

Abstract
NSMase2 is associated to the plasma membrane, whereas ASMase is predominantly lysosomal; both hydrolyze sphingomyelin (SM) to ceramide and phosphocholine. Although SM accumulated in both ASMase-/- and fro/fro (NSMase2-/-) fibroblasts, the reduction of ceramides was more dramatic in fro/fro cells. ASMase mRNA, protein and enzyme activity were substantially elevated in fro/fro fibroblasts. In contrast, NSMase2 activity was unaffected in ASMase-/- fibroblasts. ASMase-/- cells showed normal cell cycling whereas fro/fro cells grew slowly and were arrested in G1/G0 and could be corrected by transfection with smpd3 gene. This suggests two distinct subcellular pathways for SM catabolism with distinct functions.
Keywords: NSMase2, ASMase, coordination
1. Introduction
Two sphingomyelinases are major players in the regulation of sphingomyelin and ceramide levels in the cell and both have been associated with a range of biological actions from proliferation to cell death [1-5]. We took advantage of the availability of two animal models to examine possible evidence for cross regulation of the lysosomal (ASMase) and non-lysosomal (palmitoylated plasma membrane, NSMase2) forms of the enzyme. The ASMase knockout mouse generated by Horinouchi et al [6] shows a progressive neurodegenerative course and death occurs at 8 months of age. Pathological studies revealed cerebella atrophy, loss of Purkinje cells and foam cells in reticuloendothelial organs typical of a lysosomal lipid storage disease. Subsequent studies revealed an altered response to radiation-induced stress but it was concluded that ASMase is non-essential for enhancing the inflammatory signals generated by cytokines [7]. The fro/fro mouse arose from a spontaneous deletion of Smpd3 encoding NSMase2 and displays profound skeletal dysplasia, suggesting that NSMase2 plays a direct role in osteoblast development and bone formation [8]. A previous study on experimentally-derived Smpd3-/- mice concluded that the loss of NSMase2 activity led to pituitary hormone deficiency in the central nervous system and triggered a series of events that led to a reduction in circulating IGF-1 causing the observed skeletal dysplasia [9]. In general the two NSMase2-/- mouse models show similar skeletal and growth abnormalities and the phenotype is totally different from that of a lysosomal storage disease. Northern blot analysis revealed widespread expression of ASMase but that the highest level of expression of smpd3 (NSMase2) was in brain and bone, with lesser expression in skin, sternum, and trachea and very little in heart, kidney, liver, lung and spleen [10, 11]. Although there are many studies on both SMases, there have been no investigations on coordination between these two SMases. Therefore we used ASMase-/- and fro/fro mouse skin fibroblasts as a culture system to study the coordination of these two enzymes, and their effect on cell proliferation.
2. Material and methods
2.1 Standards and Reagents
Sph, DHSph, a 17-carbon analog of Sph (C17-Sph), S1P, DHS1P, a 17-carbon analog of S1P (C17-S1P), N-myristoyl (14:0), N-palmitoyl (16:0), N-oleoyl (18:1), N-stearoyl (18:0), N-arachidoyl (20:0), N-nervonoyl (24:1), N-lignoceroyl (24:0) sphingosines (ceramides (Cer)), N-palmitoyl (16:0), N-oleoyl (18:1), N-stearoyl (18:0), N-arachidoyl (20:4), N-behenoyl (22:0), N-nervonoyl (24:1), and N-lignoceroyl (24:0), and TopFluor-sphingomyelin(SM) were obtained from Avanti Polar Lipids (Alabaster, AL). The nonphosphorylated lipid standards were dissolved in methanol, whereas the sphingoid base phosphates were dissolved in methanol containing a trace amount of concentrated HCl and were stored at −20°C. [3H]Palmitic acid (43 Ci/mmol) was purchased from New England Nuclear (Boston, MA). Silica gel high performance thin-layer chromatography (HPTLC) plates were obtained from Whatman (Clifton, NJ, USA) and the protein assay kit was obtained from Bio-Rad Laboratories (Hercules, CA, USA). Chloroform, methanol, and acetic acid used for HPTLC were of ACS grade and obtained from Fisher Scientific (Pittsburgh, PA, USA). Hexamethylumbelliferyl (HMU)-phosphorylcholine was purchased from Moscerdam Substrates (Amsterdam, The Netherlands). RT-PCR primers for Smpd1, Smpd3 and 18s rRNA was obtained from Integrated DNA Technologies (Coralville, IA, USA). The antibodies for ASMase and NSMase2 were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA), beta-actin antibody and secondary antibodies anti-mouse, anti-goat and anti-rabbit were purchased from Sigma (St. Louis, MO, USA).
2.2 Generation of fro/fro (NSMase2-/-) and ASMase-/- mutant mouse fibroblast cell lines
fro/fro mouse has been maintained by breeding heterozygous (+/fro) mice together. To avoid postnatal lethality associated with fro maintenance, fro was transferred to C57BL/6N and C3H/HeN background by repeated backcrosses (>10) of heterozygous (+/fro) mice. Resulting congenic strains were maintained by breeding heterozygous mice together [12]. ASMase knock out mice (C57BL/6 strain) were obtained by propagation of heterozygous breeding pairs [13]. Mouse skin fibroblasts were isolated from sterilized ears of newly euthanized 2-week-old postnatal mice. Tissue fragments were plated in 20% bovine FCS in DMEM containing Gentamycin and colonies of fibroblasts were trypsinized and sub-cultured. The Vector pCMV 3X- FLAG (Sigma) was used to transfect Smpd3 in the cultured skin fibroblasts. Stable clones were selected on the basis of neomycin (G418-sulfate) resistance [11].
2.3 Sphingomyelinase assays
ASMase and NSMase activity was determined with the fluorimetric substrate hexamethylumbelliferyl (HMU)-phosphorylcholine as described previously [11]. Briefly, cells were harvested and washed with phosphate-buffered saline (PBS), the pellets were resuspended and lysed in 25 mM Tris– HCl, 150 mM NaCl, and 1% Triton X-100, pH 7.4. For ASMase activity assay, 50mg protein were mixed with the fluorogenic substrate HMU-phosphorylcholine, the incubation was carried out at pH 4.5, 150 mM sodium acetate buffer containing 1 mM EDTA to block any NSMase activity. NSMase2 assay was carried out at pH 7.4 reaction buffer (10mM MgCl2, 100mM Tris-HCl, pH7.4 and 0.1% Triton X-100) included 5mM fresh DTT to inhibit any ASMase activity. The HMU released was followed fluorometrically in a 96-well FLX microplate reader. The enzyme activity was calculated from the slope of the graph of intrinsic fluorescence plotted against time and standardized to mg of protein.
2.4 RT-PCR for NSMase2 and ASMase mRNA expression in cells
Total RNA was extracted from cultured fibroblasts using a Qiagen total RNA extract kit (Valencia, CA, USA). RT-PCR was executed with a RT-PCR one step kit (Qiagen) and primer pairs specific to mouse Smpd1 by using forward: 5’-TGGTTCTGGCTCTGTTTGACTCCA-3’ and backward: 5’-TCAGCTGATCTTGGCGAGACTGTT-3’; primer pairs specific to mouse Smpd3 by using forward: 5’-ACATCGATTCTCCCACCAACACCT-3’ and backward: 5’-AATTCGCACAATGCAGCTGTCCTC-3’and 18s rRNA as control, by using Forward: 5’-CCAGAGCGAAAGCATTTGCCAAGA -3’ and Backward: 5’-AATCAACGCAAGCTTATGACCCGC -3’ primers. Briefly, the reaction mixture was prepared in PCR tubes according to the kit menu and put into a Perkin Elmer GeneAMP PCR System 2400 (Perkin Elmer, Waltham, MA, USA). The programming RT-PCR procedure consisted of reverse transcription (50°C for 30 min), initial PCR activation (95°C for 15 min), then 35 cycles of 94°C for 30 s, 59°C for 30 s, and 72°C for 1 min, followed by a final extension at 72°C for 10 min, annealing temperature may change according to primer Tm. The RT-PCR amplified samples were visualized on 1.2% agarose gels using ethidium bromide.
2.5 Western blot analysis
Cell lysates from fibroblast cell cultures were subjected to SDS-gel electrophoresis. Proteins were transferred to Immobilon-P membranes (Millipore, Bedford, MA), and Western blotting carried out with antibodies according to the manufacturer’s instructions. Positive bands were detected with a chemiluminescence kit from Fisher Scientific (Pittsburgh, PA). The Western blot bands were scanned with a Bio-Rad ChemiDoc XRS (Bio-Rad, Hercules, CA).
2.6 Labeling study with TopFluor-sphingomyelin(SM)
TopFluor-SM was used at a concentration of 1 mg/mL in ethanol. It was added to cell cultures to achieve a final concentration of 0.6μg/mL. Typical labeling experiments were carried out in 100-mm Petri dishes, containing 8 mL of serum-free medium, for 24 h. Cells (3 × 106/100 mm plate) were harvested and washed three times with phosphate-buffered saline, and the pellet was extracted with methanol (1 mL). Following addition of chloroform (2 mL) and 0.01 N HCl (0.6 mL) the upper phase was discarded, the lower phase was evaporated to dryness under nitrogen, and lipids applied to HPTLC plates prior to quantification of sphingolipid fluorescence.
2.7 Analysis of lipid synthesis by HPTLC
Cells were labeled with [3H] palmitate and lipids were extracted as described previously [12]. Typical labeling experiments were carried out in 100-mm Petri dishes containing 8ml of serum-free medium for 24 h. Cells (3 × 106/100 mm plate) were harvested and washed three times with phosphate buffered saline, lipids were extracted by Chloroform-Methanol-water (2:1:0.6 v/v) partition and samples were subjected to alkaline methanolysis to remove phosphoglycerides. Lipids were applied to HPTLC plates (10 × 10 cm; LHP-K TLC plates, Whatman, Inc) and developed in chloroform: methanol: glacial acetic acid: water (70: 25: 8.8: 4.5 v/v). Lipids were visualized in iodine vapors then scraped off for quantification. Mouse brain (0.1 g) lipids were extracted by 1.2 ml H2O, 2ml methanol and 4ml chloroform, and then subjected to alkaline methanolysis to remove phosphoglycerides. Lipids were applied to HPTLC plates and developed in chloroform: methanol:glacial acetic acid:water (70:25:8.8:4.5 v/v), sphingomyelin was visualized by charring with 10% CuSO4–8% H2SO4 [14].
2.8 Lipid extraction and sample preparation for lipid quantification by LC/MS/MS and analysis of Sphingoid Bases, Sphingoid Base 1-Phosphates and Ceramides
Cellular lipids were extracted by a modified Bligh and Dyer [15] procedure with the use of 0.1N HCl for phase separation.C17-S1P (40 pmol), C17-Sph (30 pmol), and 17:0-Cer (30 pmol) were used as internal standards and were added during the initial step of lipid extraction. The extracted lipids were dissolved in methanol/chloroform (4:1, v/v), and aliquots were taken to determine the total phospholipid content as described previously [16]. Samples were concentrated under a stream of nitrogen, redissolved in methanol, transferred to autosampler vials, and subjected to consecutive LC/MS/MS analysis of sphingoid bases, ceramides, and sphingoid base 1-phosphates.
Analyses of sphingolipids were performed by combined LC/MS/MS using an automated Agilent 1100 series liquid chromatograph and autosampler (Agilent Technologies, Wilmington, DE) coupled to an API4000 Q-trap hybrid triple quadrupole linear ion trap mass spectrometer (Applied Biosystems, Foster City, CA) equipped with a TurboIonSpray ionization source. Sphingolipids were ionized via electrospray ionization (ESI) with detection via multiple reactions monitoring (MRM). Analysis of sphingoid bases and the molecular species of ceramides used ESI in positive ions with MRM analysis [17]. Standard curves for each of the sphingoid bases, sphingoid base 1-phosphates and ceramides molecular species were constructed by adding increasing concentrations of the individual analyte to 30 or 40 pmol of the corresponding structural analogs used as the internal standard. Linearity and the correlation coefficients of the standard curves were obtained by a linear regression analysis. The standard curves were linear over the range of 0.0–300 pmol of each of the sphingolipid analytes with correlation coefficients (R2) > 0.98.
2.9 Gene expression profiling
RNA was extracted from fibroblasts with the Trizol and Qiagen kit, and RNA integrity was assessed with an Agilent 2100 Bioanalyzer (Agilent, Palo Alto, CA). High-quality RNA (RNA integrity number >9.0) was used for expression microarray analysis in which 2 μg of the total RNA was processed for biotin labeled target preparation and hybridization to affymetrix mouse genome 430 2.0 Genechip expression arrays according to the Genechip Expression analysis technical manual (Affymetrix, Inc). After hybridization for 16 hours at 45°C with rotating at 60RPM, arrays were washed and stained on a GeneChip Fluidics Station (Affymetrix, Inc.) and scanned using the Gene Chip Scanner 3000 7G. The CEL intensity data extracted by GCOS (Gene Chip Operating Software) were used for data analysis. Functional analysis of statistically significant gene expression changes was performed with Ingenuity Pathways Analysis (IPA, Ingenuity Systems) and DNA-Chip Analyzer (dChip) software (www.dchip.org).
2.10 Flow Cytometric Analysis of Cell Cycle
DNA content analysis was performed on cell samples using previously established flow cytometric methods [18]. Briefly, cells grown in 6cm Petri dishes were detached by using Trypsin/EDTA for 3 min at 37°C, and then washed with PBS with 1% BSA to achieve a single cell suspension. Cells were centrifuged at 200 × g for 5 min, and then resuspended in 1ml ice-cold 70% ethanol for 1 h at 4C. Ethanol-fixed cells were spun at 200 × g for 7 minutes and then washed in PBS once. The washed pellet was stained with 1ml of a PI staining solution (0.02mg Propidium Iodide (PI), 0.2mg RNase A, and 0.1% Triton-X-100 in 1ml PBS) for 30 minutes at room temperature. Cells were immediately analyzed on a Becton, Dickinson LSRII analyzer (San Jose, CA) using a 561nm excitation source and a 630/30 emission filter. Linear Area and Width PI fluorescence data was collected as well as Forward Scatter (FSC) and Side Scatter (SSC) parameters. 10,000 single cells were collected for analysis using FlowJo analysis software (Treestar, Ashland, OR). Single cells were gated using PI-Width and Area parameters as well as FSC and SSC gates. DNA content measurements were calculated using FlowJo’s cell cycle platform and the Watson (Pragmatic) model. Percent of cells in G1, S-phase, and G2 was recorded for each sample.
2.11 Statistical Analysis
The results of LC-MS/MS analyses are from duplicate experiments run in triplicate. Statistical analyses were performed by Student’s t-test, and results were considered statistically significant when p < 0.05.
3. Results
3.1 ASMase is increased in fro/fro fibroblasts
We measured both NSMase2 and ASMase activities in fibroblasts and found NSMase2 activity to be even less than ASMase activity under optimum assay conditions (Fig.1A), NSMase2 activities in fro/fro and ASMase in ASM-/- fibroblasts were depleted as expected (Fig.1B and 1D). We also found that ASMase activity was increased in fro/fro fibroblasts, In vitro assay showed a 7-fold increase compared to control (Fig.1C), whereas NSMase2 activity was not significantly changed in ASMase-/- fibroblasts (Fig.1E). Transfection of Smpd3 into fro/fro fibroblast decreased the activity of ASMase to control levels (Fig.1F). RT-PCR and Western blot revealed increased expression of Smpd1 and ASMase in fro/fro fibroblasts, and transfection of Smpd3 into fro/fro fibroblast decreased the expression of ASMase (Fig.2A and 2C). In contrast, there was no change in expression of Smpd3 in ASMase-/- fibroblasts (Fig.2B). Gene chip expression assay supported the findings (Table 1).
Fig.1. Increased activity and expression of ASMase in fro/fro fibroblast.
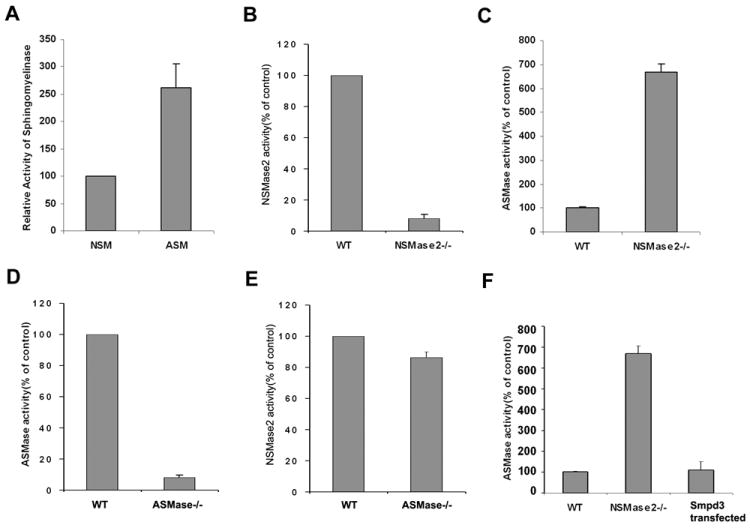
A. NSMase2 activity is minor compared to ASMase activity in mouse fibroblasts.
B. NSMase2 activity is depleted in fro/fro mouse fibroblasts.
C. Increased activity of ASMase in fro/fro mouse fibroblasts compared to its control.
D. ASMase activity is depleted in ASMase knock out mouse fibroblasts.
E. NSMase2 activity is unchanged in the ASMase knock out mouse compared to its control
F. Transfection of Smpd3 into fro/fro fibroblast decreases activity of ASMase.
SMase activity was determined by fluorometic assay, the procedures are as described in the text and results are representative of three independent experiments.
Fig.2. Increased expression of ASMase in fro/fro fibroblast.
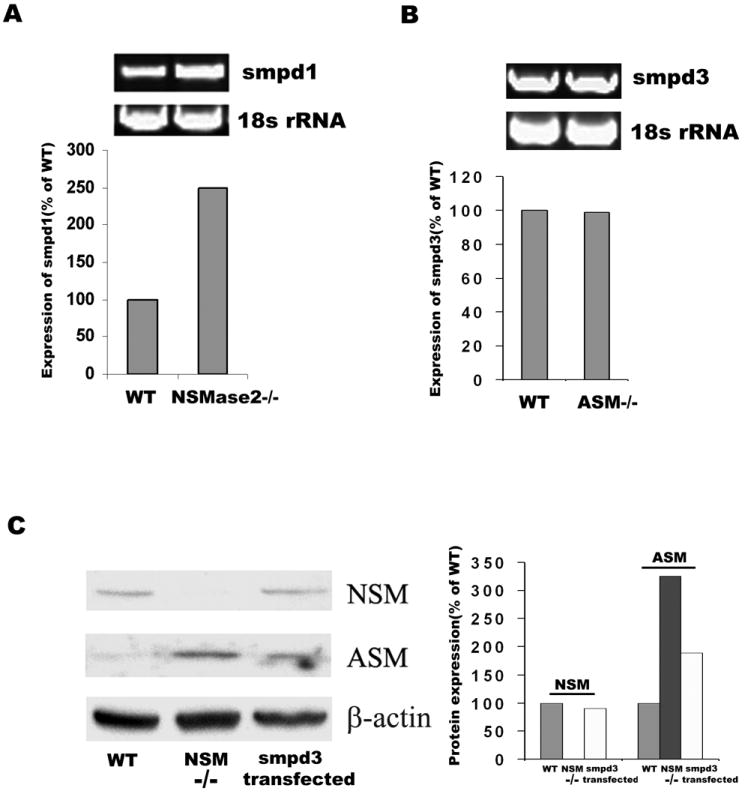
A. RT-PCR showing increased expression of Smpd1 in fro/fro fibroblasts compared to controls.
B. RT-PCR showing the same expression of Smpd3 in ASMase-/- fibroblasts compared to controls.
C. Western blot showing that the increased ASMase expression in fro/fro fibroblast can be reversed by transfection of smpd3.
Bar graphs show quantification corrected for RNA and protein, the procedures are as described in the text and results are representative of three independent experiments.
Table 1.
Affymetrix gene chip analysis showing increased expression of Smpd1 in fro/fro cells.
3.2 Regulation of sphingomyelin levels by NSMase2 and ASMase
Labeling of cells with [3H] palmitate showed accumulation of SM in both fro/fro and ASMase -/- fibroblasts (Fig.3A and 3B). However, LC/MS/MS analysis revealed that ceramide levels were about 50% in fro/fro fibroblasts (Fig.3C) compared to its control, but were less affected in ASMase -/- fibroblasts (Fig.3D). Although the expression of ASMase in terms of mRNA, protein levels and enzyme activity was increased in fro/fro fibroblasts, it did not compensate for the lack of NSMase2. The increased SM and decreased ceramides, suggested that the lysosomal localization of ASMase isolated it from the main ceramide-generating systems in fro/fro fibroblasts. LC/MS/MS analysis revealed that ceramides were reduced across the board in terms of fatty acid content and levels (Fig.3C and 3D). Since S1P is derived from ceramides through sphingosine, we also measured sphingoid bases and found a substantial reduction of S1P (reduced to 75% of control levels) in fro/fro fibroblasts (data not shown). Transfection of the fro/fro fibroblasts with Smpd3 restored the NSMase2 activity and the reduction in ceramide levels to normal (Fig.3E and 3F). LC/MS/MS analysis also showed that in fibroblasts the C16:0, C24:0 and C24:1-ceramide molecular species were the major ceramides compared to C18:0-ceramide in brain [19]. Thus the fibroblast-brain differences may be explained by differential expression of ceramide synthases [20]. In addition, analysis of fro/fro brain and calvaria revealed a 50% reduction in ceramide [11], whereas no differences in ceramides were found in ASMase-/- brain.
Fig.3. Sphingomyelin and ceramide analysis in fro/fro and ASMase-/- mouse skin fibroblasts.
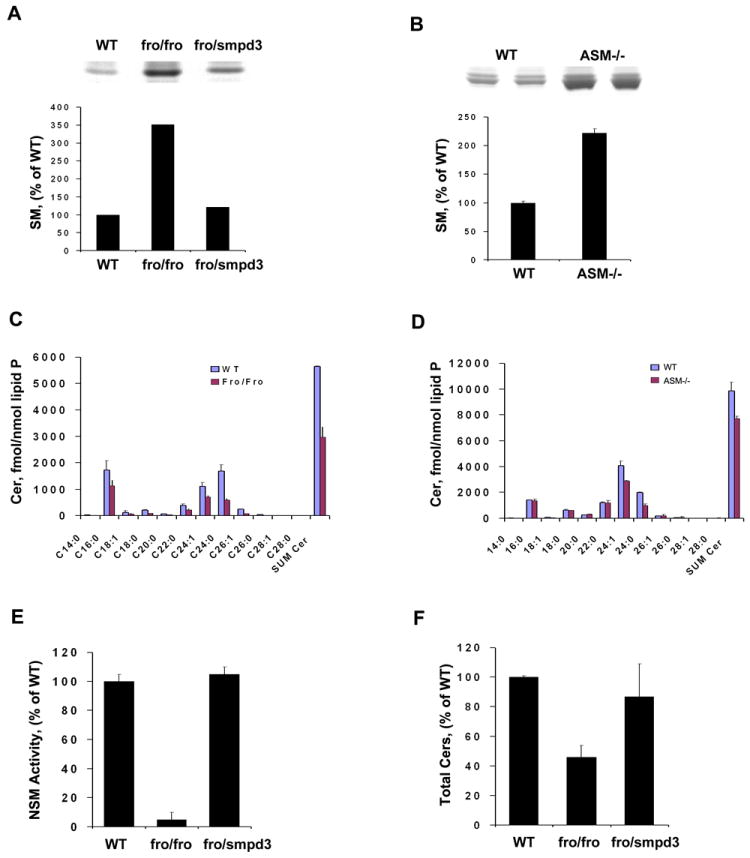
A. Autoradiogram of HPTLC of samples of normal (WT) and fro/fro [3H] palmitate-labeled sphingolipids showing increased SM in fro/fro fibroblasts. Quantification of autoradiogram showed a >3X increase of SM in fro/fro fibroblasts. Transfection of Smpd3 into fro/fro fibroblasts decreases the accumulation of SM.
B. Autoradiogram of HPTLC of samples of normal (WT) and NPD (ASM-/-) [3H] palmitate-labeled sphingolipids showing increased SM in ASMase-/- fibroblasts. Quantification showed >2X increase of SM in ASMase-/- fibroblasts.
C. Ceramide analysis by HPLC/MS/MS shows that the amount of all ceramide species was significant decreased in fro/fro compared to normal (WT) fibroblasts.
D. Ceramide analysis by HPLC/MS/MS shows that the amount of all ceramide species is less decreased in ASMase-/- compared to normal (WT) fibroblasts. C16:0, C24:0 and C24:1-ceramide molecular species were the major ceramides in mouse fibroblasts.
E and F, Recovered activity of NSMase2 and total ceramides by transfection of Smpd3 into fro/fro fibroblasts.
The procedures are as described in the text and results are representative of three independent experiments.
3.3 ASMase cannot completely compensate for NSMase2 (and vice-versa) in their respective knockout mouse fibroblasts
TopFluor-SM labeling experiments showed that SM accumulated and ceramides decreased in both fro/fro and ASMase-/- fibroblasts. The effect was less (a 75% reduction) in fro/fro cells (Lane 2) compared to 90% in ASMase-/- cells (lane 4) but clearly shows that ASMase and NSMase2 cannot compensate for each other (Fig.4).
Fig.4. SM accumulates and ceramides decrease in both fro/fro and ASMase-/- fibroblasts following loading with TopFluor-SM and HPTLC analysis.
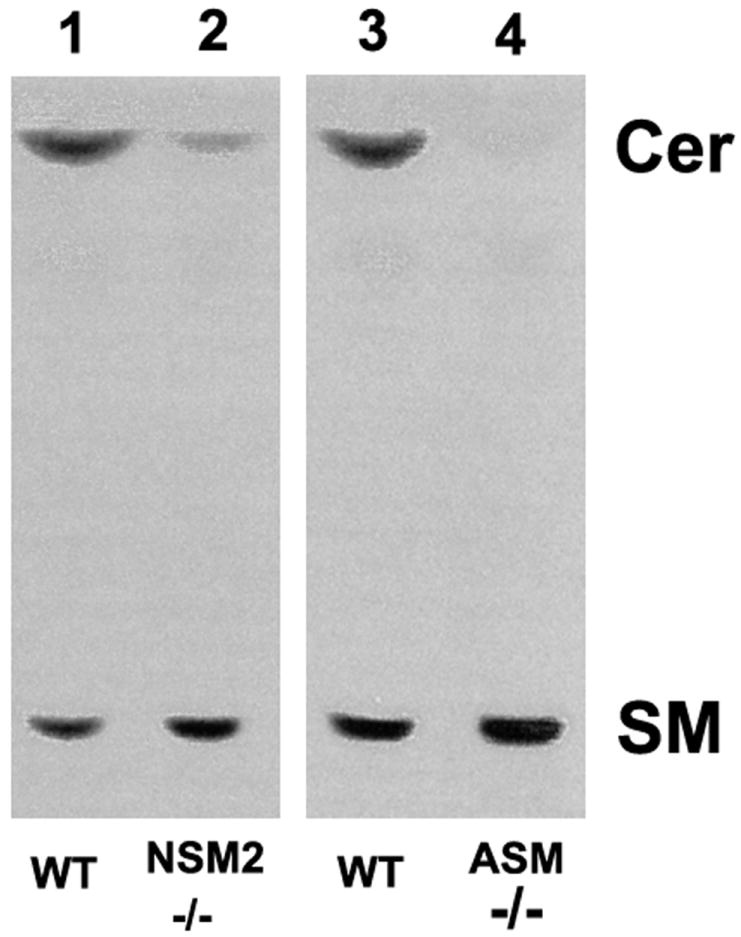
TopFluor-SM labeling of cells led to SM accumulation and ceramide decreases in both fro/fro and ASMase-/- fibroblasts compared to their controls but the effect was greater in the ASMase-/- cells.
The procedures are as described in the text and results are representative of three independent experiments.
3.4 NSMase2 and the cell cycle
The fro/fro mice showed slow growth which was mimicked in fibroblasts but was not seen in ASMase-/- fibroblasts. Cell cycle analysis by FACS showed initially that very few (only 8% compared to 42% in controls) of the fro/fro fibroblasts were in S-phase whereas 83% of cells were in G1 phase compared to only 40% in control cells (Fig.5A). This could explain why they grew slowly, had aberrant morphology and were mostly arrested in G1/Go. Cells in S-phase were increased to 35% following transfection with Smpd3 (Fig. 5A). ASMase-/- fibroblasts grew normally and were morphologically normal (Fig.5B).
Fig.5. Cell cycle abnormalities occur in fro/fro fibroblasts.

A. Cell cycle analysis of WT and fro/fro fibroblasts and the correction with Smpd3 transfection was carried out as described in the text.
B. Cell cycle analysis of WT and ASMase-/- fibroblasts showed no differences.
The procedures are as described in the text and results are representative of three independent experiments.
4. Discussion
The accumulation of SM in ASMase-/- cultured cells has been previously well documented but less attention has been paid to the role of NSMase2. NSMase 2 is one of the major intracellular regulators of ceramide levels and hydrolyzes non-lysosomal SM to ceramide but always appears to be the minor SMase by in vitro assay. However, our analysis shows that NSMase2 is involved in cell growth unlike ASMase, which could be ascribed to their working in different subcellular compartments. We now present data to support this idea. We have previously shown that stress (eg. Staurosporine or C2-ceramide) induces the translocation of NSMase2 into lipid rafts [21], and this is supported by evidence that NSMase2 is dynamically palmitoylated [22]. The fro/fro mouse (with a deletion of the part of the Smpd3 gene coding for the active site of NSMase2) has major deficits in growth and development [8, 9]. In contrast, the absence of ASMase did not affect growth rates or the cell cycle. NSMase2 absence (but not lysosomal ASMase absence) resulted in a 50% reduction in all ceramides [11], although the accumulation of SM was significant in both ASMase-/- and in fro/fro fibroblasts. Neither NSMase2 nor ASMase showed substrate specificity for a particular ceramide species (based on LC/MS/MS assay), unlike the situation with ceramide synthases [20]. NSMase2 has optimal activity at pH 7.4 and requires divalent cations such as Mg2+ or Mn2+. In contrast, ASMase has a pH optimum at pH 4.5 and is phosphate sensitive, thus ASMase and NSMase2 function in different cell compartments. ASMase undergoes an especially complex post-translation proteolytic processing [23, 24] and recent reports suggest that some ASMase could be active at the cell surface [25-27]. In addition, Aureli et al. reported that there were different patterns of SM-protein complexes in lipid rafts prepared from normal and Nieman-Pick types A disease (NPA) fibroblasts, suggesting that differences in lipid and protein compositions of cell lines determine specific lipid-protein interactions and different clustering within plasma membrane [28]. NSMase2 is activated by proteins such as FAN, and caspase 8 as well as divalent cations such as Mg2+ and Mn2+ and glutathione [29]. In contrast, required activators of ASMase include members of the Saposin family of lysosomal hydrolyase activator proteins as well as certain heat shock proteins (eg. hsp70) [30]. NSMase2 inhibitors include GW4869, whereas ASMase inhibitors include desipramine and FTY720 [31] and there is no overlap in the inhibitors.
We were surprised that the absence of NSMase2 resulted in a dramatic increase in ASMase although this did not functionally translate into a compensatory reduction of SM or an increase in ceramides back to normal levels. One possible reason is that in fro/fro SM accumulates in plasma membrane because of the lack of NSMase2 activity, leading to more SM being delivered to lysosomes through endocytic vesicles and thereby stimulating more ASMase expression. In contrast, the accumulated SM in lysosomes in ASMase-/- fibroblasts cannot be recycled to the PM, and therefore no synthesis of NSMase2 is stimulated. We have presented evidence based on [3H] palmitate labeling and HPTLC analysis that the lack of NSMase2 results in both SM accumulation and ceramide depletion in cultured fibroblasts and that this can be corrected by transfection of Smpd3 gene. In contrast, although SM also accumulates in ASMase-/- cells, the depletion of ceramide was less obvious. This difference (the relative lack of ceramide accumulation in ASMase-/- cells) was previously shown by analysis of brain lipids in the two mouse models [11]. Although the TopFluor-SM loading experiment shows that ASMase and NSMase2 activity cannot compensate for the lack of the other, and ceramides were depleted in both types of knock out cells, the LC/MS/MS analysis showed that ceramide levels in ASMase-/- fibroblasts were little decreased. This suggests that either the ceramide de novo pathway or the ceramide synthase pathway is activated in ASMase-/- fibroblasts but not in fro/fro fibroblasts. This needs to be further investigated because of the importance of SM and ceramides in many cellular and pathological processes.
Highlights.
First description of the coordination between NSMase2 and ASMase in mouse fibroblasts;
The mechanism of coordination is based on the sub-compartment localization of the two enzymes;
NSMase2 localizes mainly on the plasma membrane and is involved in cell growth, whereas ASMase has less effect on growth and is mainly lysosomal.
Acknowledgments
This work was primarily supported by USPHS Grant NS36866-37 to G.D. Mass-Spectrometric analyses were performed by Dr. Evgeny Berdyshev in the University of Chicago Core facility. The ASMase-/- mice were a generous gift from E. Schuchman (Mt. Sinai medical center, NY) and R. Wechselbaum, University of Chicago. Mouse fibroblast cultures were initiated by Sylvia Dawson and HPTLC analyses were performed by John Kilkus.
The abbreviations used are
- ASMase
acid sphingomyelinase
- Cer
ceramides
- DMEM
Dulbecco’s modified Eagle’s medium
- DMSO
dimethylsulfoxide
- fro
fragilitas ossium
- HPTLC
high performance thin-layer chromatography
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- MES
2-(N-Morpholino) ethanesulfonic acid
- NSMase2
neutral sphingomyelinase
- PM
plasma membrane
- S1P
sphingosine 1-phosphate
- SM
sphingomyelin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–50. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 2.Marchesini N, Hannun YA. Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem Cell Biol. 2004;82(1):27–44. doi: 10.1139/o03-091. [DOI] [PubMed] [Google Scholar]
- 3.Wu Bill X, Clarke Christopher J, Hannun Yusuf A. Mammalian Neutral Sphingomyelinases: Regulation and Roles in Cell Signaling Responses. 2010;12(4):320–330. doi: 10.1007/s12017-010-8120-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuchman Edward H. Acid sphingomyelinase, cell membranes and human disease: Lessons from Niemann–Pick disease. FEBS Lett. 2010;584(9):1895–1900. doi: 10.1016/j.febslet.2009.11.083. [DOI] [PubMed] [Google Scholar]
- 5.Smith Eric L, Schuchman Edward H. The unexpected role of acid sphingomyelinase in cell death and the pathophysiology of common diseases. The FASEB Journal. 2008;22:3419–3431. doi: 10.1096/fj.08-108043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horinouchi Kenichi, Erlich Shai, Perl Daniel P, Ferlinz Klaus, Bisgaier Charles L, Sandhoff Konrad, Desnick Robert J, Stewart Colin L, Schuchman Edward H. Acid sphingomyelinase deficient mice: a model of types A and B Niemann–Pick disease. Nature Genetics. 1995;10:288–293. doi: 10.1038/ng0795-288. [DOI] [PubMed] [Google Scholar]
- 7.Manthey CL, Schuchman EH. Acid sphingomyelinase-derived ceramide is not required for inflammatory cytokine signalling in murine macrophages. Cytokine. 1998;10:654–661. doi: 10.1006/cyto.1998.0344. [DOI] [PubMed] [Google Scholar]
- 8.Aubin I, Adams CP, Opsahl S, Septier D, Bishop CE, Auge N, Salvayre R, Negre-Salvayre A, Goldberg M, Guenet JL, Poirier C. A deletion in the gene encoding sphingomyelin phosphodiesterase 3 (Smpd3) results in osteogenesis and dentinogenesis imperfecta in the mouse. Nat Genet. 2005;37:803–805. doi: 10.1038/ng1603. [DOI] [PubMed] [Google Scholar]
- 9.Stoffel W, Jenke B, Block B, Zumbansen M, Koebke J. Neutral sphingomyelinase 2 (smpd3) in the control of postnatal growth and development. Proc Natl Acad Sci U S A. 2005;102:4554–4559. doi: 10.1073/pnas.0406380102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khavandgar Z, Poirier C, Clarke CJ, Li J, Wang N, McKee MD, Hannun YA, Murshed M. A cell-autonomous requirement for neutral sphingomyelinase 2 in bone mineralization. J Cell Biol. 2011;194(2):277–89. doi: 10.1083/jcb.201102051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qin J, Berdyshev E, Poirer C, Schwartz NB, Dawson G. Neutral sphingomyelinase 2 deficiency increases hyaluronan synthesis by up-regulation of Hyaluronan synthase 2 through decreased ceramide production and activation of Akt. J Biol Chem. 2012;287(17):13620–32. doi: 10.1074/jbc.M111.304857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poirier C, Berdyshev E, Dimitropoulou C, Bogatcheva N, Biddinger P, Verin A. Neutral sphingomyelinase 2 deficiency is associated with lung anomalies similar to emphysema. Mammalian Genome. 2012 doi: 10.1007/s00335-012-9419-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horinouchi K, Erlich S, Perl DP, Ferlinz K, Bisgaier CL, Sandhoff K, Desnick RJ, Stewart CL, Schuchman EH. Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat Genet. 1995;10(3):288–93. doi: 10.1038/ng0795-288. [DOI] [PubMed] [Google Scholar]
- 14.Kilkus J, Goswami R, Testai FD, Dawson G. Ceramide in rafts (detergent-insoluble fraction) mediates cell death in neurotumor cell lines. J Neurosci Res. 2003;72(1):65–75. doi: 10.1002/jnr.10549. [DOI] [PubMed] [Google Scholar]
- 15.Bligh E, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 16.Vaskovsky VE, Kostetsky EY, Vasendin IM. A universal reagent for phospholipid analysis. J Chromatogr. 1975;114:129–141. doi: 10.1016/s0021-9673(00)85249-8. [DOI] [PubMed] [Google Scholar]
- 17.Berdyshev EV, Gorshkova IA, Usatyuk P, Zhao Y, Saatian B, Hubbard W, Natarajan V. De novo biosynthesis of dihydrosphingosine-1-phosphate by sphingosine kinase 1 in mammalian cells. 2006;18(10):1779–1792. doi: 10.1016/j.cellsig.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Darzynkiewicz Z, Juan G. Current Protocols in Cytometry. 1997:7.5.1–7.5.24. doi: 10.1002/0471142956.cy0705s00. [DOI] [PubMed] [Google Scholar]
- 19.Qin J, Berdyshev E, Goya J, Natarajan V, Dawson G. Neurons and oligodendrocytes recycle sphingosine 1-phosphate to ceramide. Significance for apoptosis and multiple sclerosis. J Biol Chem. 2010;285:14134–14143. doi: 10.1074/jbc.M109.076810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pewzner-Jung Y, Ben-Dor S, Futerman AH. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)? Insights into the regulation of ceramide synthesis. J Biol Chem. 2006;281:25001–5. doi: 10.1074/jbc.R600010200. [DOI] [PubMed] [Google Scholar]
- 21.Goswami R, Ahmed M, Kilkus J, Han T, Dawson SA, Dawson G. Differential regulation of ceramide in lipid-rich microdomains (rafts): antagonistic role of palmitoyl:protein thioesterase and neutral sphingomyelinase 2. J Neurosci Res. 2005;81(2):208–17. doi: 10.1002/jnr.20549. [DOI] [PubMed] [Google Scholar]
- 22.Tani M, Hannun YA. Neutral sphingomyelinase 2 is palmitoylated on multiple cysteine residues. Role of palmitoylation in subcellular localization. J Biol Chem. 2007;282(13):10047–56. doi: 10.1074/jbc.M611249200. [DOI] [PubMed] [Google Scholar]
- 23.Jenkins RW, Idkowiak-Baldys J, Simbari F, Canals D, Roddy P, Riner CD, Clarke CJ, Hannun YA. A novel mechanism of lysosomal acid sphingomyelinase maturation: requirement for carboxyl-terminal proteolytic processing. J Biol Chem. 2011;286(5):3777–88. doi: 10.1074/jbc.M110.155234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jenkins RW, Canals D, Idkowiak-Baldys J, Simbari F, Roddy P, Perry DM, Kitatani K, Luberto C, Hannun YA. Regulated secretion of acid sphingomyelinase. J Biol Chem. 2010;285:35706–18. doi: 10.1074/jbc.M110.125609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galvan C, Camoletto PG, Cristofani F, Van Veldhoven PP, Ledesma MD. Anomalous surface distribution of glycosyl phosphatidyl inositol-anchored proteins in neurons lacking acid sphingomyelinase. Mol Biol Cell. 2008;19(2):509–22. doi: 10.1091/mbc.E07-05-0439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scandroglio F, Venkata JK, Loberto N, Prioni S, Schuchman EH, Chigorno V, Prinetti A, Sonnino S. Lipid content of brain, brain membrane lipid domains, and neurons from acid sphingomyelinase deficient mice. J Neurochem. 2008;107(2):329–38. doi: 10.1111/j.1471-4159.2008.05591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tam C, Idone V, Devlin C, Fernandes MC, Flannery A, He X, Schuchman E, Tabas I, Andrews NW. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J Cell Biol. 2010;189(6):1027–38. doi: 10.1083/jcb.201003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aureli M, Prioni S, Mauri L, Loberto N, Casellato R, Ciampa M, Chigorno V, Prinetti A, Sonnino S. Photoactivable sphingosine as a tool to study membrane microenvironments in cultured cells. J Lipid Res. 2010;51(4):798–808. doi: 10.1194/jlr.M001974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marchesini N, Hannun YA. Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem Cell Biol. 2004;82:27–44. doi: 10.1139/o03-091. [DOI] [PubMed] [Google Scholar]
- 30.Kirkegaard T, Roth AG, Petersen HT, Jaattela M. Hsp70 stabilizes lysosomes and reverts Niemann Pick disease-associated lysosomal pathology. Nature. 2010;463:549–554. doi: 10.1038/nature08710. [DOI] [PubMed] [Google Scholar]
- 31.Dawson G, Qin J. Gilenya (FTY720) inhibits acid sphingomyelinase by a mechanism similar to tricyclic antidepressants. Biochem Biophys Res Commun. 2011;404:321–323. doi: 10.1016/j.bbrc.2010.11.115. [DOI] [PMC free article] [PubMed] [Google Scholar]