

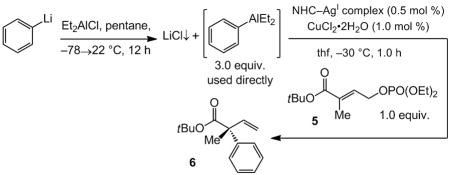
Catalytic enantioselective allylic substitution (EAS) reactions[i] are among the most versatile classes of transformations in organic chemistry: such processes convert readily available achiral substrates to enantiomerically enriched products bearing a stereogenic center adjacent to a readily functionalizable alkene. We have devised chiral amino acid-based[ii] and bidentate N-heterocyclic carbene (NHC) Cu complexes[iii] that promote EAS processes with dialkylzinc reagents and generate quaternary carbon stereogenic centers[iv] with high site- and enantioselectivity. Catalytic enantioselective Cu-free additions of alkylmagnesium halides to allylic halides,[v] as well as dialkylzinc and trialkylaluminum reagents[vi] to phosphates, have also been developed. Nonetheless, significant and compelling problems remain unaddressed. Catalytic EAS transformations involving aryl nucleophiles and which deliver quaternary carbon centers are notoriously scarce, and protocols that involve hetero-arylmetals do not exist (including those that furnish tertiary C–C bonds). The only reported cases of aryl additions by catalytic EAS that generate quaternary carbons correspond to Si-substituted alkenes and involve diarylzinc reagents,[vii] which are relatively difficult to prepare in high purity and offer only one of the two aryl units. Such considerations underline a shortcoming in the state-of-the-art: the absence of EAS methods that deliver quaternary carbon stereogenic centers through reactions with mono-arylmetal reagents, in particular those that are easily accessible and atom-economical.[i]
Herein, we introduce the first set of efficient catalytic EAS processes that involve the use of aryllithium or hetero-aryllithium reagents and generate quaternary carbon stereogenic centers (Scheme 1). Reactions proceed with exceptional site-(up to >98% SN2′) and high enantioselectivity (up to >98:2 e.r.); transformations are complete within three hours with ≤3.0 mol % of a chiral NHC–Cu complex derived from commercially available and air-stable CuCl2•2H2O.
Scheme 1.

Practical, site- and enantioselective synthesis of versatile small molecules with an allylic all-carbon quaternary stereogenic center by Cu-catalyzed allylic substitutions through the use of mono-aryl- or mono-hetero-arylmetal reagents; LG = leaving group.
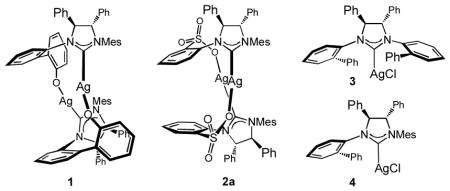
We began by developing a protocol that only requires aryllithium reagents, and decided to take advantage of the higher propensity of an aryl unit of an (aryl)dialkylaluminum to transfer to copper;[viii] reaction of an aryllithium with a commercially available and inexpensive dialkylaluminum halide would deliver the desired reagent.[ix] Thus, EAS with phenyl(diethyl)aluminum, synthesized and used in situ from phenyllithium, and allylic phosphate 5 was investigated (Table 1). The ability of four representative chiral NHC–Cu complexes, obtained from bidentate 1 and 2a[x] and monodentate 3 and 4,[xi] to promote the C–C bond forming processes was examined. Initial studies indicated that, although all reactions proceed efficiently (Ph:Et transfer = >98:2), the Cu complex derived from sulfonate-bearing 2a furnishes the highest site- and enantioselectivity.[xii] It should be noted that PhMgCl can also be used to access 6 with similar efficiency and selectivity (>98% conv., >98% SN2′, 89.5:10.5 e.r.).[xiii] Reaction of the Grignard reagent with Et2AlCl is performed in dioxane so that [xiv] residual magnesium halide (MgCl2•dioxane) can be removed.
Table 1.
Initial examination of various chiral NHC complexes.[a]
| ||||
|---|---|---|---|---|
| Entry | NHC–AgI | Ph:Et addition[b] | SN2′:SN2[b] | e.r.[c] |
| 1 | 1 | >98:2 | >98:2 | 86:14 |
| 2 | 2a | >98:2 | >98:2 | 91:9 |
| 3 | 3 | >98:2 | 56:44 | 89:11 |
| 4 | 4 | >98:2 | 83:17 | 58:42 |
Reactions were performed under N2 atmosphere; >98% conversion in all cases.
Determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures.
Determined by GLC analysis; see the Supporting Information for details.
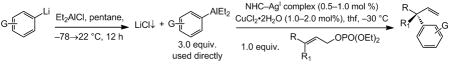
An assortment of aryllithium reagents, either purchased or prepared by well-established procedures through metal-halogen exchange with the corresponding bromides, can be used in site-selective (>98% SN2′), efficient (81–98% yield) and enantioselective reactions (Table 2).[xv] Cu-catalyzed additions with α,β-unsaturated esters (entries 1–4, Table 2) afford the EAS product in up to 91.5:8.5 e.r.; reactions with Si-substituted allylic phosphates deliver the desired allylsilanes in up to 97:3 e.r. (entries 5–6, Table 2). All transformations proceed with >98% SN2′ selectivity. The reactions in entries 5–6, affording Si-substituted quaternary carbons, are substantially more efficient than those previously reported involving diarylzincs.[iiic] For example, with (pOMeC6H4)2Zn, 2.5 mol % 2a and 24 hours of reaction time are needed (vs. 1.0 mol % and three hours needed in entry 6), and the desired product is isolated in 88:12 e.r. (vs. 97:3 e.r. with the arylaluminum reagent). Furthermore, the (CuOTf)2•C6H6 salt required for reactions with diarylzincs must be handled under rigorously inert conditions as opposed to the air-stable CuCl2•2H2O utilized here.
Table 2.
Synthesis and in situ use of arylaluminum reagents in Cu-catalyzed enantioselective allylic substitution reactions.[a]
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Aryllithium | Substrate [R; R1] | NHC–AgI; mol % | t (h) | SN2′:SN2[b] | Yield [%][c] | e.r.[d] |
| 1 | PhLi | CO2tBu; Me | 2a; 0.5 | 0.5 | >98:2 | 98 | 91:9 |
| 2 | PhLi | CO2tBu; Et | 2a; 0.5 | 3.0 | >98:2 | 96 | 91.5:8.5 |
| 3 | pOMeC6H4Li | CO2tBu; Me | 2a; 0.5 | 1.0 | >98:2 | 87 | 90.5:9.5 |
| 4 | pCF3C6H4Li | CO2tBu; Me | 1; 0.75 | 1.5 | >98:2 | 88 | 83:17 |
| 5 | PhLi | SiMe2Ph; Me | 2a; 1.0 | 3.0 | >98:2 | 81 | 96:4 |
| 6 | pOMeC6H4Li | SiMe2Ph; Me | 2a; 1.0 | 3.0 | >98:2 | 97 | 97:3 |
Reactions were performed under N2 atmosphere; >98% conversion in all cases.
Determined through analysis of 400 MHz 1H NMR spectra of unpurified mixtures.
Yields of isolated purified products.
Determined by GLC or HPLC analysis; see the Supporting Information for details.
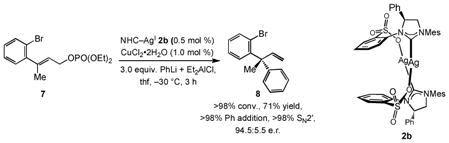
Although the products obtained through the transformations shown in Table 2 can, in principle, be accessed by reactions of the related aryl-substituted substrates with alkylmetal reagents, such processes remain largely unexplored (the only reported variant involves dialkylzinc reagents[iid]). Regardless of impending future developments, diaryl-substituted products, such as 8 [Eq. (1)], can only be accessed by an effective catalytic arylmetal addition. Another notable feature of the Cu-catalyzed EAS in Eq. (1) is that a higher enantiomeric purity is obtained with modified complex 2b; when 2a is used in the latter instance, 26% ethyl addition product is observed (vs. <2% with 2b) and 8 is formed in 90:10 e.r. Similarly, the EAS in entry 4 of Table 2 requires complex 1, since with 2a, the desired product is isolated in 78.5:21.5 e.r. (vs. 83:17 e.r.).
 |
(1) |
Next, we probed the possibility of catalytic enantioselective additions of heterocyclic units. Efficient and site-selective deprotonation of furan (Scheme 2) can be followed by subjection of the resulting 2-(furyl)lithium to Et2AlCl to afford (2-furyl)Al(Et)2. The resulting hetero-arylaluminum can be utilized without isolation or purification in a highly group-(<2% Et addition), site- (>98% SN2′) and enantioselective (>98:2 e.r.) process, converting allylic phosphate 5 to α-furyl ester 9 within one hour with 0.5 mol % 2c. When 2a is utilized in this case, 9 is obtained with equally high efficiency and site-selectivity (>98% conv., >98% SN2′, >98% furyl addition) but with diminished enantiomeric purity (90.5:9.5 vs. >98:2 e.r.).
Scheme 2.

Synthesis and in situ utilization of (2-furyl)AlEt2 in a highly group-, site- and enantioselective NHC–Cu-catalyzed enantioselective allylic substitution reaction, affording an all-carbon quaternary stereogenic center.
The data in Table 3 illustrate that furyl- and thienyl(diethyl)aluminum[xvi] participate in efficient (>98% conv. in 1.0 h with 0.5–1.5 mol % chiral NHC–AgI complex) reactions that afford products with high enantioselectivity (86.5:13.5 to >98:2 e.r.) and yield (86–98% after purification). Substrates containing an aryl substituent or those that bear a carboxylic ester (entry 11, Table 3) or a silyl unit (entries 6 and 14, Table 3) undergo notably facile and selective transformations. Reactions with 3-furyl- and 3-thienyl(diethyl)aluminum begin with readily available (bromo)heterocyclic precursors, which are converted to the derived (hetero-aryl)aluminum reagents through a simple metal-halogen exchange procedure.[xvii]
Table 3.
Synthesis and in situ use of heterocyclic aluminum reagents in Cu-catalyzed enantioselective allylic substitution reactions.[a]
| ||||||
|---|---|---|---|---|---|---|
| Entry | Heterocycle | Substrate [R] | mol % 2a | SN2′:SN2[b] | Yield [%][c] | e.r.[d] |
| 1 | 2-furyl | Ph | 0.5 | >98:2 | 93 | >98:2 |
| 2 | 2-furyl | oBrC6H4 | 0.5 | >98:2 | 86 | >98:2 |
| 3 | 2-furyl | oMeOC6H4 | 0.5 | >98:2 | 95 | >98:2 |
| 4 | 2-furyl | oMeC6H4 | 1.0 | >98:2 | 98 | 98:2 |
| 5 | 2-furyl | pNO2C6H4 | 0.5 | >98:2 | 96 | >98:2 |
| 6 | 2-furyl | SiMe2Ph | 1.0 | >98:2 | 91 | 86.5:13.5 |
| 7[e] | 3-furyl | Ph | 1.0 | >98:2 | 90 | 97:3 |
| 8 | 2-thienyl | Ph | 0.5 | >98:2 | 98 | 96:4 |
| 9 | 2-thienyl | oBrC6H4 | 1.5 | >98:2 | 98 | 98:2 |
| 10 | 2-thienyl | pNO2C6H4 | 0.5 | >98:2 | 96 | 94:6 |
| 11 | 2-thienyl | CO2tBu | 0.5 | >98:2 | 94 | 87.5:12.5 |
| 12[e] | 3-thienyl | Ph | 1.0 | >98:2 | 94 | 94:6 |
| 13[e] | 3-thienyl | oBrC6H4 | 1.0 | >98:2 | 89 | 97:3 |
| 14[e] | 3-thienyl | SiMe2Ph | 1.0 | >98:2 | 95 | 94:6 |
See Table 2.
The corresponding 3-bromofuran or 3-bromothiophene used as starting materials (treatment with nBuLi in Et2O); see the Supporting Information for experimental details.
The final stage involved reactions of allylic phosphates with two alkyl substituents, a particularly challenging class of substrates vis-à-vis obtaining exceptionally high enantioselectivities (Scheme 3). Transformations proceed readily to afford the EAS products in 88–97% yield and with >98% site selectivity. Reactions with a methyl and an n-alkyl group (cf. sporochnol and 11–12, 78.5:21.5–91:9 e.r.) are typically less enantioselective than those bearing a larger cyclohexyl unit (cf. 13–16, 91:9–98:2 e.r.). The enantiotopic faces of the alkenes in substrates in Scheme 3 are difficult to differentiate due to similar size and nearly identical electronic attributes of their alkyl substituents. Products are therefore obtained with lower enantioselectivities compared to aryl-, silyl-, and carboxylic ester-substituted alkenes in Scheme 2 or Table 3.
Scheme 3.
NHC–Cu-catalyzed enantioselective additions of aryl and hetero-aryl moieties to substrates that bear two alkyl substituents. All reactions performed under the conditions shown for EAS reaction leading to sporochnol, except for 15–16. [a] With 1.0 mol % NHC–AgI 2a and 2.0 mol % CuCl2•2H2O. [b] With 1.0 mol % NHC–AgI 2a and 2.0 mol % CuCl2•2H2O, −50 °C, 3 h.
The transformation with allylic phosphate 10 and the aryl(diethyl)aluminum bearing a p-methoxyaryl unit affords R-(−)-sporochnol. The same natural product was previously synthesized by a process involving an aryl-substituted allylic phosphate with di(2-methyl-2-pentenyl)Zn, promoted by 10 mol % of an amino acid-based chiral Cu complex.[iia] Although the earlier version is more enantioselective (91:9 e.r. vs. 78.5:21.5 e.r.), the EAS depicted in Scheme 3 is superior on several fronts. First, 1.0 mol % of the NHC–Cu complex is required for a reaction that is complete in one hour at −30 °C versus 10 mol % of the amino acid–Cu complex for 48 hours at −78 °C (lower temperature required for maximum enantioselectivity). Second, the combination of an aryllithium and commercially available Et2AlCl, which is used in situ, is considerably more practical than the preparation, purification and use of the aforementioned dialkylzinc reagent. Third, catalytic EAS with an arylmetal reagent requires allylic phosphate 10, which is easily accessed in one step from geraniol (vs. Horner-Emmons olefination and reduction of ester, followed by phosphate formation).
In brief, we address a longstanding shortcoming in an important class of enantioselective C–C bond forming reactions. The present investigations introduce efficient and highly selective Cu-catalyzed EAS reactions that provide access to numerous versatile molecules, containing aryl- or hetero-aryl-substituted quaternary carbon stereogenic centers, and which cannot be easily prepared by any alternative protocols.[xviii] Moreover, the investigations outlined herein offer additional evidence regarding the unique ability of sulfonate-bridged bidentate NHC–based metal complexes to provide an effective solution to difficult problems in catalysis.
Supplementary Material
Footnotes
The NIH (GM-47480) provided financial support. Y. L. is grateful for an AstraZeneca graduate fellowship. Mass spectrometry facilities at Boston College are supported by the NSF (CHE-0619576).
References & Footnotes
- i.For reviews on catalytic enantioselective allylic substitution (EAS) reactions with “hard” C-based nucleophiles, see: Hoveyda AH, Hird AW, Kacprzynski MA. Chem Commun. 2004:1779–1785. doi: 10.1039/b401123f.Yorimitsu H, Oshima K. Angew Chem Int Ed. 2005;44:4435–4439. doi: 10.1002/anie.200500653.Alexakis A, Bäckvall JE, Krause N, Pàmies O, Diéguez M. Chem Rev. 2008;108:2796–2823. doi: 10.1021/cr0683515.
- ii.a) Luchaco-Cullis CA, Mizutani H, Murphy KE, Hoveyda AH. Angew Chem Int Ed. 2001;40:1456–1460. doi: 10.1002/1521-3773(20010417)40:8<1456::AID-ANIE1456>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]; b) Murphy KE, Hoveyda AH. J Am Chem Soc. 2003;125:4690–4691. doi: 10.1021/ja0300618. [DOI] [PubMed] [Google Scholar]; c) Kacprzynski MA, Hoveyda AH. J Am Chem Soc. 2004;126:10676–10681. doi: 10.1021/ja0478779. [DOI] [PubMed] [Google Scholar]; d) Murphy KE, Hoveyda AH. Org Lett. 2005;7:1255–1258. doi: 10.1021/ol047331r. [DOI] [PubMed] [Google Scholar]
- iii.a) Larsen AO, Leu W, Oberhuber CN, Campbell JE, Hoveyda AH. J Am Chem Soc. 2004;126:11130–11131. doi: 10.1021/ja046245j. [DOI] [PubMed] [Google Scholar]; b) Van Veldhuizen JJ, Campbell JE, Giudici RE, Hoveyda AH. J Am Chem Soc. 2005;127:6877–6882. doi: 10.1021/ja050179j. [DOI] [PubMed] [Google Scholar]; c) Kacprzynski MA, May TL, Kazane SA, Hoveyda AH. Angew Chem Int Ed. 2007;46:4554–4558. doi: 10.1002/anie.200700841. [DOI] [PubMed] [Google Scholar]
- iv.a) Christophers J, Baro A, editors. Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis. Wiley-VCH; Weinheim: 2006. [Google Scholar]; b) Cozzi PG, Hilgraf R, Zimmermann N. Eur J Org Chem. 2007:5969–5994. [Google Scholar]
- v.For Cu-free allylic alkylations with alkylmagnesium halides, affording all-carbon quaternary stereogenic centers, see: Lee Y, Hoveyda AH. J Am Chem Soc. 2006;128:15604–15605. doi: 10.1021/ja067456m.For a closely related study with derivatives of bidentate NHC complexes originally designed in these laboratories, see: Jackowski O, Alexakis A. Angew Chem Int Ed. 2010;49:3346–3350. doi: 10.1002/anie.201000577.
- vi.For Cu-free allylic alkylation reactions promoted by NHC–ZnII and NHC–AlIII complexes, see: Lee Y, Li B, Hoveyda AH. J Am Chem Soc. 2009;131:11625–11633. doi: 10.1021/ja904654j.
- vii.a) Ref. 3c. For catalytic EAS reactions that involve arylmetals but deliver tertiary C–C bonds, see: Selim KB, Yamada K-i, Tomioka K. Chem Commun. 2008:5140–5142. doi: 10.1039/b809140d.Selim KB, Matsumoto Y, Yamada K-I, Tomioka K. Angew Chem Int Ed. 2009;48:8733–8735. doi: 10.1002/anie.200904676.Falciola CA, Alexakis A. Chem Eur J. 2008;14:10615–10627. doi: 10.1002/chem.200801309.Polet D, Rathgeb X, Falciola CA, Langlois JB, Hajjaji SE, Alexakis A. Chem Eur J. 2009;15:1205–1206. doi: 10.1002/chem.200801879.
- viii.For the more favored transfer of an aryl unit versus an alkyl group of an Al-based reagent, see: Mole T, Surtees JR. Aus J Chem. 1964;17:310–314.Bumagin NA, Ponomaryov AB, Beletskaya IP. Tetrahedron Lett. 1985;26:4819–4822.Lu BZ, Jin F, Zhang Y, Wu X, Wald SA, Senanayake CH. Org Lett. 2005;7:1465–1468. doi: 10.1021/ol0501020.Merino E, Melo RPA, Ortega-Guerra M, Ribagorda M, Carreño MC. J Org Chem. 2009;74:2824–2831. doi: 10.1021/jo900109p.Gao H, Knochel P. Synlett. 2009:1321–1325.
- ix.For use of this procedure in Cu-catalyzed enantioselective conjugate additions, see: May TL, Brown MK, Hoveyda AH. Angew Chem Int Ed. 2008;47:7358–7362. doi: 10.1002/anie.200802910.For a related study involving a relatively limited substrate range and Feringa-type phosphoramidite ligands, see: Hawner C, Li K, Cirriez V, Alexakis A. Angew Chem Int Ed. 2008;47:8211–8214. doi: 10.1002/anie.200803436. Conjugate additions with hetero-arylaluminum reagents have not been reported.
- x.For reports on the utility of sulfonate-based bidentate NHC–Cu complexes in enantioselective formation of quaternary carbon stereogenic centers, see: Brown MK, May TL, Baxter CA, Hoveyda AH. Angew Chem Int Ed. 2007;46:1097–1100. doi: 10.1002/anie.200604511.b) Ref. 3c; c) Ref. 9a; Brown MK, Hoveyda AH. J Am Chem Soc. 2008;130:12904–12906. doi: 10.1021/ja8058414.e) Ref. 6 Guzman-Martinez A, Hoveyda AH. J Am Chem Soc. 2010;132:10634–10637. doi: 10.1021/ja104254d.Gao F, McGrath KP, Lee Y, Hoveyda AH. J Am Chem Soc. 2010;132 doi: 10.1021/ja106829k. ASAP.
- xi.For C1-symmetric chiral NHC–Cu complexes and their utility in conjugate additions resulting in C–C, C–Si or C–B bonds, respectively, see: Lee K-s, Hoveyda AH. J Org Chem. 2009;74:4455–4462. doi: 10.1021/jo900589x.Lee K-s, Hoveyda AH. J Am Chem Soc. 2010;132:2898–2900. doi: 10.1021/ja910989n.O’Brien JM, Lee K-s, Hoveyda AH. J Am Chem Soc. 2010;132:10630–10633. doi: 10.1021/ja104777u.
- xii.When Me2AlCl or (iBu)2AlCl are used, alkyl (vs. aryl) transfer products are generated (12% and 10%, respectively) and 6 is formed in 73:27 and 88:12 e.r., respectively.
- xiii.When PhMgBr is used, site- and enantioselectivity remain the same but efficiency is diminished (68% vs. >98% conv. with PhMgCl).
- xiv.Metal halides can be detrimental to enantioselectivity. For example, when the reaction in entry 2 of Table 1 is performed with an added equiv. of LiCl, the desired product is obtained in ~70:30 e.r. Arylaluminum solutions are allowed to stand for 30 min. – 1.0 h. to permit precipitation of LiCl to occur prior to the supernatant being utilized. See the Supporting Information for experimental details.
- xv.Reactions with ortho-substituted arylaluminums are efficient and site-selective (>98% SN2′) but furnish products of lower enantiomeric purity. For example, when (diethyl)o-methylphenylaluminum is used with the allylic phosphate in entries 1–4 of Table 2, the EAS product is obtained in 69.5:30.5 e.r. In the corresponding conjugate addition processes, such arylaluminums give rise to improved e.r. values (see Ref. 9a).
- xvi.For use of furyl- and thienylaluminum reagents in enantioselective additions to carbonyls, see: Wu KH, Chuang DW, Chen CA, Gau HM. Chem Commun. 2008:2343–2345. doi: 10.1039/b802441c.Biradar DB, Zhou S, Gau HM. Org Lett. 2009;11:3386–3389. doi: 10.1021/ol901193q.
- xvii.As the example below indicates, EAS with N-containing heterocyclic substrates proceed readily but are hampered by non-selective initial hetero-aryl-lithium formation, affording a difficult-to-separate mixture of products. See the Supporting Information for details. Use of the pyridine-containing aluminum reagents leads to <2% conversion. Studies to improve the above transformations are in progress.
- xviii.Ni-catalyzed reactions of styrenes with ethylene furnish enantiomerically enriched products with a benzylic vinyl unit at an all-carbon quaternary stereogenic center. See: Smith CR, Lim HJ, Zhang A, RajanBabu TV. Synthesis. 2009:2089–2100. doi: 10.1055/s-0029-1216826.and references cited therein.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.