

Abstract
The p53 tumor suppressor gene is highly mutated in human cancers. Individuals who inherit one p53 mutant allele are susceptible to a wide range of tumor types, including breast cancer and sarcoma. We recently generated p53 knockout rats through gene targeting in embryonic stem cells. Here we show that rats homozygous for the null allele are prone to early onset spontaneous sarcomas and lymphoma with high incidence of metastases. Heterozygous rats are also highly predisposed to cancer, but with a delayed onset and a wider spectrum of tumor types compared with homozygotes. Importantly, up to 20% of female heterozygotes developed breast cancer and about 70% of the tumors were positive for estrogen receptor. Exposing p53-deficient rats to a low dose of the carcinogen diethylnitrosamine dramatically decreased the latency for sarcoma development and survival time compared with equivalently treated wild-type rats. These unique features make this knockout line a valuable model for investigating human malignancy and in vivo carcinogenicity of chemicals and therapeutic compounds.
Introduction
Loss or mutation of the p53 tumor suppressor gene is the most frequently observed genetic lesion in human cancers (1). Mutant p53 alleles that encode no or dysfunctional p53 protein arise both in somatic cells during sporadic human tumorigenesis and in the germline (Li–Fraumeni cancer syndrome) (2). A number of mouse models have been developed to investigate the role of p53 in mammalian tumorigenesis (3). Constitutive inactivation of p53 through gene targeting has allowed for in vivo analysis of the effects of p53 activity loss. p53-null mice have been generated by several different groups, and, despite subtle differences in the targeting strategies employed, all strains exhibit very similar phenotypes with relatively uniform tumor spectra (4–6). Among the types of tumors observed in the p53-deficient mice, lymphomas, usually of the T-cell type, were most common, although p53 is less frequently mutated in human lymphomas (7). Soft tissue sarcomas and osteosarcomas appear at a relatively lower frequency but malignant carcinomas are rarely observed in the p53-deficient mice. The spectrum of lesions differed from that predicted by the Li–Fraumeni syndrome, which has been linked to a variety of germline p53 mutations. Li–Fraumeni families are prone to various types of tumors, with breast cancer, soft tissue sarcomas, osteosarcomas, brain tumors and leukemias being identified as the most frequently observed cancers (2). Interestingly, the high rate of mammary tumor development was found in the BALB/c background but not in other mouse strains (8). A notable absence of tumors of epithelial origin in most p53-deficient mice suggests that the mouse model may not be an exact mimic of the human syndrome, possibly due to species differences.
As rats are physiologically more similar to humans than are mice, rat models are more appropriate for studying human diseases and testing the pharmacodynamics and toxicity of potential therapeutic compounds (9). Gene targeting in mouse embryonic stem (ES) cells has been used to generate various mouse strains for the study of diverse human genetic diseases and cancer, but the inability to establish germline-competent ES-cell lines from other species had prevented broader application of this technology. Recently, we developed the 3i/2i culture system that enabled the derivation of germline-competent rat ES cells and generated a p53 knockout rat via homologous recombination in rat ES cells (10). It was demonstrated that rats homozygous for a null p53 allele were developmentally normal but highly susceptible to early onset of cancer. The most frequently observed tumor type in p53-null rats was hemangiosarcoma whereas existing p53-deficient mouse models predominantly develop lymphomas (11). A similar observation was made in another p53 knockout rat generated via N-ethyl-N-nitrosourea (ENU)-mediated random mutagenesis (12). However, due to the low number of animals followed in that study, a complete tumor spectrum was not presented.
Here we report a complete assessment of tumor development in our p53-deficient rat models. We observed a more human-like tumor spectrum in our models, particularly in the p53 heterozygous rats, than what has been reported in p53-deficient mice or ENU-generated p53 knockout rats. Spontaneous breast cancer, which is frequently seen in Li–Fraumeni females but rarely in most p53-deficient mice, appear in some p53 heterozygous rats, making them a better predisposition model to mimic the cognate human syndrome. Furthermore, p53-deficient rats were extremely susceptible to carcinogen-induced tumorigenesis. Thus, p53-deficient rats provide a sensitive model system for mechanistic study of cognate human syndromes and assessment of the carcinogenic potential of novel compounds.
Materials and methods
Animals
Generation of rats lacking one or both alleles of p53 by gene targeting of DAc8 rat ES cells has been described previously (10). Rats that were p53 heterozygous were intercrossed to produce rats with no, one or two p53-null alleles. These rats were designated wild-type, heterozygotes (p53 +/-) or homozygotes (p53 -/-), respectively, and were monitored for spontaneous and carcinogen-induced tumor development. These p53-mutant rats have been deposited to the Rat Resource and Research Center in Missouri. All procedures were performed according to investigator’s protocols approved by the University of Southern California Institutional Animal Care and Use Committee.
Tumor analysis
Animals that died or were sacrificed were subjected to complete necropsy. Tumor samples were surgically removed and fixed in 10% neutral buffered formalin and processed for histological examination. The paraffin blocks were sectioned at 5 µm, and the sections were stained with hematoxylin and eosin.
Immunohistochemistry
For imunohistochemical analysis, sections were mounted on charged slides, dried overnight at 55°C and deparaffinized. Rehydrated sections were subjected to antigen unmasking in 10mM sodium citrate buffer, pH 6.0, at a subboiling temperature for 10min. Once cooled, sections were exposed to 3% hydrogen peroxide, rinsed and blocked with 5% normal goat serum. Then, tissue sections were probed with antibodies specific for the following protein targets: Vimentin (Ab-2), Pancytokeratin (Ab-3), CD3 (SP7, T-cell marker), CD31/PECAM-1 (endothelial cell marker), CD68 (Ab-3, macrophage marker), and CD79a/mb-1 (Ab-1, B-cell marker). All antibodies were purchased from Lab Vision (Thermo Scientific, Kalamazoo, MI).
Carcinogen-induced tumorigenesis
Thirteen p53 wild-type rats, 14 p53 heterozygotes and 12 p53 homozygotes were intraperitoneally injected with diethylnitrosamine (DEN) (20mg/kg body weight; Sigma–Aldrich) weekly for 5 weeks. Rats were monitored daily over the course of the experiment. Thirteen wild-type rats, 16 p53 heterozygotes and 11 p53 homozygotes not treated with the carcinogen were monitored in parallel with the treated rats. Dead animals were examined by necropsy. Liver and lung tissues were fixed and stained as described above and examined for histopathologic lesions.
Results
Spontaneous tumorigenesis in p53-deficient rats
Forty p53 homozygous and 66 heterozygous animals were examined for tumors twice each week. Although initially healthy, rats homozygous for the p53 deficiency were highly predisposed to malignancy. By the age of 2.5 months, approximately half of the homozygotes had developed tumors and almost all of these animals had died of tumors by 6 months of age. Compared with wild-type rats, the p53 heterozygotes also had a greatly accelerated rate of tumorigenesis. By 8 months of age, half of the heterozygotes had developed tumors (33 out of 66) and most rats (90.9%) that survived to 12 months of age had succumbed to tumors or death (Figure 1A). All control littermates (38 rats) with wild-type p53 alleles survived to the end of the study with the exception of two rats that were euthanized upon recognition of subcutaneous masses. Both of them had developed undifferentiated subcutaneous sarcoma on their flanks and were more than 10 months old, which is consistent with the observations of incidental tumor development in Sprague–Dawley rats of this age (13).
Fig. 1.
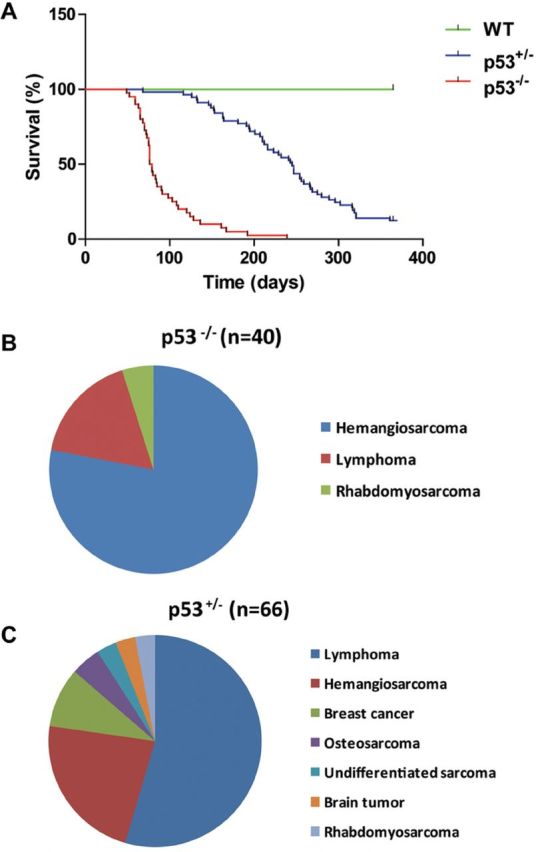
Tumor incidence in p53-deficient rats. (A) Survival curves of wild-type, p53 heterozygous (p53+/-) and p53 homozygous (p53-/-) rats. Forty p53-/- homozygous rats, 66 p53+/- heterozygous rats and 38 wild-type littermate controls were monitored twice weekly for the development of tumors over a period of 365 days. Moribund rats or rats with obvious tumors were euthanized, necropsied and the tumor tissue was examined by histopathology. Kaplan–Meier analysis, P < 0.0001. (B and C) Pie charts of the tumor types identified in homozygous (B) and heterozygous (C) mutant animals. Numbers in parentheses indicate the total number of tumors analysed.
Histopathological examination revealed a significant difference in tumor spectra between the p53 homozygous and heterozygous rats (Figure 1B and 1C). Almost 80% of p53 homozygotes developed liver hemangiosarcoma and a large proportion thereof displayed pulmonary metastases. These hemangiosarcomas stained positive for factor VIIIra by immunohistochemistry, confirming an endothelial origin of these neoplastic cells (Figure 2A and 2B). Lymphoma, a dominant tumor type observed in p53 homozygous mice, was found in 18% of homozygous mutant rats, and extensive CD3 or CD79a immunoreactivity indicated the lymphomas were of either T- or B-cell origin. One animal with hemangiosarcoma also had a lymphoma in its thymus. No carcinomas were observed among the homozygotes.
Fig. 2.
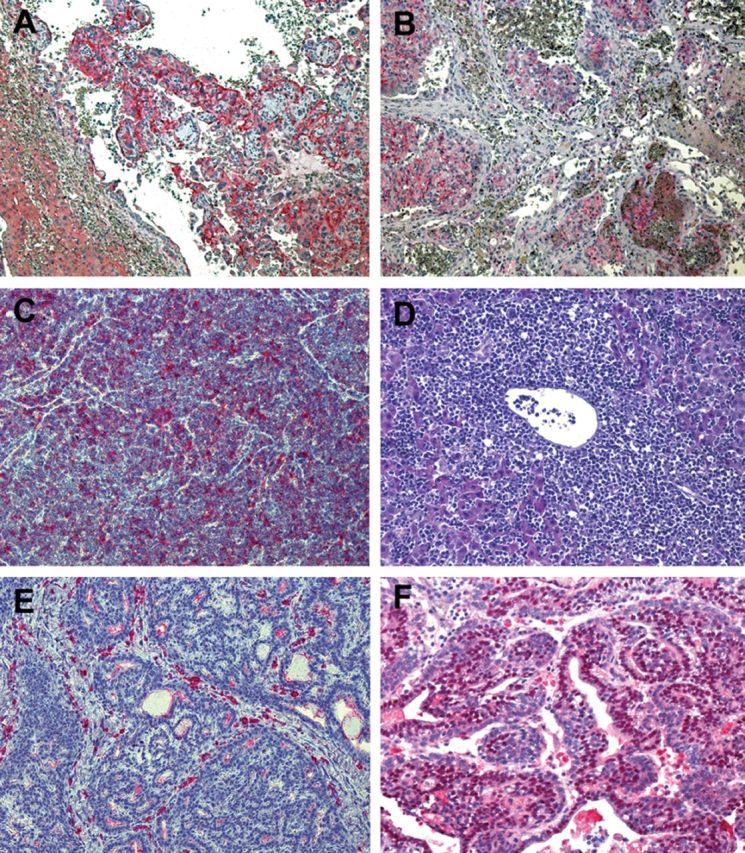
Histopathology of representative tumors from p53-deficient rats. (A) Liver hemangiosarcoma stained with endothelium marker CD31/PECAM-1 showing tumor tissue of endothelial origin and large blood-filled spaces. (B) Immunoreactivity of CD31/PECAM-1 in intravascular pulmonary metastasis of liver hemangiosarcoma. (C) Thymic lymphoma from a p53 heterozygous rat with monotonous tumor cells positive for T-cell marker CD3. (D) Extensive infiltration of malignant lymphocytes into the liver of a rat with thymic lymphoma. (E) Ductal breast carcinoma in a heterozygous p53-mutant rat is stained negative for estrogen receptor. (F) Breast carcinoma showing strong nuclear estrogen receptor immunopositivity.
In contrast, the heterozygous rats showed a much higher rate of development of lymphoma than sarcoma, mainly of T-cell origin, accounting for 55% of the tumor incidence in heterozygous rats (Figure 2C). Enlarged spleens and livers in rats with lymphoma were frequently observed as a result of diffuse infiltration by malignant lymphocytes (Figure 2D). Interestingly, although lymphoma was the predominant type of spontaneous tumor in heterozygous rats, it was often observed in rats older than 7 months of age. About 25% of the heterozygotes developed hemangiosarcoma. Interestingly, among the tumors diagnosed in the heterozygous rats, 6 breast cancers were found in 31 female animals, and 4 of them showed immunoreactivity for estrogen receptor α(Figure 2E and F). Several osteosarcomas could be classified based on their histological appearance showing osteocytes within lacunae. Rhabdomyosarcomas that arose from the muscles of the legs and pelvis were positive for MyoD-1 (a skeleton marker) immunostaining and locally invasive. A large tumor was found in the region of the left adrenal gland in one heterozygous rat. The tumor cells were relatively uniform and organized along thin fibrovascular septa, as is typically observed in masses of endocrine origin. The animal also presented with hemangiosarcoma in its liver. The breast and endocrine cancers in combination with the hard and soft tissue sarcomas represent a spectrum of cancers similar to those of Li–Fraumeni syndrome in humans (14). Three heterozygous rats that died or were euthanized due to hind limb paralysis had no evidence of cancer based on macroscopic and histopathologic examination of tissues.
Carcinogen-induced tumorigenesis
To determine whether germline reduction of p53 gene dosage confers higher sensitivity of rats to carcinogen, animals of all three p53 genotypes were exposed to the carcinogen DEN, which is a particularly potent inducer of liver hemangiosarcoma and hepatocellular carcinoma (HCC). Low dose levels of DEN (20mg/kg) were administered weekly for 5 weeks to 13 wild-type rats, 14 heterozygotes and 12 homozygotes, beginning at 6 weeks of age. The animals were observed until moribund. The p53-deficient rats were dramatically more susceptible to the effects of DEN than were wild-type controls (Figure 3A). All homozygous rats treated with DEN died of liver hemangiosarcoma within 3 months of initial exposure. The untreated homozygotes survived significantly longer, although most of them developed spontaneous hemangiosarcoma at the end of the observation period (Figure 3B). Heterozygous rats also displayed significantly greater susceptibility to DEN-induced hemangiosarcoma and shorter survival time than did untreated controls. Only 4 of 16 untreated heterzygotes displayed obvious liver hemangiosarcoma, whereas 9 of 14 DEN-treated heterzygotes died of the disease (Figure 3C). Interestingly, only the latency of hemangiosarcoma development was significantly reduced in DEN-treated p53-deficient rats, consistent with the selective carcinogenic effects of DEN. In contrast, none of the wild-type rats developed liver hemangiosarcoma during the observation period of DEN treatment. With the dose of 20mg/ml and 5-week treatment protocol, which efficiently induced liver hemangiosarcoma, no HCCs were found in the three p53 genotypes at the end of the observation period.
Fig. 3.
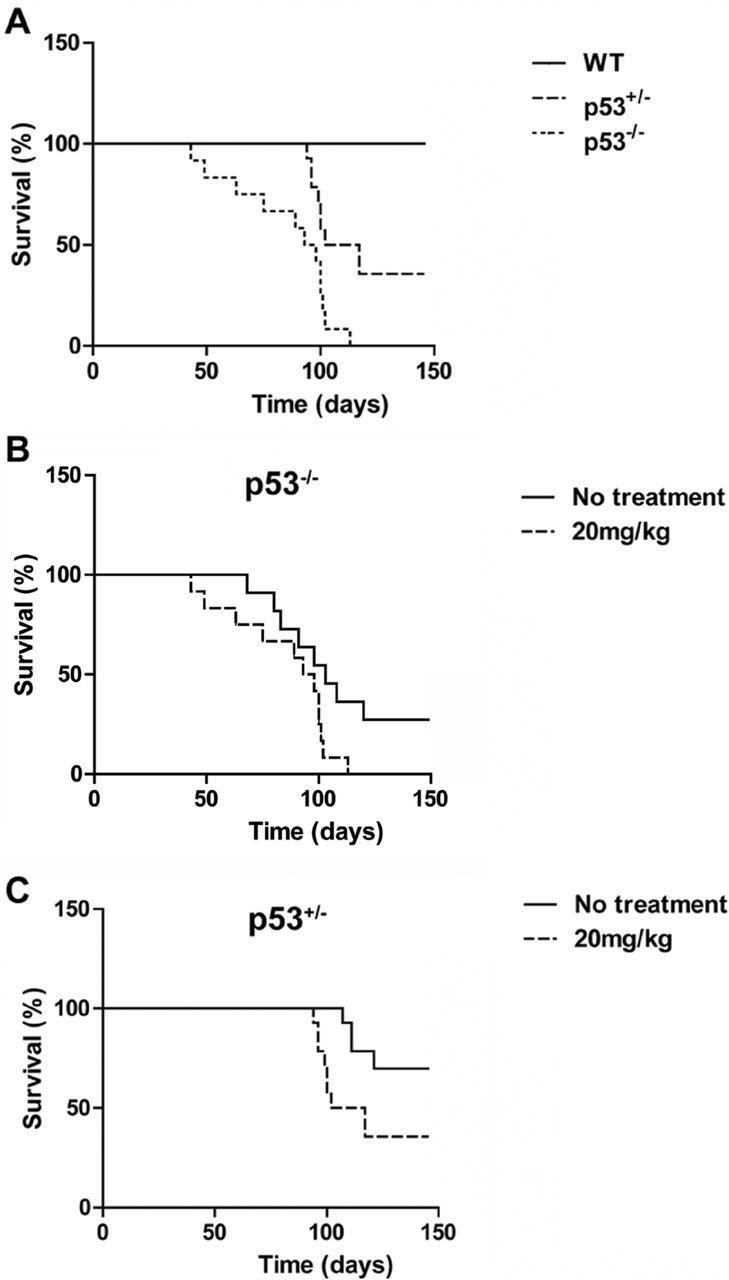
Tumor development in carcinogen-treated p53-deficient rats. (A) 13 wild-type, 14 heterozygous and 12 homozygous rats were intraperitoneally administered with DEN (20mg/kg) for 5 weeks. Comparison of the survival times of the three groups revealed an obvious difference between wild-type and p53-deficient animals. Kaplan–Meier analysis, P < 0.0001. (B) Comparison of the survival times between non-treated and DEN-treated p53 homozygous rats. Kaplan–Meier analysis, P < 0.05. (C) Comparison of the survival times between non-treated and DEN-treated p53 heterozygous rats. Kaplan–Meier analysis, P < 0.01.
The major cause of death in rats with hemangiosarcoma is tumor rupture and hemorrhagic shock. Multiple and large space-occupying hemangiosarcomas were found in both homozygous and heterozygous animals. Their body cavities were often filled with uncoagulated blood and lung metastasis was frequently evident. DEN-treated heterozygous liver displayed toxic lesions consisting of fatty change, coagulative necrosis, anisokayosis of hepatocyte nuclei, chronic hepatic inflammation and fibrosis. In contrast, the wild-type rats equivalently treated with DEN showed only a similar degree of liver toxicity with no evidence of hemangiosarcoma development.
Discussion
The rat is the preferred model organism in many fields of biomedicine, including cancer research, due to its having a physiopathology similarity to that of humans. Although mouse knockout models have been extremely powerful tools for identifying important cancer-related genes, there are discrepancies between the human disease phenotypes and those observed in mouse models. For example, mouse Apc knockouts mostly develop tumors in the small intestine whereas humans carrying Apc mutation suffer from familial colon cancer. Interestingly, rats with Apc deficiency develop colon cancer (15). Furthermore, mouse models that lack the same gene but in a different strain background display important differences, emphasizing the need for complementary mammalian mutant models in different species to enable in vivo phenotypic comparison. The higher tolerance of rats to genetic modification enables researchers to knockout genes that used to cause embryonic lethality in mice. For example, although knockout of the tumor suppressor gene Brca2 was limited by low viability in mice, the Brca2 -/- rats are viable and the vast majority survive past 1 year of age (16). With their relatively large size, which facilitates experimental and surgical interventions, these genetically modified rats may serve as more physiologically appropriate models for the study of human cancer biology.
Most techniques for genetic manipulation in mice, including homologous recombination and conditional mutations, depend upon the culture and manipulation of ES cells. Before the development of germline-competent rat ES-cell lines in 2008 (17), several technologies had already been devised to enable generation of rat models of human disease, including chemical mutagenesis using ENU, mobile DNA technology and nuclease-based technology. Despite the high mutagenicity of ENU and transposon-based mobile DNA technology, mapping mutations responsible for a particular phenotype and distinguishing neutral mutations from causative mutations are typically difficult and time-consuming (18,19). The development of nuclease technology with the use of Zinc finger nucleases and Transcription Activator-Like Effector Nucleases enabled the transition from random to targeted mutagenesis in generating knockout rats. However, validating constructs encoding specific Zinc finger nucleases, as well as systematically archiving the mutant lines, remains a challenge. By using a specific culture medium containing 3 or 2 differentiation inhibitors (3i or 2i medium), we recently showed that true pluripotent rat ES cells could be isolated and propagated in vitro. Furthermore, successful gene targeting by homologous recombination in these cells results in the generation of a targeted p53 gene knockout rat (10). Gene targeting in ES cells via homologous recombination offers the unique advantage of a low rate of non-specific mutation in the genome, which seems to occur with relatively high frequency in Zinc finger nuclease-targeted animals and even more in ENU-mutagenesis, in which more than 1000 mutations per genome need to be introduced in order to identify the gene of interest (20).
Similar to p53 knockout mice, our rat model exhibited accelerated cancer rates and developed a large number of sarcomas and lymphomas. Interestingly, whereas p53 homozygous mice predominantly develop lymphoma, our p53 homozygous rats predominantly developed meta stasizing hemangiosarcoma, similar to the ENU-mutated p53 homozygous rats. The majority of spontaneous tumors developed in our heterozygous rats were lymphomas, whereas osteosarcomas predominated in p53 heterozygous mice. Lymphoma is infrequently observed in ENU-mutated p53 heterozygous rats, possibly due to the low numbers of animals followed in that study (9 versus 62 in this study). In addition, when compared with p53 knockout mouse models, heterozygous rats had a more-rapid rate of tumor onset. These differences reflect the species-specific phenotypes in different rodent models. The tumor spectrum of the p53 knockout mice is similar to the incidence of spontaneous tumors in mice. Accordingly, in Harlan Sprague–Dawley and Wistar Han rats, the common spontaneous tumors are lymphoma, sarcoma and mammary tumors, which is consistent with the tumor spectrum observed in our p53 knockout rat models (21,22). A unique feature of our model is that the tumors originated from diverse tissues, including epithelial organs. As breast tumors are important component tumors of Li–Fraumeni syndrome but are infrequently observed in p53 knockout mice, the p53 knockout rats might serve as a better model for this disease.
The delayed latency for tumorigenesis in p53 heterozygous rats suggested that they might be used as a platform to assess the carcinogenic potential of chemicals. We tested their response to a low, chronic dosing of the carcinogen DEN. Compared with another hepatic carcinogen, dimethylnitrosamine, which induces high incidences of liver hemangiosarcoma, DEN mainly causes HCC when high doses (70mg/kg) were chronically administered to rats. Treatment of three genotypes of rats with a low dose of DEN (20mg/kg) failed to induce HCC development during the observation period. Instead, p53 heterozygous animals showed a dramatically accelerated development of liver hemangiosarcoma, whereas none of the wild-type rats displayed an observable tumor at the end of the study. These results indicate that the effects of a carcinogen could be readily monitored against p53-deficient background of spontaneous tumors and that these rats might be a sensitive test animal for some carcinogenesis procedures.
In summary, our results demonstrate that the p53-deficient rats are highly cancer prone, which is compatible with existing mouse models, as well as rat models developed with alternative targeting techniques in this species. Thanks to a wealth of detailed physiological and pharmacological phenotypic knowledge for rats, the value of p53 knockout rats extends beyond analysis of their particular tumor phenotypes to mapping of these traits to signaling contexts. These rats can be bred to other transgenic rat strains to examine the cooperative effects of p53 deficiency and other oncogenic mutations which might not have been fully appreciated in knockout mouse models.
Funding
National Institutes of Health (R01OD010926 to Q.-L.Y).
Acknowledgements
We thank Gohar Saribekyan for technical assistance.
Glossary
Abbreviations:
- DEN
diethylnitrosamine
- ENU
N-ethyl-N-nitrosourea
- ES
embryonic stem
- HCC
hepatocellular carcinoma
Footnotes
Conflict of Interest Statement: None declared.
References
- 1. Hollstein M., et al. (1991). p53 mutations in human cancers. Science 253 49–53 [DOI] [PubMed] [Google Scholar]
- 2. Malkin D., et al. (1990). Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250 1233–1238 [DOI] [PubMed] [Google Scholar]
- 3. Donehower L.A., et al. (2009). 20 years studying p53 functions in genetically engineered mice. Nat. Rev. Cancer 9 831–841 [DOI] [PubMed] [Google Scholar]
- 4. Donehower L.A., et al. (1992). Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356 215–221 [DOI] [PubMed] [Google Scholar]
- 5. Harvey M., et al. (1993). Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat. Genet. 5 225–229 [DOI] [PubMed] [Google Scholar]
- 6. Jacks T., et al. (1994). Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 4 1–7 [DOI] [PubMed] [Google Scholar]
- 7. Krug U., et al. (2002). Tumor suppressor genes in normal and malignant hematopoiesis. Oncogene 21 3475–3495 [DOI] [PubMed] [Google Scholar]
- 8. Kuperwasser C., et al. (2000). Development of spontaneous mammary tumors in BALB/c p53 heterozygous mice. A model for Li-Fraumeni syndrome. Am. J. Pathol. 157 2151–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abbott A. (2004). Laboratory animals: the Renaissance rat. Nature 428 464–466 [DOI] [PubMed] [Google Scholar]
- 10. Tong C., et al. (2010). Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature 467 211–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang G., et al. (2011). Beyond knockout rats: new insights into finer genome manipulation in rats. Cell Cycle 10 1059–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van B.R., et al. (2011). Homozygous and heterozygous p53 knockout rats develop metastasizing sarcomas with high frequency Am. J. Pathol. 179 1616–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ikezaki S., et al. (2011). Natural occurrence of neoplastic lesions in young sprague-dawley rats. J. Toxicol. Pathol. 24 37–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olivier M., et al. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. (2010);2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Amos-Landgraf J.M., et al. (2007). A target-selected Apc-mutant rat kindred enhances the modeling of familial human colon cancer. Proc. Natl. Acad. Sci. U.S.A. 104 4036–4041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cotroneo M.S., et al. (2007). Characterizing a rat Brca2 knockout model. Oncogene 26 1626–1635 [DOI] [PubMed] [Google Scholar]
- 17. Li P., et al. (2008). Germline competent embryonic stem cells derived from rat blastocysts. Cell 135 1299–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Izsvák Z., et al. (2010). Generating knockout rats by transposon mutagenesis in spermatogonial stem cells. Nat. Methods 7 443–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen Y., et al. (2000). Genotype-based screen for ENU-induced mutations in mouse embryonic stem cells. Nat. Genet. 24 314–317 [DOI] [PubMed] [Google Scholar]
- 20. Wienholds E., et al. (2003). Efficient target-selected mutagenesis in zebrafish. Genome Res. 13 2700–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dinse G.E., et al. (2010). Comparison of NTP historical control tumor incidence rates in female Harlan Sprague Dawley and Fischer 344/N Rats. Toxicol. Pathol. 38 765–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Son W.C., et al. (2010). Profile of early occurring spontaneous tumors in Han Wistar rats. Toxicol. Pathol. 38 292–296 [DOI] [PubMed] [Google Scholar]