YF, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koh KP, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bacher U, et al. Mutations of the TET2 and CBL genes: novel molecular markers in myeloid malignancies. Ann Hematol. 2010;89:643–652. doi: 10.1007/s00277-010-0920-6. [DOI] [PubMed] [Google Scholar]
- 18.Quivoron C, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 19.Moran-Crusio K, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118:4509–4518. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ko M, et al. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci USA. 2011;108:14566–14571. doi: 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ko M, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haffner MC, et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget. 2011;2:627–637. doi: 10.18632/oncotarget.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wossidlo M, et al. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun. 2011;2:241. doi: 10.1038/ncomms1240. [DOI] [PubMed] [Google Scholar]
- 25.Gu TP, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–610. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- 26.Pastor WA, Huang Y, Henderson HR, Agarwal S, Rao A. The GLIB technique for mapping 5-hydroxymethylcytosine. Nat Protoc. 2012;7:1909–1917. doi: 10.1038/nprot.2012.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pastor WA, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayatsu H, Wataya Y, Kai K, Iida S. Reaction of sodium bisulfite with uracil, cytosine, and their derivatives. Biochemistry. 1970;9:2858–2865. doi: 10.1021/bi00816a016. [DOI] [PubMed] [Google Scholar]
- 29.Hayatsu H, Shiragami M. Reaction of bisulfite with the 5-hydroxymethyl group in pyrimidines and in phage DNAs. Biochemistry. 1979;18:632–637. doi: 10.1021/bi00571a013. [DOI] [PubMed] [Google Scholar]
- 30.Frommer M, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Y, et al. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS ONE. 2010;5:e8888. doi: 10.1371/journal.pone.0008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weber M, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 33.Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- 34.Wu H, et al. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679–684. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams K, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ficz G, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 37.Xu Y, et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell. 2011;42:451–464. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matarese F, Carrillo-de Santa Pau E, Stunnenberg HG. 5-Hydroxymethylcytosine: a new kid on the epigenetic block? Mol Syst Biol. 2011;7:562. doi: 10.1038/msb.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Booth MJ, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–937. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- 40.Song CX, et al. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nat Methods. 2012;9:75–77. doi: 10.1038/nmeth.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu M, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the Mammalian genome. Cell. 2012;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kornberg SR, Zimmerman SB, Kornberg A. Glucosylation of deoxyribonucleic acid by enzymes from bacteriophage-infected Escherichia coli. J Biol Chem. 1961;236:1487–1493. [PubMed] [Google Scholar]
- 43.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]